Original Contribution
OXIDATIVE DAMAGE TO MITOCHONDRIAL DNA IN ATRIAL MUSCLE OF
PATIENTS WITH ATRIAL FIBRILLATION
PO-HANLIN,*†SHIH-HUANG LEE,‡§ CHIA-PINGSU,*and YAU-HUEIWEI*§
*Department of Biochemistry and Center for Cellular and Molecular Biology, National Yang-Ming University, Taipei 112, Taiwan, Republic of China;†Department of Medicine, Taipei Medical University, Taipei 110, Taiwan, Republic of China;‡Wu Ho
Shih Shing Kong Memorial Hospital, Taipei 111, Taiwan, Republic of China; and§Department of Medicine, National Yang-Ming University, Taipei 112, Taiwan, Republic of China
(Received 29 October 2002; Revised 1 July 2003; Accepted 31 July 2003)
Abstract—Atrial fibrillation (AF) is the most common cause of arrhythmia and is an aging-related disease encountered
in clinical practice. The electrophysiological remolding with Ca2⫹ overloading and cellular structure changes were
found in cardiomyocytes of AF patients. In previous studies, increased oxidative stress and oxidative damage was found in cardiomyocytes during the ischemia/reperfusion injury. Besides, mitochondrial DNA (mtDNA) deletion and mtDNA proliferation occur frequently in affected tissues of patients with certain degenerative diseases and during aging of the human. However, it remains unclear whether high oxidative stress and alteration of mtDNA play a role in the pathophysiology of AF. In this study, we first screened for large-scale deletions of mtDNA in the atrial muscle of AF patients by long-range polymerase chain reaction (PCR). The results showed that large-scale deletions between nucleotide positions 7900 and 16500 of mtDNA occurred at a high frequency. Among them, the 4977 bp deletion was the most frequent and abundant one, and the mean proportion of mtDNA with the 4977 bp deletion in the atrial muscle of the patients with AF was 3.75-fold higher than that of the patients without AF (p⬍ .005). Furthermore, quantitative PCR was performed to evaluate lesions in mtDNA caused by oxidative damage. We found that the degree of mtDNA damage in the patients with AF was greater than that of the patients without AF (3.29 vs.1.60 per 10 kb, p⬍ .0005). The 8-OHdG, which is one of the most common products of oxidative damage to DNA, was also found at a higher frequency in mtDNA of patients with AF as compared with those without AF. In addition, the mtDNA content was found to increase significantly in the patients with AF (p⫽ .0051). The level of mtDNA lesion and the mtDNA content was positively correlated (r⫽ 0.44). These results suggest that oxidative injury and deletion of mtDNA in cardiac muscle are increased in the patients with AF, which may contribute to the impairment of bioenergetic function of mitochondria and induction of the oxidative vicious cycle involved in the pathogenesis of atrial myopathy in AF. © 2003 Elsevier Inc.
Keywords—Atrial fibrillation, Mitochondria, Oxidative damage, Mitochondrial DNA, Deletion, Free radicals
INTRODUCTION
Atrial fibrillation (AF) is the most common sustained arrhythmia and is an aging-related disease encountered in clinical practice [1]. Previous studies of the mecha-nism underlying AF suggested that it is associated with electrical remolding and changes in cellular structures, which reveal intracellular Ca2⫹ overloading and disor-ganization of organelles, such as the disruption of
sarco-meres, deposition of glycogen, and aggregation of ab-normal mitochondria [2,3]. The clinical hallmarks in electrical remolding of AF are the shortness of the re-fractory periods and rapid activation of atrial cardiomy-ocytes [4]. This rapidity of depolarization and repolar-ization may increase the energy demand of mitochondria because the cardiomyocytes have to maintain a normal cell membrane potential. Besides working hard to supply more energy via oxidative metabolism, mitochondria are activated to meet the energy need for increased uptake of Ca2⫹ from the Ca2⫹-overloaded cytoplasm [5]. In a previous study, the Ca2⫹-mediated oxidative damage to mitochondria was found in the ischemia/reperfusion in-Address correspondence to: Prof. Yau-Huei Wei, Department of
Biochemistry, School of Life Science, National Yang-Ming University, Taipei, Taiwan 112, Republic of China; Tel:⫹886 (2) 28267118; Fax:
⫹886 (2) 28264843; E-Mail: [email protected].
Printed in the USA. All rights reserved 0891-5849/03/$–see front matter doi:10.1016/j.freeradbiomed.2003.07.002
jury of cardiomyocytes [6]. Thus, these changes to mi-tochondria may increase oxidative stress in the atrial muscle of patients with AF.
Mitochondria are not only the power plant of human cells but also the important biological source and target of reactive oxygen species (ROS) and free radicals. With the increase of age and excess exogenous stress like AF, the free radicals and ROS can overwhelm the antioxidant system and result in damage to cellular constituents such as lipids, proteins, and DNA. Human mitochondrial DNA (mtDNA) is a naked double-stranded circular DNA molecule with 16569 bp in size, which is usually at-tached to the inner membrane of mitochondria. Under enhanced oxidative stress, mtDNA is extremely vulner-able to oxidative damage and mutation because of poor proofreading and inefficient DNA repair during replica-tion [5,7]. Although the excision repair system for short patch of modified bases was recently found in mitochon-dria, DNA damages are still accumulated due to the lack of ability to repair bulky DNA lesions and insufficient turnover or repair to eliminate defective mtDNA mole-cules [8,9]. Accumulation of somatic mtDNA mutations has been documented to contribute to human aging and progressive organ dysfunction in some degenerative dis-eases [10]. The unusual structures of hot-regions flanking large-scale deletions in mtDNA have been well charac-terized. The nucleotide position (np) 7900 to 16500 of human mtDNA was demonstrated as the region with high frequency of large-scale deletion [7]. A common 4977 bp deletion of mtDNA spanning np 8470/8482 to 13447/13459 has been reported frequently in aging hu-man tissues with increased oxidative stress [7,10,11]. Moreover, both mitochondrial mass and mtDNA copy number are varied with the cell type and are subjected to changes with cellular physiological conditions. Increased synthesis of mitochondrial respiratory enzymes and overproliferation of mitochondria are often observed in somatic tissues under oxidative stress and in the affected tissues of patients with mitochondrial myopathies [12].
In a previous study, atrial myopathy was documented as a dominant factor in the recurrence of chronic AF [13]. Based on this and other lines of evidence, we hypothesized that increased oxidative injury and alter-ations of mtDNA may involve in the underlying patho-physiology in atrial cardiomyocytes of AF. In this study, we used the mtDNA as a reporter to test whether AF is associated with increased oxidative stress. First, we screened for large-scale deletions of mtDNA in atrial muscle of patients with AF. The results showed that the 4977 bp deletion occurred at a high frequency in atrial muscle of AF patients. A semi-quantitative PCR method was then employed to estimate the proportion of mtDNA with the 4977 bp deletion. Moreover, quantitative poly-merase chain reaction (QPCR) was performed to assess
mtDNA damage, which is considered a good biomarker for chronic oxidative stress. We also assessed oxidative damage to mtDNA by performing PCR on atrial muscle DNA samples that had been treated with hOGG1, which cleaves 8-hydroxyl 2⬘-deoxyguanosine (8-OHdG) in ox-idative lesions of DNA. The alteration of the mtDNA copy number in the atrial muscle of patients with AF was also investigated.
MATERIALS AND METHODS Patients and preparation of samples
During opening heart surgery and before extracorpo-ral circulation, right atrial appendages were excised with consent from 14 patients with chronic AF (ⱖ3 month) and 26 patients with sinus rhythm. There was no differ-ence in age, sex, and hemodynamics between the patients with and without AF. The right atrial appendage was cut into three pieces and placed into different vials, which were stored immediately in a liquid nitrogen tank.
Screening for large-scale mtDNA deletion
Total cellular DNA was extracted from one piece (30 –50 mg) of frozen muscle. Large-scale deletions of mtDNA were detected by a long-range PCR technique [14] followed by electrophoresis of a portion of the PCR products on a 1.5% agarose gel. Oligonucleotide primers used for amplification of mtDNA are listed in Table 1.
Analysis of 4977 bp deletion of mtDNA
The incidence of the specific 4977 bp deletion of mtDNA was determined by the method described by Lee et al. [15]. A 524 bp DNA fragment was amplified by PCR with primers L5 and H2 (see Table 1) from mtDNA with the 4977 bp deletion. In order to ascertain that this band represented the mtDNA with 4977 bp deletion, we performed the primer-shift PCR (see Fig. 2A) to confirm the existence of the deletion. The proportion of the
Table 1. Nucleotide Sequences of the Primers Designed for Polymerase Chain Reaction Amplification of Human mtDNA L1 (110–130) 5⬘-CACCCTATGTCGCAGTATCTG-3⬘ L2 (3304–3323) 5⬘-AACATACCCATGGCCAACCT-3⬘ L3 (3726–3745) 5⬘-CTCATATGAAGTCACCCTAG-3⬘ L4 (7901–7920) 5⬘-TGAACCTACGAGTACACCGA-3⬘ L5 (8150–8169) 5⬘-CCGGGGGTATACTACGGTCA-3⬘ L6 (8285–8304) 5⬘-CTCTAGAGCCCACTGTAAAG-3⬘ L7 (8344–8363) 5⬘-ACCAACACCTCTTTACAGTG-3⬘ L8 (12279–12298) 5⬘-AACAGCTATCCATTGGTCTT-3⬘ H1 (3836–3817) 5⬘-GGCAGGAGTAATCAGAGGTG-3⬘ H2 (13650–13631) 5⬘-GGGGAAGCGAGGTTGACCTG-3⬘ H3 (13595–13576) 5⬘-CTTGTCAGGGAGGTAGCGAT-3⬘ H4 (13928–13905) 5⬘-CTAGGGTAGAATCCGAGTATGTTG-3⬘ H5 (16228–16208) 5⬘-GTTGAGGGTTGATTGCTGTAC-3⬘ H6 (16540–16521) 5⬘-GTGGGCTATTTAGGCTTTAT-3⬘
mtDNA with 4977 bp deletion was determined with a semi-quantitative PCR method on each sample with se-rial dilutions [15].
Quantitative PCR for assessment of oxidative DNA damage
Total cellular DNA was isolated from another piece of frozen muscle sample with addition of 2 mM butylated hydroxytoluene in the tissue lysate. The final DNA pellet was dried by a slow stream of argon gas to minimize oxidation artifacts and prevent DNA damage [16].
In chronic ROS-exposed or aging tissue cells, mtDNA molecules with extensive and persistent damages are accu-mulated, which can serve as a useful biomarker for ROS-associated diseases [8,9,17–19]. Oxidative damage to mtDNA can be assessed by a QPCR method, which is based on the assumption that DNA polymerase can be inhibited by DNA lesions, including oxidative damages such as strand breaks, base modification (e.g., thymine glycol, cyclopurine deoxynucleosides, etc.) and apurinic sites [8,17–19]. These lesions reduce the efficiency of PCR amplification of a target DNA sequence. We showed in a previous study that the segment between np 7900 and 16500 of mtDNA bears unusual structures and that most large-scale deletions have occurred in this region [7]. We thus employed the method described by Ballinger et al. [18,19] to analyze for oxidative damage in this region of mtDNA. Briefly, oxidative damage to mtDNA was quanti-fied by comparing the relative efficiency of amplification of a large fragment of DNA (by primer pair L4-H6, np 7901 to 16540) that was normalized with the amplification of a smaller fragment (by primer pair L3-H1, np 3726 to 3836), which had a statistically negligible probability of containing damaged nucleotides. We used the mtDNA from the 143B cell line (wild type mtDNA) as the control. The long-range PCR was carried out by denaturation of DNA at 95°C for 2 min, followed by 26 cycles of 95°C denaturation for 15 s and 68°C primer extension for 10 min, and a final incuba-tion at 72°C for 10 min to complete the reacincuba-tion. The level of DNA lesions was calculated as the efficiency of ampli-fication of a patient’s (damaged) sample (Ad) relative to the amplification of the undamaged control (A0), which was expressed as a relative amplification ratio (Ad/A0). Assum-ing a random distribution of DNA lesions and usAssum-ing the Poisson equation [f(x)⫽ exx/x!, where is the average level of mtDNA lesions] for the undamaged DNA template, the average level of lesions per mtDNA strand was then determined by ⫽ ⫺ln Ad/A0.
Analysis of 8-OHdG in mtDNA
The most common oxidative product of DNA is 8-OHdG, which is detectable in mtDNA of human tissue cells [8,20]. Under enhanced oxidative stress, the amount
of 8-OHdG in mtDNA was found to increase although the base excision repair (BER) system of oxoguanine glycosylase and DNA ligase are present in mitochondria [8,9]. Human oxoguanine glycosylase 1 (hOGG1) can recognize 8-OHdG and catalyze the removal of 8-OHdG through cleavage of the phosphodiester bond in modified DNA.
It has been shown that 8-OHdG is one of the impor-tant oxidative DNA lesions related to mitochondrial dys-function and aging [8,20]. Unlike other DNA base mod-ifications such as thymine glycol and other bulky oxidized products, 8-OHdG does not distort the structure of DNA and exerts only slight inhibition of Taq DNA polymerase [8,20]. However, if an 8-OHdG is present at a specific region of the DNA template, digestion of the 8-OHdG by hOGG1 can break the DNA template at the lesion site and block the amplification of this region by DNA Taq polymerase. In this study, we performed PCR to amplify different fragments of mtDNA on each sam-ple with and without hOGG1 pretreatment (see Fig. 4). Therefore, the greater the proportion of mtDNA in our samples contained 8-OHdG, the less intact mtDNA was left after hOGG1 digestion, which resulted in a lower yield of the PCR product. Thus, attenuation in the effi-ciency of PCR would be more quickly detected in the medium-sized PCR fragment than in the larger PCR fragment. The reaction condition for hOGG1 digestion of mtDNA was as follows: 100 ng of DNA sample (1l) was incubated at 37°C for 1 h in a 20l reaction buffer, which contained 1 unit of hOGG1, 20 mM Tris-HCl (pH 8.0), 1 mM dithiothreitol, 1 mM ethylenediaminetet-raacetic acid (EDTA), and 0.1 mg/ml bovine serum al-bumin. The untreated control of each DNA sample was also incubated under identical conditions except no hOGG1 was included in the reaction mixture.
Determination of mtDNA content
The content of mtDNA relative to nuclear DNA (nDNA) was quantified by slot blot hybridization. Two micrograms of total cellular DNA was transferred into a reaction buffer (10 mM EDTA, 0.4 N NaOH, pH 8.2) for denaturation, followed by blotting and binding to a piece of Hybond N⫹ nylon membrane (Amersham) under ul-traviolet light illumination. Hybridization with a [␣-32P] dCTP-labeled 28S rRNA gene probe and an mtDNA D-loop probe (np 16455–1462) was carried out at 65°C for 4 h in a rapid hybridization buffer. The membrane was washed successively with 2⫻ saline sodium citrate (SSC)/0.1% sodium dodecyl sulfate (SDS), 1 ⫻ SSC/ 0.1% SDS, 0.5⫻ SSC/0.1% SDS, and 0.1 ⫻ SSC/0.1% SDS solution at 37°C, 45°C, 50°C, and 65°C each for 15 min, respectively. The washed membrane was air-dried and exposed to a phosphorimager screen to measure the
intensity of the target DNA. The mtDNA/nDNA ratio was determined for each sample as an index of the relative content of mtDNA.
Data analysis
The statistical significance was determined by inde-pendent Student’s two-tailed t-test for the difference in the measured values between AF and non-AF groups with respect to the frequency of 4977 bp deletion of mtDNA, frequency of DNA lesions and mtDNA copy number. To compare the incidence of 4977 bp deletion between AF patients and patients without AF, we used the2test to perform statistical analysis. Difference with a p-value⬍.05 was considered significant.
RESULTS
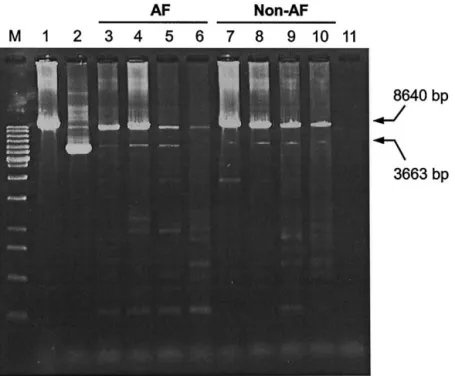
Using the long-range PCR, we found variable lengths of DNA fragments amplified from atrial mus-cle DNA of the AF patients, encompassing the se-quence between np 7901 and 16540 of mtDNA (Fig. 1). These large-scale deletions reveal that the mito-chondrial genome was damaged and resulted in the accumulation of large-scale mtDNA deletions in this postmitotic tissue. No mtDNA deletions were found to
predominate except that a common 3663 bp PCR product was obtained from mtDNA with 4977 bp deletion, which occurred at a high frequency in pa-tients with and without AF.
In order to ascertain the existence of the 4977 bp deletion in mtDNA, primers L5 and H2 were used and the appearance of a 524 bp PCR product indicated the presence of this specific deletion. The primer-shift PCR method was performed to confirm this mtDNA deletion (Fig. 2A). Four PCR products of 802 bp, 667 bp, 524 bp, and 389 bp in size were respectively amplified from mtDNA with the 4977 bp deletion. This specific deletion was detected in the atrial muscle of all 14 patients with AF (100%), and in 20 of 24 subjects without AF (83.3%). There was no difference in the incidence of 4977 bp deletion in atrial muscle between the two groups. The proportion of 4977 bp mtDNA was then analyzed for the atrial muscles that had been confirmed to harbor this specific deletion. The mean proportion of mtDNA with 4977 bp deletion in the patients without AF was 0.18⫾ 0.15%, and that in the patients with AF was 0.68 ⫾ 0.53%. This result showed that the level of the 4977 bp deleted mtDNA in the atrial muscle of patients with AF was 3.75 times higher than that of the patients without AF (p ⬍ .005, Fig. 2B).
Fig. 1. Gel electrophorectogram of polymerase chain reaction (PCR) products amplified from mitochondrial DNA (mtDNA) of atrial muscles of atrial fibrillation (AF) and non-AF patients by using the primers L4 and H6. PCR was performed as described in Materials and Methods and the products were separated on a 1.5% agarose gel and detected under ultraviolet transillumination after ethidium bromide staining. The 3663 bp DNA fragment indicates the PCR products amplified from mtDNA with 4977 bp deletion. Lane 1 denotes the PCR product from wild-type mtDNA of the143B cell line, lane 2 represents the PCR product amplified from the skin fibroblasts of a chronic progressive external ophthalmoplegia patient who had been proved to harbor the 4977 bp deletion of mtDNA. Lanes 3– 6 indicate PCR products from mtDNA’s of four patients with AF and lanes 7–10 denote another four patients without AF. Lane 11 is the negative control (blank) and lane M stands for the 1000 bp size marker.
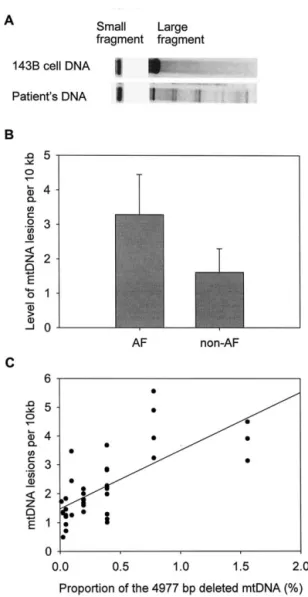
On the other hand, we assessed the oxidative damage to mtDNA in atrial muscles of the patients with and without AF. Using the QPCR technique, we found that the level of mtDNA lesions in atrial muscle was signif-icantly higher in the patients with AF (3.29 per 10 kb) compared with that (1.60 per 10 kb) of the patients without AF (p ⬍ .0005, Fig. 3). This result clearly indicates that oxidative damage to mtDNA is concur-rently enhanced in atrial muscle of patients with AF. The correlation between the proportion of mtDNA with 4977 bp deletion and the level of mtDNA lesion was also examined in the atrial muscle. The result showed that in both groups of patients, the proportion of mtDNA with 4977 bp deletion was positively correlated with the level of mtDNA damage in the atrial muscle (r⫽ 0.71, Fig. 3C).
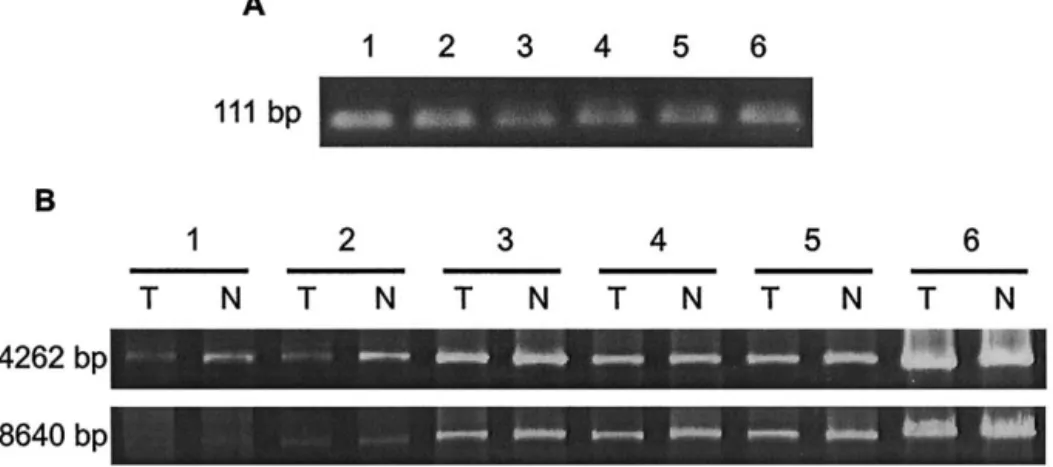
There are many kinds of oxidative damages in DNA and one of them is 8-OHdG. This modified nucleobase is an indicator of oxidative damage to DNA [20]. In the analysis of a 4262 bp mtDNA segment generated by PCR, we found that the difference in band intensity between the atrial DNA samples with and without hOGG1 pretreatment was more pronounced in the group of patients with AF (Fig. 4). This attenuated PCR effi-ciency indicated that there was a high level of 8-OHdG in the atrial DNA of patients with AF. However, this difference in DNA band intensity in the group of patients Fig. 2. Effect of atrial fibrillation (AF) on the accumulation of the 4977
bp deleted mitochondrial DNA (mtDNA). (A) The primer-shift poly-merase chain reaction (PCR) demonstrated the existence of the 4977 bp deleted mtDNA in the atrial muscle of the patient. The PCR products of 802 bp, 667 bp, 524 bp, and 389 bp DNA fragment were amplified with the primer pairs L5-H4, L6-H4, L5-H2, and L6-H2, respectively. (B) The bar graph shows that the mean proportion of the 4977 bp deleted mtDNA in the atrial muscle of the patients with AF is signif-icantly higher than that of the patients without AF (*p⬍ .005).
Fig. 3. Quantitative polymerase chain reaction (PCR) analysis of the level of mitochondrial DNA (mtDNA) lesions. (A) [␣-32P]dATP was incorporated in the products of quantitative PCR and the intensity of the DNA band was measured by a phosphorimager screen. (B) The bar graph shows that the level of oxidative damage to mtDNA of the patients with atrial fibrillation (AF) (3.29 per 10 kb) is greater than that of the patients without AF (1.60 per 10 kb). *Significant difference in the measured value as compared with the control (*p⬍ .0005). (C) The scatter plot shows that the proportion of the 4977 bp deleted mtDNA is positively correlated with the level of mtDNA lesions in all patients (r
with AF was overlooked in the larger (8640 bp) PCR fragments. These results suggest that a variety of oxida-tive lesions were accumulated in mtDNA of the atrial muscles of the AF patients examined, which is consistent with the results obtained by QPCR.
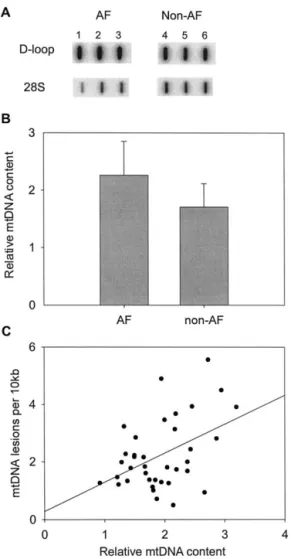
Furthermore, we determined the mtDNA content of the atrial muscle by slot blot. The average mtDNA/ nDNA ratio in the atrial muscle of the patients with AF was 2.26, which is significantly higher than the ratio (1.70) of the patients without AF (p ⫽ .0051, Fig. 5). Further analysis showed that the level of the mtDNA lesion was positively correlated with the mtDNA content in the atrial muscle of AF patients (r ⫽ 0.44, Fig. 5C).
DISCUSSION
In this study, we demonstrated that 4977 bp deletion and lesions of mtDNA and mtDNA content were in-creased in the atrial muscle of the patients with AF. These findings have provided strong evidence to support the notion that oxidative injury in the cardiac muscle is enhanced in the patients with AF.
Recently, oxidative stress was shown to associate with myocardial dysfunction and contribute to disease progression of the heart [21]. Mitochondria have been well recognized as the major source of ROS and the main target of ROS-mediated damage. The 4977 bp deletion of mtDNA frequently occurs in aging human tissues and
affected tissues of patients with degenerative diseases. AF is considered as an aging- related cardiac arrhythmia, which occurs at a high incidence in the elderly people and the incidence is increased with age [1]. In this study, we recruited 14 AF patients with a mean age of 61⫾ 10 years and 26 non-AF patients with a comparable mean age (61 ⫾ 11 years). No significant difference in the incidence of the 4977 bp deletion was found between these two groups, but the proportion of mtDNA with this deletion was significantly higher in the patients with AF than that in the patients without AF. This observation suggested that mild oxidative stress is developed under the normal aging process, but an excess of ROS is generated in the cardiomyocytes of the patients with AF. This notion of increased oxidative damage to mtDNA of AF patient was first assessed by the QPCR technique. Moreover, a higher level of 8-OHdG in mtDNA of the atrial muscles of AF patients was confirmed by the observation of attenuation in the efficiency of PCR am-plification of a medium-sized (4262 bp) PCR fragment from the mtDNA sample with hOGG1 pretreatment (Fig. 4). However, we also noticed that there was no signifi-cant difference in the DNA band intensity of a PCR-amplified 8640 bp mtDNA fragment between atrial DNA samples with and those without hOGG1 treatment. We conjectured that this was due to the fact that the larger the PCR fragment to be amplified, the more oxidative DNA lesions are present in the mtDNA template to inhibit the Fig. 4. Gel electrophorectogram of polymerase chain reaction (PCR) products amplified from mitochondrial DNA (mtDNA) of atrial muscles of atrial fibrillation (AF) and non-AF patients by using the primer pairs (A) L3/H1 (111 bp) and (B) L8/H6 (4262 bp), and L4/H6 (8640 bp). PCR was performed as described in Materials and Methods and the products were separated on a 1.5% agarose gel and detected under ultraviolet transillumination after ethidium bromide staining. Samples 1 and 2 denote the PCR products amplified from mtDNA of two patients with AF, samples 3 to 5 represent another three patients without AF. Sample 6 denotes the PCR product from the wild-type mtDNA of the143B cell line. (A) Similar intensities of the PCR products (111 bp) indicate that the concentrations of the six DNA samples were comparable. (B) The lanes marked with “T” indicate that the DNA sample was pretreated with hOGG1 for 1 h before conduction of PCR and the lanes with “N” represent that without treatment with hOGG1. In the gel electrophorectogram of medium-sized PCR products (4262 bp), the difference in the intensity between “T” and “N” is prominent in samples 1 and 2, and the difference is less conspicuous in samples 3–5, and no visible difference was noted in sample 6. In the gel electrophorectogram of large fragment (8640 bp), the difference in the samples 1 and 2 between “T” and “N” was less pronounced. In contrast, the difference was a little more perceptible in the samples 3–5 between “T” and “N” compared with that in the electrophorectogram of the 4262 bp PCR products. This result indicates that there was a greater amount of 8-OHdG in each of the DNA samples from the two patients with AF compared to those without AF.
function of Taq DNA polymerase. Therefore, when a large PCR fragment was to be amplified from mtDNA of AF patients, other oxidative lesions could have masked the influence of hOGG1 digestion on the intactness of mtDNA template. These findings are consistent with the results obtained by QPCR, and may suggest that a high frequency of different oxidative DNA damages exist in mtDNA of AF patients. Taken together, our results con-sistently showed that oxidative stress in the atrial muscle was indeed significantly higher in the patients with AF compared with controls.
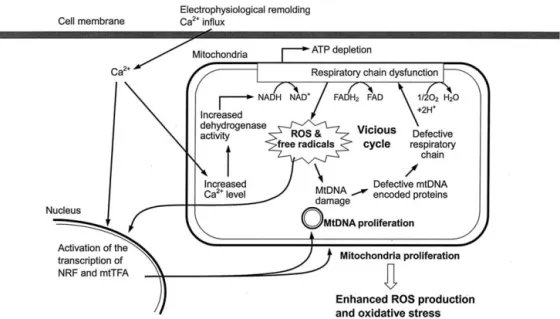
In previous studies, AF was demonstrated as a pro-gressive disease and that disease progression is resulted from cumulative electrophysiological remolding and cel-lular structure changes [2,4,13]. Electrophysiological re-molding with the Ca2⫹overloading is considered as one of the early events in the perpetuation of AF [2,22,23] and Ca2⫹-mediated oxidative injury was proposed as a pathophysiological factor of AF [22]. We hypothesized that mitochondrial oxidative stress plays a critical role in the progression of AF after initial Ca2⫹overloading (Fig. 6). Due to a high Ca2⫹ concentration in the cytoplasm, mitochondria would take up huge amounts of Ca2⫹from the cytoplasm to maintain the intracellular ion homeosta-sis [5]. Under normal physiological conditions, appropri-ate increase of Ca2⫹ level in the mitochondrial matrix can enhance adenosine triphosphate (ATP) synthesis via increased activity of pyruvate and ␣-keto glutarate de-hydrogenases [24]. However, accumulation of a large amount of Ca2⫹ in the mitochondrial matrix in the car-diomyocytes of AF patients eventually alter the mito-chondrial membrane potential to decline ATP synthesis and produce excess ROS and inhibit antioxidant mech-anism and elicit damages to lipids, proteins and DNA [5,6,9,25]. This is consistent with our observation that the levels of 4977 bp deletion (Fig. 2B) and oxidative lesions of mtDNA (Fig. 3B and Fig. 4) in the atrial muscle were increased in AF patients. In addition, 4977 bp deletion, which removes genes encoding ATPase subunits 6 and 8 and subunits 3, 4, and 5 of NADH dehydrogenase, would disrupt aerobic metabolism to fa-cilitate electron leakage from the respiratory chain to generate free radicals and ROS, like•O2⫺and H2O2[26]. The ROS generated from mitochondria harboring deleted mtDNA would further damage mtDNA to increase the frequency of oxidative lesions. As shown in Figs. 3 and 4, we found a high incidence of variable oxidative dam-ages to mtDNA in atrial muscle of patients with AF. These mtDNA lesions from atrial muscle of AF patients not only affect its replication but also gene expression [8]. An inadequate synthesis of polypeptides encoded by mtDNA in turn causes defects in the aerobic metabolism and increase in ROS generation. High levels of ROS eventually lead to large-scale deletions during mtDNA replication [7,9,11,14]. Figure 3C clearly demonstrated that the proportion of the 4977 bp deletion correlated well with the level of oxidative mtDNA lesions (r ⫽ 0.71). The results obtained in this study led us to suggest that a mitochondrial oxidative vicious cycle is induced to cause progressive damage to the components such as mtDNA in the cardiomyocytes of patients with AF [26]. Furthermore, we found an increase in mtDNA content in the atrial muscle of patients with AF. The increase in ROS production from the defective respiratory chain is thought to play a role in the increase of mitochondrial Fig. 5. Increase of mitochondrial DNA (mtDNA) content in the atrial
muscle of atrial fibrillation (AF) patients. (A) Slot blot analysis for the determination of mtDNA content. Total DNA was hybridized with [32P]-labeled DNA probes, which were synthesized by polymerase chain reaction (PCR) amplification of part of the D-loop region of mtDNA and the nuclear 28S rRNA gene, respectively. A typical scan of the slot blots for the patients with AF (blots 1–3) and the patients without AF (blots 4 – 6) is shown. (B) Comparison of the relative mtDNA content (mtDNA/nuclear DNA ratio) in the atrial muscle between the patients with AF and those without AF. The two bars represent the mean⫾ SD of the results obtained from 14 AF patients and 26 patients without AF (*p⫽ .0051 vs. non-AF). (C) The scatter plot shows that relative mtDNA content is positively correlated with the level of mtDNA lesions in all patients (r⫽ 0.44).
mass and mtDNA content [10,27]. It was suggested that the increase in the copy number of mtDNA is the result of a feedback mechanism that compensates for defective mitochondria bearing an impaired respiratory chain. In-deed, we observed a concurrent increase in the mtDNA content and the level of oxidative lesions to mtDNA in atrial muscle of all patients (Fig. 5C). It is worth noting that an increase in Ca2⫹ concentration could result in accelerated mtDNA replication and mitochondrial bio-genesis [28]. Both the ROS and Ca2⫹ may act as the second messengers to trigger the expression of nuclear respiratory factors (NRF-1 and NRF-2) and drial transcription factors (mtTFA) to induce mitochon-drial biogenesis and mtDNA proliferation [10,27,28]. Because of the high oxidative stress and elevated Ca2⫹ level in mitochondria, the mtDNA molecules are rapidly damaged and mitochondria tend to lose their normal bioenergetic function. Moreover, the functionally com-promised mitochondria generate large amounts of ROS in atrial muscle of AF patients. As a result, the compen-satory proliferation of mitochondria and mtDNA was not able to rescue the cell under the stressful conditions, but instead accelerated the oxidative vicious cycle to damage various cellular components, particularly mtDNA [26]. Although several types of repair enzymes or turnover of mtDNA in mitochondria may dispose of some of these damages, a portion of the defective mtDNA molecules in atrial muscle can escape repair or degradation so as to accumulate progressively under enhanced oxidative
stress environment [9]. These may explain our observa-tions that 4977 bp deletion and oxidative lesions of mtDNA and the copy number of the mitochondrial ge-nome were increased concurrently with the oxidative injury in cardiomyocytes of patients with AF. The chronic oxidative damage may lead to morphological changes of the affected cardiomyocytes, which are man-ifested as the overproliferation of vacuolated and swollen mitochondria and derangement of other organelles [29]. This notion is consistent with the previous reports of the structural changes of atrial cardiomyocytes in the pa-tients with AF [3,30].
In this study, we provided novel evidence that in-creased oxidative stress does not only occur in the isch-emia/reperfusion injury [6], but also exists in the atrial muscle with altered electrical activity. We conclude that oxidative vicious cycle caused by dysfunctional mito-chondria underlies the disease progression of AF. In clinical practice, pharmacological cardioversion from AF to sinus rhythm is effective only in the initial stage of AF [23]. In addition, less effort to restore the sinus rhythm and high recurrence rate in the patients with chronic AF are the limitation of the antiarrhythmia therapy. The time course and the progression of AF may determine the therapeutic efficiency of successful result from AF back to sinus rhythm [23]. Thus, it is of clinical significance to develop efficient antioxidant therapies to block oxidative injury as well as oxidative lesions and deletions of mtDNA in atrial muscle of patients with AF.
Fig. 6. A schematic illustration of the role of oxidative injury, mitochondrial (mtDNA) damage and mitochondrial dysfunction in the pathophysiology of atrial fibrillation (AF). Oxidative stress initiated by the electrophysiological remolding with Ca2⫹overloading leads to dysfunction of the respiratory chain and adenosine triphosphate depletion, which increase the production of reactive oxygen species (ROS) and free radicals and trigger the oxidative vicious cycle. The ROS and Ca2⫹will then induce the expression of nuclear respiratory factors and mitochondrial transcription factors, which will increase the biogenesis of mitochondria and mtDNA prolifer-ation. The proliferated mitochondria generate more and more ROS to accelerate the vicious cycle, which may result in the disease progression and structural change of cardiomyocytes in the atrial muscle of AF patients.
Acknowledgements — This work was supported by grants (No.
NSC89-2815-C-010-036R-B and NSC 90-2320-B-010-079) from the National Science Council, Taiwan, Republic of China.
REFERENCES
[1] Feinberg, W. M.; Blackshear, J. L.; Laupacis, A.; Kronmal, R.; Hart, R. G. Prevalence, age distribution, and gender of patients with atrial fibrillation. Analysis and implications. Arch. Intern.
Med. 155:469 – 473; 1995.
[2] Ausma, J.; Wijffels, M.; Thone, F.; Wouters, L.; Allessie, M.; Borgers, M. Structure changes of atrial myocardium due to sus-tained fibrillation in the goat. Circulation 96:3157–3163; 1997. [3] van Wagoner, D. R.; Nerbonne, J. M. Molecular basis of electrical
remolding in atrial fibrillation. J. Mol. Cell. Cardiol. 32:1101– 1117; 2000.
[4] Yu, W. C.; Lee, S. H.; Tai, C. T.; Tsai, C. F.; Hsieh, M. H.; Chen, C. C.; Ding, Y. A.; Chang, M. S.; Chen, S. A. Reversal of atrial electrical remodeling following cardioversion of long-standing atrial fibrillation in man. Cardiovasc. Res. 42:470 – 476; 1999. [5] Jahngir, A.; Ozcan, C.; Holmuhamedov, E. L.; Terzic, A.
In-creased calcium vulnerability of senescent cardiac mitochondria: protective role for a mitochondrial potassium channel opener.
Mech. Ageing Dev. 122:1073–1086; 2001.
[6] Murata, M.; Akao, M.; O’Rourke, B.; Marban, E. Mitochondrial ATP-sensitive potassium channels attenuate matrix Ca2⫹ over-load during simulated ischemia and reperfusion: possible mech-anism of cardioprotection. Circ. Res. 89:891– 898; 2001. [7] Hou, J. H.; Wei, Y. H. The unusual structures of the hot-regions
flanking large-scale deletions in human mitochondrial DNA.
Bio-chem. J. 318:1065–1070; 1996.
[8] Wallace, S. S. Oxidative DNA damage and repair: biological consequences of free radical-damage DNA bases. Free. Radic.
Biol. Med. 33:1–14; 2002.
[9] Kopsidas, G.; Kovalenko, S. A.; Heffernan, D. R.; Yarovaya, N.; Kramarova, L.; Stojanovski, D.; Borg, J.; Islam, M. M.; Cara-gounis, A.; Linnane, A. W. Tissue mitochondrial DNA changes. A stochastic system. Ann. N. Y. Acad. Sci. 908:226 –243; 2000. [10] Fukagawa, N. K.; Li, M.; Liang, P.; Russell, J. C.; Sobel, B. E.;
Absher, P. M. Aging and high concentrations of glucose poten-tiate injury to mitochondrial DNA. Free Radic. Biol. Med. 27: 1437–1443; 1999.
[11] Berneburg, M.; Grether-Beck, S.; Kurten, V.; Ruzicka, T.; Briviba, K.; Sies, H.; Krutmann, J. Singlet oxygen mediates the UVA-induced generation of the photoaging-associated mitochon-drial common deletion. J. Biol. Chem. 274:15345–15349; 1999. [12] Lee, H. C.; Yin, P. H.; Lu, C. Y.; Chi, C. W.; Wei, Y. H. Increase of mitochondria and mitochondrial DNA in response to oxidative stress in human cells. Biochem. J. 348:425– 432; 2000. [13] Everett, T. H.; Li, H.; Mangrum, J. M.; McRury, I. D.; Mitchell,
M. A.; Redick, J. A.; Haines, D. E. Electrical, morphological, and ultrastructural remodeling and reverse remodeling in a canine model of chronic atrial fibrillation. Circulation 102:1454 –1460; 2000.
[14] Lim, P. S.; Cheng, Y. M.; Wei, Y. H. Large-scale mitochondrial DNA deletions in skeletal muscle of patients with end-stage renal disease. Free Radic. Biol. Med. 29:454 – 463; 2000.
[15] Lee, H. C.; Yin, P. H.; Yu, T. N.; Chang, Y. D.; Hsu, W. C.; Kao, S. Y.; Chi, C. W.; Liu, T. Y.; Wei, Y. H. Accumulation of mitochondrial DNA deletions in human oral tissues— effects of betel quid chewing and oral cancer. Mutat. Res. 493:67–74; 2001.
[16] Lee, H. C.; Lu, C. Y.; Fahn, H. J.; Wei, Y. H. Aging- and smoking-associated alteration in the relative content of mitochon-drial DNA in human lung. FEBS Lett. 441:292–296; 1998. [17] Yakes, F. M.; van Houten, B.. Mitochondrial DNA damage is
more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci.
USA 94:514 –519; 1997.
[18] Ballinger, S. W.; Patterson, C.; Yan, C. N.; Doan, R.; Burow, D. L.; Young, C. G.; Yakes, F. M.; Van Houten, B.; Ballinger, C. A.; Freeman, B. A.; Runge, M. S. Hydrogen peroxide- and peroxynitrite-induced mitochondrial DNA damage and dysfunc-tion in vascular endothelial and smooth muscle cells. Circ. Res.
86:960 –966; 2000.
[19] Ballinger, S. W.; Patterson, C.; Knight-Lozano, C. A.; Burow, D. L.; Conklin, C. A.; Hu, Z.; Reuf, J.; Horaist, C.; Lebovitz, R.; Hunter, G. C.; McIntyre, K.; Runge, M. S. Mitochondrial integrity and function in atherogenesis. Circulation 106:544 –549; 2002. [20] Cheng, K. C.; Cahill, D. S.; Kasai, H.; Nishimura, S.; Loeb, L. A.
8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G3 T and A 3 C substitutions. J. Biol. Chem. 267:166– 172; 1992.
[21] Douglas, B. S.; Wilson, S. C. Mitochondrial oxidative stress in heart failure “oxygen wastage” revisited. Circ. Res. 86:119 –120; 2000.
[22] Carnes, C. A.; Chung, M. K.; Nakayama, T.; Nakayama, H.; Baliga, R. S.; Piao, S.; Kanderian, A.; Pavia, S.; Hamlin, R. L.; McCarthy, P. M.; Bauer, J. A.; van Wagoner, D. R.. Ascorbate attenuates atrial pacing-induced peroxynitrite formation and elec-trical remodeling and decreases the incidence of postoperative atrial fibrillation. Circ. Res. 89:32e–38e; 2001.
[23] Allessie, M. A.; Boyden, P. A.; Camm, A. J.; Kleber, A. G.; Lab, M. J.; Legato, M. J.; Rosen, M. R.; Schwartz, P. J.; Spooner, P. M.; Van Wagoner, D. R.; Waldo, A. L. Pathophysiology and prevention of atrial fibrillation. Circulation 103:769 –777; 2001. [24] Territo, P. R.; French, S. A.; Dunleavy, M. C.; Evans, F. J.; Balaban, R. S. Calcium activation of heart mitochondrial oxida-tive phosphorylation: rapid kinetics of mVO2, NADH, and light scattering. J. Biol. Chem. 276:2586 –2599; 2001.
[25] Suleiman, M. S.; Halestrap, A. P.; Griffiths, E. J. Mitochondria: a target for myocardial protection. Pharmacol. Ther. 89:29 – 46; 2001.
[26] Wei, Y. H.; Lee, H. C. Oxidative stress, mitochondrial DNA mutation and impairment of antioxidant enzymes in aging. Exp.
Biol. Med. 227:671– 682; 2002.
[27] Angela, M. S.; Lezzaa, V. P.; Antonella, C.; Flavio, F.; Jacopo, V.; Giorgio, F.; Palmiro, C.; Maria, N. G. Increased expression of mitochondrial transcription factor A and nuclear respiratory fac-tor-1 in skeletal muscle from aged human subjects. FEBS Lett.
501:74 –78; 2001.
[28] Wu, H.; Kanatous, S. B.; Thurmond, F. A.; Gallardo, T.; Isotani, E.; Bassel-Duby, R.; Williams, R. S. Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science 296:349 –352; 2002.
[29] Szabados, E.; Fischer, G. M.; Toth, K.; Csete, B.; Nemeti, B.; Trombitas, K.; Habon, T.; Endrei, D.; Sumegi, B. Role of reactive oxygen species and poly-ADP-ribose polymerase in the develop-ment of AZT-induced cardiomyopathy in rat. Free Radic. Biol.
Med. 26:309 –317; 1999.
[30] Thijssen, V. L.; Ausma, J.; Liu, G. S.; Allessie, M. A.; van Eys, G. J.; Borgers, M. Structural changes of atrial myocardium during chronic atrial fibrillation. Cardiovasc. Pathol. 9:17–28; 2000.