Prediction of B-cell linear epitopes with a combination of support vector machine classification and amino acid propensity identification
Hsin-Wei Wang1, Ya-Chi Lin1, Tun-Wen Pai1,2§, Hao-Teng Chang3,4,5§
1Department of Computer Science and Engineering, 2Center of Excellence for Marine
Bioenvironment and Biotechnology, National Taiwan Ocean University, Keelung, Taiwan, R.O.C.
3Graduate Institute of Molecular Systems Biomedicine, 4Graduate Institute of Clinical
Medical Science, 5Graduate Institute of Basic Medical Science & Ph.D. Program for Aging, China Medical University, Taichung, Taiwan, R.O.C.
§Corresponding author: Dr. Hao-Teng Chang, Graduate Institute of Molecular
Systems Biomedicine, College of Medicine, China Medical University, No. 91,
Hsueh-Shih Road, Taichung, 40402, Taiwan, R.O.C. TEL:+886-4-22052121 ext 7721,
FAX:+886-4-22333641, E-mail: [email protected]
§Corresponding author: Dr. Tun-Wen Pai, Department of Computer Science and
Engineering & Center of Excellence for Marine Bioenvironment and Biotechnology, National Taiwan Ocean University, No. 2, Peining Road, Keelung, 20224, Taiwan, R.O.C. TEL: +886-2-24622192 ex. 6618, FAX: +886-2-24623249, E-mail:
Keywords: Linear epitope, antigenicity, support vector machine, machine learning,
Abstract
Epitopes are antigenic determinants that are useful because they induce B cell
antibody production and stimulate T cell activation. Bioinformatics can enable rapid, efficient prediction of potential epitopes. Here, we designed a novel B-cell linear epitope prediction system called LEPS, Linear Epitope Prediction by Propensities and
Support Vector Machine, that combined physico-chemical propensity identification
and support vector machine (SVM) classification. We tested the LEPS on four datasets: AntiJen, HIV, a newly generated PC, and AHP, a combination of these three datasets. Peptides with globally or locally high physico-chemical propensities were first identified as primitive linear epitope (LE) candidates. Then, candidates were classified with the SVM based on the unique features of amino acid segments. This reduced the number of predicted epitopes and enhanced the positive prediction value (PPV). Compared to four other well-known LE prediction systems, the LEPS
achieved the highest accuracy (72.52%), specificity (84.22%), PPV (32.07%), and Matthews correlation coefficient (10.36%). The LEPS is freely available for academic use at http://LEPS.cs.ntou.edu.tw.
Introduction
Epitopes, also called antigenic determinants, are clusters of amino acid segments located on the surfaces of an antigen. Epitopes can elicit the immune response and are recognized by specific antibodies [1]. Basically, B-cell epitopes are categorized into two types: linear and conformational. Linear epitopes (LEs) are composed of contiguous amino acid residues within a continuous stretch of a primary protein sequence. Conformational epitopes (CEs) consist of amino acids that are dispersed among discontinuous regions, but become aggregated on the protein surface [2, 3]. In general, over 90% of B-cell epitopes are discontinuous [4, 5]; thus, CEs play critical roles in biological and biomedical applications, including the prevention and
neutralization of pathogen infections, and the design of therapeutic drugs. However, the prediction and identification of CEs within a protein depend on resolved three-dimensional structural information. One major, generally accepted concept is that
conformational epitopes cannot be properly formed without binding to a corresponding antibody [6]. Therefore, antigen-antibody co-crystallographic
information is a major concern in CE prediction. On the other hand, because CEs are discontinuous epitopes, it is difficult to design a peptide that forms the same
conformation as the predicted CE. Thus, CEs that are predicted by computational analysis may not be verifiable in biochemical experiments, except with the co-crystallographic approach. Although B-cell LEs occupy a small part of the entire epitope group, they are important in biochemistry [7], virology [8], immunology [9], and vaccine research [10]. Therefore, research and development of accurate
computational approaches for LE prediction remains a critical challenge in
bioinformatics and computational biology [6]. Most published B-cell LE predictors have been based on the characteristics of amino acids, like hydrophobicity, surface accessibility, mobility, protrusion area, physico-chemical properties, antigenicity, and pocket characteristics [1, 3, 11-16]. For example, BcePred [16], BEPITOPE [17], PEOPLE [11], VaxiJen [18], and LEP [12] are bioinformatics tool that use various mathematical approaches to predict LEs according to the physico-chemical
propensities of amino acids. Nevertheless, in 2005, Blythe and Flower led a group that evaluated the physico-chemical propensities of amino acids to predict LEs in proteins; they reported that even the best physico-chemical propensity scales available
instead of using the antigenicity scale alone, LE prediction may be improved by integration with other computational approaches.
Several machine learning computational methods have been applied to improve the accuracy of LE prediction. For example, BepiPred combined a hydrophilicity scale with a hidden Markov model [20]; BCPred [21] and FBCPred [22] employed SVM with a subsequent kernel; Söllner and Mayer utilized a molecular operating
environment with the decision tree and nearest neighbour approaches [6]. However, these machine learning approaches were mostly set to predict peptides of fixed lengths. It is difficult to analyze true LEs, because they generally range from 8-20 amino acid residues in length [11, 23-25]. Epitopes with fixed lengths are not typically sufficient to represent the whole region of antigenic determinants. To
overcome the drawbacks of training and/or predicting fixed length epitopes, ABCPred used two artificial neural network methods, the feed-forward network and the
recurrent neural network, for the prediction of B-cell LEs [26]. Both networks were used with different window lengths from 10 to 20 amino acids and a two-residue interval.
Although bioinformatists have expended great effort on developing LE predictors, there remains much room for improvement. Theoretically, an epitope identified by experimental immunological or biochemical methods must possess biological antigenicity that can induce antibody production in animals. However, when computational skills are used for the prediction, some experimentally identified epitopes could be missed or ignored. This generated the interesting study of how to retrieve the unpredictable epitopes and enhance their antigenicity score in silico. In 2008, LEP was developed for predicting LEs based on physico-chemical
propensities combined with a mathematical morphology approach. LEP could retrieve some of the LEs that were locally embedded in the noise signals of the antigenic index [12]. We reasoned that prediction accuracies could be further improved, and retain the advantage of variable length conditions, by combining the LEP with machine learning technologies.
As mentioned above, the machine learning methods used in previous LE prediction methods were often trained to predict epitopes with fixed lengths. Chen’s study showed that the frequencies of occurrence for some amino acid pairs in the epitope dataset were significantly higher than in non-epitope datasets, or vice versa [23]. We noticed this important statistical feature and applied it to enhance the performance of
LE prediction systems. Hence, in order to explore the statistical advantages of verified epitopes and retain the antigenic characteristics of candidate peptides, we decided to extend the concept of amino acid pairs from Chen’s study, which only considered peptides with 2 residues.
In this study, we developed a novel B-cell LE prediction system called LEPS (Linear
Epitope Prediction by Propensities and Support Vector Machine). We adopted the
library for SVM (LIBSVM) tool and trained it to recognize features of amino acid segments (AASs) with lengths from 2 to 4 residues. Then, SVM was used to
characterize those patterns as epitope and non-epitope clusters [27]. Accordingly, the LEPS approach first performed physico-chemical propensities and mathematical morphology approaches, and then used the AAS features to cluster the predicted LE candidates and remove the less probable LEs.
Materials and Methods
Testing datasets and Predictors
Four datasets were used in this study. The AntiJen dataset was recommended at an international meeting sponsored by the National Institute for Allergy and Infectious Disease [6] and contained 171 protein sequences with 691 verified, non-overlapping epitopes [19]. The HIV dataset was a collection of the antigenic
determinants located on 10 HIV proteins with 54 non-overlapping, verified epitopes [28]. The PC dataset, generated in this study, was a collection of 12 protein sequences with 98 non-overlapping, verified epitopes (Table 1). In order to balance out the variation of each dataset in quantity and antigen diversity, these three datasets were merged into one, comprehensive dataset called the “AHP dataset”. These datasets were analyzed with different LE predictors, including the BepiPred [20], ABCPred [26], BCPred [21], and FBCPred [22], to compare performances with that of the LEPS developed here.
System flow
The proposed system was divided into three main steps (Fig. 1a). The first step retrieved primitive epitope candidates from a query protein sequence with LEP [12], which was developed in our previous work and was used with the default settings.
improve prediction accuracies. In the final step, the predicted epitope residues were highlighted in the query sequence and visualized in a predicted structure. The virtual structure was generated from Modeller 9.9, based on homologous protein structure modeling approaches [29].
Training datasets and SVM model
The process of training the SVM model comprised two major steps (Fig. 1b). The first step (step 1b) evaluated the statistical characteristics that determined the frequencies of occurrence of AASs with various lengths from an independent B-cell epitope dataset (Bcipep [30]) and a non-epitope dataset (Chen [23]). The second step (step 2b) produced a SVM model that recognized the epitopes and non-epitopes of the Chen dataset based on the statistical features derived from step 1b.
The Bcipep dataset comprised 1230 experimentally verified, B-cell, and
non-redundant LEs with lengths that ranged from 3 to 56 residues that were identified in over 1000 antigen proteins. This dataset was used in step 1b to analyze the statistical characteristics associated with the frequencies of occurrence of AASs of 2 to 4 residues in length that represented epitopes.
The Chen dataset contained 872 epitopes and 872 epitopes. All epitopes and non-epitopes within this dataset were restricted to a length of 20 residues. These verified epitopes were retrieved from the Bcipep dataset by applying a ‘‘truncation-extension treatment’’. That is, when the length of an LE was longer than 20 residues, an equal number of superfluous residues were truncated from both the N- and C- termini to preserve the central 20 residues. Conversely, when the length of an LE was shorter than 20 residues, an equal number of residues were added to both the N- and C- termini until the epitope comprised 20 residues. On the other hand, the 872 non-epitopes were generated by randomly selecting peptide segments from the Swiss-Prot database [31], with the stipulation that none was the same as any of the 872 epitopes. The 872 non-epitopes were used to analyze the statistical characteristics of AASs for non-epitopes in step 1b. After determining the statistical features that were associated with frequencies of occurrence, the proposed system applied these features (step 2b) to produce a SVM model in a 5-fold cross-validation on the Chen dataset.
For LE verification, we considered the statistical features to be AASs of 2 (AAS ), 3 2 (AAS ), and 4 (3 AAS ) residues in length for both epitopes and non-epitopes. For 4
2
AAS , 400 possible combinations of residue pairs were analyzed for occurence
frequencies within both the epitope and non-epitope datasets. The epitope index (Epidex ) of the ii2 th pattern (
2
i
AAS ) was calculated by taking logrithm value of the
ratio of the number of AAS among all epitopes i2 AASs compared to the same ratio in 2 the non-epitope AASs group with the following equation: 2
2 2 2 2 2 log i i i ( 1, 2,...400) i i i i f f Epidex i f f
where fi2 and fi2 were the numbers of AAS in the epitope and non-epitope i2 datasets, respectively, and
2 i i f
and 2 i i f
denoted the total number of 2i
AAS in
the corresponding dataset. Finally, the values of Epidex were normalized to the i2 range of [0, 1] to avoid dominance of any individual Epidex in the classifier i2 learning processes.
There were a total of 8000 and 160,000 possible combinations for AAS and 3 AAS , 4 respectively. A large portion of AAS or 3 AAS did not appear in the non-epitope 4 dataset; this would cause a problem, because it could lead to a zero in the denominator. Hence, the definitions of Epidex and i3
4
i
Epidex were modified from the definition for
2
i
Epidex , and the corresponding epitope indexes for 3
AAS and 4
AAS were defined as
follows:
l l l
i i i i
Epidex f
f ,
where l was equal to 3 or 4. Again, the values of Epidex and i3
4
i
Epidex were
normalized to the range of [0, 1].
SVM features and model selection
In this study, we adopted the SVM as a learning method to classify epitope and non-epitope peptides. We employed the open source LIBSVM toolbox for executing this
classification. In LIBSVM, each instance in the training set possessed one target value (class label) and several features (attributes). In the testing set, only the features were required for each instance. The objective of SVM was to generate a model from the training set that facilitated the prediction of the target value of each instance in the testing set. In this study, a peptide corresponded to an instance and the target value (1 or -1) represented whether that peptide was an epitope. Each peptide contained three feature values based on Epidex , i2
3
i
Epidex , and 4
i
Epidex . For example, a 20-mer
peptide was decomposed into 19 AAS subsegments, and the corresponding epitope i2 index of this peptide was obtained by taking the average of 19 Epidex from the i2 corresponding AAS . Similarly, the feature values of i2 Epidex and i3 Epidex could be i4 obtained by calculating the averages of 18 Epidex and 17 i3
4
i
Epidex subsegments,
respectively.
The Chen dataset was used to construct a SVM model based on three feature values and the target values of each epitope and non-epitope. There were four common kernel functions provided by LIBSVM, including linear, polynomial, radial basis function (RBF), and sigmoid. We examined these four kernel functions with a 5-fold cross-validation. The training dataset was equally divided into 5 different subsets; four of the subsets were used for training the model and the last one was used for testing the model. These processes were repeated five times with each individual subset used as the testing subset. Here, the RBF kernel was selected as the default kernel function, because it provided the best cross-validation accuracy with the training data. Subsequently, the RBF kernel function was applied to train the whole testing dataset for constructing the final SVM classifier in the LEPS.
Performance measurement
To evaluate the performance of the LEPS at the level of the amino acid residue, five indicators were used to measure effectiveness at the default settings. These
indicators were: (1) sensitivity (SEN), defined as the percentage of epitopes that were correctly predicted as epitopes; (2) specificity (SPE), defined as the percentage of non-epitopes that were correctly predicted as non-epitopes; (3) positive predictive
value (PPV), defined as the probability that a predicted epitope was, in fact, an
and (5) Matthews correlation coefficient (MCC), which was a measure of the predictive performance that incorporated both SEN and SPE into a single value between -1 and +1 [26]. These parameters were calculated with the following equations: (1) Sensitivity TP TP FN (2) Specificity TN TN FP (3) Accuracy TP TN TP FP TN FN (4) PPV TP TP FP (5) MCC TP TN FP FN (TP FP)(TP FN)(TN FP)(TN FN)
where TP represented the true positive; TN, the true negative; FP, the false positive; and FN, the false negative.
Results and Discussion
A new linear epitope dataset: PC
The new dataset, called the PC dataset (collected by Pai and Chang),
contained 12 sequences that did not overlap with other datasets. It was generated and analyzed in this study. The experimental epitopes in the PC dataset were identified with the peptide scan methodology, a conventional method for epitope determination. The average length of the identified epitopes in the PC dataset was 18.9 residues. This was considered a practical length for an epitope to be used in peptide vaccine
development or antibody generation. The average epitope lengths in the HIV and AntiJen datasets were 26.4 and 16.3 residues, respectively. All sequences in the PC dataset were analyzed with the LEPS, and the predicted and experimentally verified epitopes are listed in Table 1.
The performance of LEPS
The epitope information collected from the PC, AntiJen, and HIV datasets were utilized to verify the performance of LEPS. The PC dataset was described in the previous section. The original AntiJen dataset comprised 3619 epitopes, of which
3168 were found in the Swiss-Port database. As in our previous report, we
regenerated the original AntiJen dataset by removing the repeated epitopes [12]. The HIV dataset focused on one infectious pathogen and was recognized as a useful tool in the field of HIV immunology [28]. The AHP dataset combined these three datasets to balance the variations in each dataset including variations in epitope length and the physico-chemical properties of antigens. With these 4 datasets, we compared the performance of five LE predictors, including LEPS, BepiPred [20], ABCPred [26], BCPred [21], and FBCPred [22].
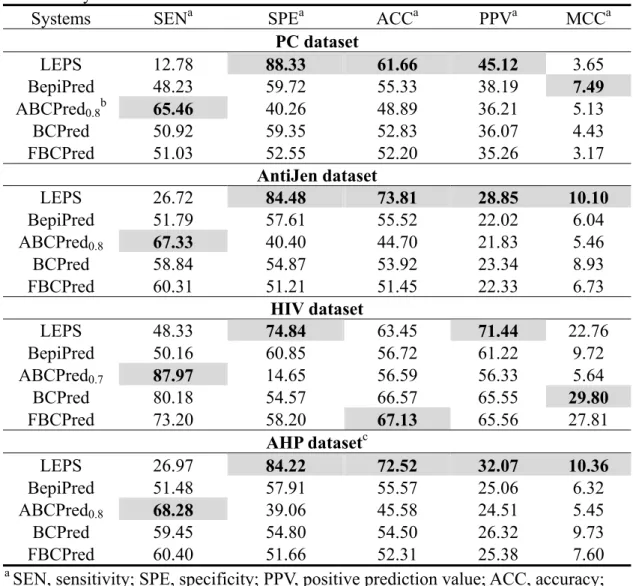
As expected, LEPS provided favorable results in all four datasets (Fig. 2). Table 2 shows that LEPS displayed the best specificity (SPE), with values of 88.33%, 84.48%, 74.84%, and 84.22% in the PC, AntiJen, HIV, and AHP datasets, respectively.
Moreover, LEPS showed the best PPVs, with values of 45.12%, 28.85%, 71.44%, and 32.07% in the PC, AntiJen, HIV, and AHP datasets, respectively. The PPV indicated the rate of identifying real epitopes among all positive predicted candidates. It is one of the most important factors in conducting vaccine development. Reduction of the false positive candidates can improve the effectiveness and efficiency of identifying the real epitopes. Therefore, the LEPS will outperform the other predictors in terms of biological experiment cost-effectiveness. In the field of computational science,
prediction accuracy is one of the most concerned factors for system evaluation. Except in the HIV dataset, LEPS displayed the best ACCs, with values of 61.66%, 73.81%, and 72.52% for the PC, AntiJen, and AHP datasets, respectively. These results showed that LEPS displayed excellent performance for LE prediction. The LEPS also showed the best performance in the MCC for the AntiJen and AHP datasets (10.10% and 10.36%), and the MCC was only a little lower (22.76%) than BCPred (29.80%) and FBCPred (27.81%) for the HIV dataset. Taken together, LEPS
displayed excellent performance in SPE and PPVs for all four datasets; it also showed the best or equivalent ACCs for all datasets. However, it showed relatively low SEN compared to the other predictors, mainly due to less number of predicted LEs.
The LEPS platform
The LEPS provides a user-friendly interface for biologists to predict linear epitope candidates (Fig. 3a). LEPS will accept either FASTA format or text, and the default parameters were set as indicated. In this system, several physicochemical propensities can be dynamically modified by users, including secondary structures, hydropathy,
surface accessibility, flexibility, polarity, and other factors. The scanning window size for each parameter is also adjustable. After executing the prediction, the overall antigenicity of the query protein and the predicted LE candidates are displayed. For example, Fig. 3b shows the LEs in HIV integrase predicted by LEPS. Seventeen candidates were initially predicted by LEP based on the global and local distributions of antigenicity. These candidates were further filtered by SVM selection, with only 9 remaining candidates. Within these 9 epitope candidates, number 1 (residue 5-19), number 2 (residue 41-50), numbers 7 and 8 (residue 227-239, and residue 243-247), and number 9 (residue 261-266) overlapped with the experimental epitopes at residues 1-16, residues 42-55, residues 228-252, and residues 262-271, respectively. To verify the surface conditions of the predicted LEs within the query protein sequence, a protein structure was simulated based on homologous modeling approaches. This structure can be viewed and analyzed by clicking on the button labeled ‘predicted structure’.
Visualization of the predicted LEs on 3D structures
Predicted structures of the query sequences can be rendered by Jmol
(http://www.jmol.org/) in LEPS, and the corresponding PDBs and PyMOL script files (http://www.pymol.org/) are downloadable by request. For example, Figure 4 shows the simulated structure of HIV integrase as predicted by Modeller, with the predicted epitope segments displayed in yellow solid spheres. Because there is a high
probability that true epitopes will be exposed on the protein surfaces for binding with antibodies, visualization of the predicted LEs on 3D structures can facilitate the selection of suitable epitopes from predicted candidates according to their surface distributions. Figure 5 shows an example of the experimentally verified epitopes and predicted epitopes for the 10 kDa chaperonin protein in the AntiJen dataset. The yellow spheres in both Fig. 5a and 5b show the true and predicted epitope atoms, respectively. The position of the remaining protein is shown in red and blue solid balls in the two simulated structures. In both cases, most of the epitope residues are located on the protein surface.
Acceptability of low sensitivities
challenge that biological experiments would not cover all the true epitopes within an individual antigen. Peptide scanning data could only identify potential epitopes that were recognized by a specific antibody. However, different antibodies to the same antigen might recognize different epitopes. These biological variations caused low coverage of epitopes within an antigen [32]. This situation implies that the
sensitivities of a LE predictor should generally be low. Alternatively, a LE predictor might ubiquitously predict more epitopes to regain the sensitivities accompanying with the reduction of specificities. This will definitely lead to higher experimental costs in general. Nevertheless, to persuade biologists to conduct in vitro experiments on the predicted potential LEs, the accuracy and MCC values could provide balanced statistics for evaluating the performance of a prediction system.
In this study, LEPS displayed high accuracy, MCC, specificity, and PPV, although the sensitivity was a little low. However, the reduced sensitivity was offset by the high PPV. Therefore, the LEPS provides a high probability of success for molecular biologists in predicting and selecting functional epitopes effectively and efficiently.
Acknowledgments
This work was supported by National Science Council, Taiwan (NSC-98-2311-B-039-003-MY3 and NSC-99-2627-B-039-002 to H.T. Chang, and NSC 99-2627-B-019-007
and NSC98-2221-E-019-031-MY2 to T.W. Pai), and by Taiwan Department of Health
References
[1] D. R. Davies and G. H. Cohen, "Interactions of protein antigens with antibodies," Proc Natl Acad Sci U S A, vol. 93, pp. 7-12, Jan 9 1996.
[2] M. H. Van Regenmortel, "Immunoinformatics may lead to a reappraisal of the nature of B cell epitopes and of the feasibility of synthetic peptide vaccines," J
Mol Recognit, vol. 19, pp. 183-7, May-Jun 2006.
[3] D. J. Barlow, et al., "Continuous and discontinuous protein antigenic determinants," Nature, vol. 322, pp. 747-8, Aug 21-27 1986.
[4] D. C. Benjamin, "B-cell epitopes: fact and fiction," Adv Exp Med Biol, vol. 386, pp. 95-108, 1995.
[5] A. D. Vinion-Dubiel, et al., "Antigenic diversity among Helicobacter pylori vacuolating toxins," Infect Immun, vol. 69, pp. 4329-36, Jul 2001.
[6] J. A. Greenbaum, et al., "Towards a consensus on datasets and evaluation metrics for developing B-cell epitope prediction tools," J Mol Recognit, vol. 20, pp. 75-82, Mar-Apr 2007.
[7] O. Serup Andersen, et al., "Identification of a linear epitope in sortilin that partakes in pro-neurotrophin binding," J Biol Chem, vol. 285, pp. 12210-22, Apr 16 2010.
[8] J. Xiang, et al., "Expression and characterization of recombinant VP19c protein and N-terminal from duck enteritis virus," Virol J, vol. 8, p. 82, 2011. [9] A. Lanza, et al., "Controversial role of antibodies against linear epitopes of
desmoglein 3 in pemphigus vulgaris, as revealed by semiquantitative living cell immunofluorescence microscopy and in-cell ELISA," Int J Immunopathol
Pharmacol, vol. 23, pp. 1047-55, Oct-Dec 2010.
[10] M. Yadav, et al., "Identification of major antigenic peptide of filarial glutathione-S-transferase," Vaccine, vol. 29, pp. 1297-303, Feb 1 2011. [11] A. J. Alix, "Predictive estimation of protein linear epitopes by using the
program PEOPLE," Vaccine, vol. 18, pp. 311-4, Sep 1999.
[12] H. T. Chang, et al., "Estimation and extraction of B-cell linear epitopes predicted by mathematical morphology approaches," J Mol Recognit, vol. 21, pp. 431-41, Nov-Dec 2008.
[13] H. T. Chang, et al., "A reinforced merging methodology for mapping unique peptide motifs in members of protein families," BMC Bioinformatics, vol. 7, p. 38, 2006.
[14] P. Haste Andersen, et al., "Prediction of residues in discontinuous B-cell epitopes using protein 3D structures," Protein Sci, vol. 15, pp. 2558-67, Nov 2006.
[15] T. W. Pai, et al., "REMUS: a tool for identification of unique peptide segments as epitopes," Nucleic Acids Res, vol. 34, pp. W198-201, Jul 1 2006.
[16] S. Saha and G. P. S. Raghhava, "BcePred: Prediction of Continuous B-Cell Epitopes in Antigenic Sequences Using Physico-chemical Properties.," LNCS, vol. 3239, pp. 197-204, 2004.
[17] M. Odorico and J. L. Pellequer, "BEPITOPE: predicting the location of continuous epitopes and patterns in proteins," J Mol Recognit, vol. 16, pp. 20-2, Jan-Feb 2003.
[18] I. A. Doytchinova and D. R. Flower, "VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines," BMC
[19] C. P. Toseland, et al., "AntiJen: a quantitative immunology database
integrating functional, thermodynamic, kinetic, biophysical, and cellular data,"
Immunome Res, vol. 1, p. 4, Oct 6 2005.
[20] J. E. Larsen, et al., "Improved method for predicting linear B-cell epitopes,"
Immunome Res, vol. 2, p. 2, 2006.
[21] Y. El-Manzalawy, et al., "Predicting linear B-cell epitopes using string kernels," J Mol Recognit, vol. 21, pp. 243-55, Jul-Aug 2008.
[22] Y. El-Manzalawy, et al., "Predicting flexible length linear B-cell epitopes,"
Comput Syst Bioinformatics Conf, vol. 7, pp. 121-32, 2008.
[23] J. Chen, et al., "Prediction of linear B-cell epitopes using amino acid pair antigenicity scale," Amino Acids, vol. 33, pp. 423-8, Sep 2007.
[24] L. Florea, et al., "Epitope prediction algorithms for peptide-based vaccine design," Proc IEEE Comput Soc Bioinform Conf, vol. 2, pp. 17-26, 2003. [25] C. G. Roberts, et al., "Prediction of HIV peptide epitopes by a novel
algorithm," AIDS Res Hum Retroviruses, vol. 12, pp. 593-610, May 1 1996. [26] S. Saha and G. P. Raghava, "Prediction of continuous B-cell epitopes in an
antigen using recurrent neural network," Proteins, vol. 65, pp. 40-8, Oct 1 2006.
[27] C. C. Chang and C. J. Lin, LIBSVM: a library for support vector machine, 2001.
[28] B. T. M. Korber, et al., "HIV Immunology and HIV/SIV Vaccine Databases," Los Alamos National Laboratory, Theoretical Biology and Biophysics, Los Alamos, New Mexico. LA-UR 04-8162., 2003.
[29] N. Eswar, et al., "Comparative protein structure modeling using Modeller,"
Curr Protoc Bioinformatics, vol. Chapter 5, p. Unit 5 6, Oct 2006.
[30] S. Saha, et al., "Bcipep: a database of B-cell epitopes," BMC Genomics, vol. 6, p. 79, 2005.
[31] B. Boeckmann, et al., "The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003," Nucleic Acids Res, vol. 31, pp. 365-70, Jan 1 2003.
[32] S. E. Caoili, "Benchmarking B-cell epitope prediction for the design of peptide-based vaccines: problems and prospects," J Biomed Biotechnol, vol. 2010, p. 910524.
[33] M. Sachsamanoglou, et al., "Antigenic profile of human recombinant PrP: generation and characterization of a versatile polyclonal antiserum," J
Neuroimmunol, vol. 146, pp. 22-32, Jan 2004.
[34] L. L. Argiro, et al., "Identification of a candidate vaccine peptide on the 37 kDa Schistosoma mansoni GAPDH," Vaccine, vol. 18, pp. 2039-48, Apr 3 2000.
[35] A. W. Burks, et al., "Mapping and mutational analysis of the IgE-binding epitopes on Ara h 1, a legume vicilin protein and a major allergen in peanut hypersensitivity," Eur J Biochem, vol. 245, pp. 334-9, Apr 15 1997.
[36] S. J. Liu, et al., "Immunological characterizations of the nucleocapsid protein based SARS vaccine candidates," Vaccine, vol. 24, pp. 3100-8, Apr 12 2006. [37] X. Cui, et al., "Identification and evaluation of an infertility-associated ZP3
epitope from the marsupial brushtail possum (Trichosurus vulpecula),"
Vaccine, vol. 28, pp. 1499-505, Feb 10 2010.
[38] M. Mueller, et al., "Antigenic characterization of recombinant hemagglutinin proteins derived from different avian influenza virus subtypes," PLoS One, vol. 5, p. e9097, 2010.
[39] A. N. da Silva, et al., "Identification of continuous human B-cell epitopes in the envelope glycoprotein of dengue virus type 3 (DENV-3)," PLoS One, vol. 4, p. e7425, 2009.
[40] R. A. Stetler, et al., "HSP27: mechanisms of cellular protection against neuronal injury," Curr Mol Med, vol. 9, pp. 863-72, Sep 2009.
[41] T. Senger, et al., "Identification of B-cell epitopes on virus-like particles of cutaneous alpha-human papillomaviruses," J Virol, vol. 83, pp. 12692-701, Dec 2009.
[42] C. D. Kelly-Cirino and N. J. Mantis, "Neutralizing monoclonal antibodies directed against defined linear epitopes on domain 4 of anthrax protective antigen," Infect Immun, vol. 77, pp. 4859-67, Nov 2009.
[43] T. U. Consortium, "The Universal Protein Resource (UniProt) in 2010,"
Nucleic Acids Res, vol. 38, pp. D142-8, Jan 2010.
Table 1: Epitopes predicted in the PC dataset after analysis with LEPS
Antigen:Length
(UniProt IDa) LEPS predicted Epitopes Experimental Epitopes Ref.
PrP:253 (P04156) M1ANLGCWML9 S143DYEDRYYRENMHRYPN159 Y218ERESQAYYQRGS230 R37YPGQG42 Q52GG54 Q91GGGT95 N100KPSKPKTNMKHMA113 G123GLGGYMLG131 H140FGSDY145 Q160VYYRPMD167 F198TETD202 [33] [33] [33] [33] [33] [33] [33] [33] GAPDH:338 (P20287) AA421KVGINGAFLKNTVDV10 30 V31SVNDPFIDL40 K48RDSTHGTFPGEVSTENGKLKVNG KL73 C78ERDPANIPWDKDGA92 A108QAHIKNNRAK118 S123APSADAPM131 V136NENSYEKS144 V148SNASCTTN156 K163VIHDKFEIV172 V188VDGPSSKLWRDGRGAM204 A210STGAAKAVG219 L225NGKLT230 R235VPTPDVSV243 R249LGKGASYEE258 F287VGSTSSS294 I302SLNNNF308 Y315DNEFGY321 I329THMHKVDHA338 V31SVNDPFIDLEYM43 G58EVSTENGKLKVNGKLISVHCERDP82 G100VFTTIDKAQAHIKN114 K163VIHDKFEIVE173 S268GPLKGILEYTEDEVVSSDFVG289 [34] [34] [34] [34] [34] Ara h 1:626 (P43238) KQ4726QEPDDLKSSPYQKKTENPC54 38 P75RGHTGTTNQRSPPGERTRGRQPG DYDDDRRQPRREEGGRWGPAGPRE REREEDWRQPREDWRRPSHQQPR KIRPEGREGEQEWGTPGSHVREETSR NN173 K381SVSKKGSEEEGDI394 K472EQQQRGRREEEEDEDEEEEGSNR EV497 P587QSQSQSPSSPEKESPEKEDQEEEN QGGKGP617 K26SSPYQKK33 Q48EPDDLKQKA57 E66YDPRCVY73 E90RTRGRQPGDYDDDRR105 R108REEGGRW115 E124REEDWRQ131 E134DWRRPSHQQPRKIRPEG151 P295GQFEDFF302 Y312LQGFSRN319 F325NAEFNEIRR334 Q345EERGQRR352 D393ITNPINLRE402 N409NFGKLFEVK418 G463NLELV468 R498RYTARLKEG507 E525LHLLGFGIN534 H539RIFLAGDKD548 I551DQIEKQAKDLAFPGSGE568 [35] [35] [35] [35] [35] [35] [35] [35] [35] [35] [35] [35] [35] [35] [35] [35] [35] [35]
SARS N:422 (Q19QW0) H60GKEEL65 T77NSGPDDQ84 L140NTPKDHIGTRNPNNN155 A36RPKQRRPQGLPNNTASWFT55 A156ATVLQLPQGTTLPKGFYAEGSRGG180 T266KQYNVTQAFGRRGP280 N286FGDQDLIRQGTDYK300 K356HIDAYKTFPPTEPKKDKKK375 R386QKKQPTVTLLPAADMDDFSRQLQN410 [36] [36] [36] [36] [36] [36] ZP3:399 (O77685, residue 24-422) T31QSPAPGSSFSP42 P124NLSQ128 T31QSPAPGSSFSPPPVVA47 Q71AAELTLGPSACAPVPAEPLSK92 H101ECGSELQMTPDSLIYSTVLHY122 L126SQSPLVLRSSP137 G156IQPTWVPFHSTLSREQ172 D251SSSIFISPRPG262 V291TATDQAPSPLN302 A311DEWLPVEGPRD322 Q346EPGNPSEFEADLMLGPLVLSEAENGP372 [37] [37] [37] [37] [37] [37] [37] [37] [37] AIV-H4:511 (A3KF09, residue17-527) Q17NYTGNPVIC26 S169DGNAYP175 D107TCYPFDVPEYQSLR121 F137QWNTVKQNGKSGACKRANVNDFFNRLNWLVK SDGNAYPLQNLTKINNGDYARLYIWGVHHPSTDT202 N206LYKNNPGRVTVSTK220 T224SVVPNIGSGPLVRGGQSGRVSXYWTIV250 V257FNTIGNLIAPRGHYKLNNQKKSTILNTAIPIGSCV SKCHTDKGSLSTTKPFQNISRIAVGDCPRYVKQGSL KLATGMRNIPEKASRGLFGAI349 D455SEMNKLFERVRRQL469 A473EDKGNGCFEIFHKCDNN490 N512RFQIQGVKLTQGYM526 [38] [38] [38] [38] [38] [38] [38] [38] AIV-H5:568 (A5HNY9) E284LEYGNCNTKC294 A25NNSTEQVDTIMEKNVTVTHAQDILEKTHNGKL57 E85FLNVPEWSYIVEKINPANDLCYP108 C151PYQGRSSFFRNVVW165 D199AAEQTRLYQNPTTY213 R223SKVNGQSGRMEFFWTILKPNDAINFESNGNFIAP ENAYKIV273 L472RDNAKELGNGCFEFYHR489 [38] [38] [38] [38] [38] [38] AIV-H12:527 (C7FPM3, residue 1-527) T35LIEQNVPVT44 D31TVNTLIEQNVPVTQVEELVH51 K127YERVKMFDFTKWNVTYTGTSKACNNTSNQGSF YRSMRWLTLKSGQFPVQTDEY180 F190TWAIHHPPTSDEQVKLYKNPNSLSSVTTDEINRS FRPNIGPRPL234 Q238QGRMDYYWAVLKPGQTV255 T259NGNLIAPEYGHLITGKSHGRILKNDLPIGQCTTEC 294 T310SKHYIGKCPKYIPS324 R334NVPQAQDRGLFGAIAGFIEG354 I430TDIWAYNAELLVLLENQKTLDEHDANVRNLHDR VR465 G478CFEILHKCDDGCMDTIKNGT498 Q502DYEEESKLERQRINGVKLEENSTYK527 [38] [38] [38] [38] [38] [38] [38] [38] [38] [38] DEN-3 E-glycoprotein:493 (D2JWZ8, residue 281-773) S533QEGA537 W669YKKGSSI676 L707NSLG711 T331QLATLRKLCIEGKI345 D351SRCPTQGEAVLPEEQDPNY370 Q411YENLKYTVIITVHTGDQHQVGNETQGVTAEITP QASTTE450 L476LTMKNKAWMVHRQW490 Q526EVVVLGSQEGAMHT540 [39] [39] [39] [39] [39]
O. tsutsugamushi 47-kDa antigen:466 (Q53246) L245KKGEKIR252 H21SKSLLNQKAVLPQQKSDMHIN42 T65NIGISLNNKVSKYQQEV82 V97TNENVIAGR106 Y145ATFGDSNQS154 V173TNGIISSKGRDMG186 F193IQTNAAIHM202 H201MGSFGGPMF210 I233PSNTVLEAV242 L245KKGEKIRRG254 L333LRNGKSMTLKCKIIANK350 Q357SNDQSLVVN366 L373TPDLVKKYNITSA386 [40] [40] [40] [40] [40] [40] [40] [40] [40] [40] [40] [40] HPV L1 protein:510 (A8BQ01) V122GRGQPL128 R326AQGHNNGMCW336 V416PPPPSASL424 K440PTPPKTPTDP450 G497TPPPTSKRKRV508 D41VYVTRTNVYYHGGSSRLLTVGHPYYSIKKSNNK VAVPKV80 V90KLPDPNKFGLPDADLYDPDTQRLLWACVGVEVG RGQPLGV130 T205TIEDGDMVET215 D219ICTNTCKYPDYLKMAAEPY238 G235DSMFFSLRREQMFTRHFFNRGGKMGDTIPD285 S350TNVSLCATEA360 F370KEYLRHMEEYDLQFIFQLCKITLTPEIMAY400 P450YASLTFWDVDLSESFSMDLD470 [41] [41] [41] [41] [41] [41] [41] [41] Bacillus anthracis, PA domain III and IV:248 (P13423, residue 488-735) N538PSDPLETTKPDMT551 N720PNYK724 R532RIAAVNPSDPLETTKPDMT551 A596ELNATNIYTVL607 I620RDKRFHYDRNNIAVGADES639 L692NISSLRQDGKT703 L716YISNPNYKVNVYAVTKENT735 [42] [42] [42] [42] [42] aBecause some of the epitopes in the PC dataset were partial antigen fragments, the serial numbers for the residues in each epitope were assigned according to the sequence information retrieved from the UniProt database [43]. The overlapping amino acids between the experimentally verified and predicted epitopes are shown in bold.
Table 2. Comparison of the performances of LEPS, BepiPred, ABCPred, BCPred, and FBCPred systems.
Systems SENa SPEa ACCa PPVa MCCa
PC dataset LEPS 12.78 88.33 61.66 45.12 3.65 BepiPred 48.23 59.72 55.33 38.19 7.49 ABCPred0.8b 65.46 40.26 48.89 36.21 5.13 BCPred 50.92 59.35 52.83 36.07 4.43 FBCPred 51.03 52.55 52.20 35.26 3.17 AntiJen dataset LEPS 26.72 84.48 73.81 28.85 10.10 BepiPred 51.79 57.61 55.52 22.02 6.04 ABCPred0.8 67.33 40.40 44.70 21.83 5.46 BCPred 58.84 54.87 53.92 23.34 8.93 FBCPred 60.31 51.21 51.45 22.33 6.73 HIV dataset LEPS 48.33 74.84 63.45 71.44 22.76 BepiPred 50.16 60.85 56.72 61.22 9.72 ABCPred0.7 87.97 14.65 56.59 56.33 5.64 BCPred 80.18 54.57 66.57 65.55 29.80 FBCPred 73.20 58.20 67.13 65.56 27.81 AHP datasetc LEPS 26.97 84.22 72.52 32.07 10.36 BepiPred 51.48 57.91 55.57 25.06 6.32 ABCPred0.8 68.28 39.06 45.58 24.51 5.45 BCPred 59.45 54.80 54.50 26.32 9.73 FBCPred 60.40 51.66 52.31 25.38 7.60
a SEN, sensitivity; SPE, specificity; PPV, positive prediction value; ACC, accuracy;
MCC, Matthews correlation coefficient, unit, %
b The subscripts of ABCPred denote threshold values according to the highest
accuracy.
Fig. 1 The design of LEPS. (a) Step 1a: Primitive epitope candidates with globally and locally high antigenicity were extracted by calculating weighting coefficients for various physic-chemical propensities of each amino acid. After the filtering process with the SVM classifier (step 2a), predicted epitopes were highlighted (step 3a) in the query sequence and the simulated structure. (b) Step 1b: 1230 experimentally verified epitopes and 872 non-epitopes were analyzed to determine the statistical
characteristics of AASs. Step 2b: Subsequently, epitope indexes of 872 epitopes and 872 non-epitopes were used to train the SVM model to predict candidate epitopes based on the statistical characteristics defined in step 1b.
Fig. 2 Comparison of the performances of LEPS, BepiPred, ABCPred, BCPred, and FBCPred systems. The best performance for each indicator is marked with a star.
Fig. 3 The LEPS server. (a) Users can input a query sequence and manually adjust the weight and window size of each propensity. (b) The output information of HIV integrase predicted by LEPS shows 17 candidates, and only 9 candidates were retained after SVM filtration. The final predicted epitope segments are labeled in yellow at the bottom.
Fig. 4 The predicted LEs of HIV integrase mapped onto a simulated 3D structure. The predicted epitopes are labeled in yellow and the selected epitopes (number 1 and number 3) are shown in yellow spheres.
Fig. 5 The experimental and predicted epitopes of 10 kDa chaperonin. The structural surfaces display the true epitopes (a) and predicted epitopes (b) in yellow spheres. The red and blue spheres represent the remainder of the protein. Both figures were created with PyMOL.
Figure 1
Protein Sequence
Generation of Epitope Candidates by Antigenicity Analysis High Antigenic Fragments Local Peak Fragments + SVM Classifier Epitope Candidates Filter Out Less Probable
Candidates
Visualization of the Predicted Epitopes Query Sequence Predicted Structure +
Step 1: Statistical analysis of AASs
Bcipep Dataset (1230 epitopes) Chen Dataset (872 non-epitopes) + Chen’s Dataset (872 epitopes + 872 non-epitopes) 3 Epitope Indexes (Epidex2, 3, 4) + = SVM Classifier Step 2: SVM Training (a) (b)