383
Journal of Orgunometallic Chemistry, 371 (1989) 383-392 Elsevier Sequoia S.A., Lausanne - Printed in The Netherlands JOM 09877
Reinvestigation of the reaction of acid chloride
with Wilkinson’s catalyst: crystal and molecular structure
of ci&RhCl 2( PPh,) 2( COC,H,) complex
Ju-Yeh Shie, Ying-Chih Lin *, and Yu Wang
Department of Chernistty, National Taiwan University, Taipei, (Taiwan, R. 0. C.)
(Received November 28th, 1988; in revised form February llth, 1989)
Abstract
The interactions of Wilkinson’s catalyst RhCl(PPh,), (1) with some acid chlo- rides are reported. The less known cis-complex obtained directly from oxidative addition is the subject of this study. Reaction of CH,CHClC(O)Cl with 1 initially gives the cis-Rh(II1) acyl complex, RhCl,(PPh,),(COCHClCH,) (cis-2d), which then transforms to the corresponding truns-isomer. The 31P NMR spectra of this cis-complex show the virtual coupling for the two magnetically inequivalent PPh, ligands with the coupling constant of 15 Hz. No such virtual coupling was observed in the trans-isomer. Another five-coordinated cis-complex Cl,Rh(COC,H,)(PPh,) 2 was isolated from the reaction of CH,CH,COCl with 1 at low temperature and its crystal structure has also been determined. The cis-complex crystallizes in the monoclinic space group P?l/c in a cell having dimensions of a = 13.329(3), b = 14.644(3) c = 19.712(3) A and /3 = 99.52(l) O. Some 2954 unique reflections with I > 2.5 o(l) were used in the refinement to give final discrepancy indices of R = 0.045 and R, = 0.038. The shortest Rh-C separation 1.95(l) A, known for a single bond, might be the reason why the isolation and crystallization of this complex is possible.
Introduction
The Wilkinson catalyst is effective in the decarbonylation of aldehydes [l] and acid halides [2] which is a useful and important synthetic method for organic compounds. There are some reports on the decarbonylations catalyzed by RhCl(PPh,), under relatively mild conditions. Various products are observed when differing acid chlorides or aldehydes are used. An olefin is the product if the acid halide or aldehyde has a /&hydrogen. If the aldehyde or acid chloride has no P-hydrogen the alkane or alkyl chloride is the product of the decarbonylation. 0022-328X/89/%03.50 6 1989 Elsevier Sequoia S.A.
The mechanism of the decarbonylation [3] has been proposed as follows: RhCl(PPh,), + RhCl(PPh,), + PPh, (I) RhCl(PPh,), + RCOCl + cis-RhCl,(PPh,),(COR) (cis-2) cis-RhCl,(PPh,),(COR) + tran~-RhCl~(PPh~)~(C0R) ( trans-2) truns-RhC12(PPh&COR) + RhCl,(PPh,),(CO)R (3)
The stoichiometric decarbonylation reaction begins with the oxidative addition of acid chloride to RhCl(PPh,), to afford a cis-RhCl,(PPh,),(COR), followed by the conversion of the c&isomer to the trczns-isomer. However, it was not certain in complexes with PPh, ligands, whether the migration of the carbonyl group to form the alkyl complex occurs exclusively from the truns-isomer. Recently, Pignolet and co-workers were able to isolate a cis-Rh(II1) acyl complex by use of a chelating phosphine ligand but found no decarbonylation ability by such a c&complex even upon warming to 190 o C [4]. However, for a complex without chelating ligand, the detailed structure of cis-/ truns-isomers remains somewhat clouded. Preliminary results from single crystal X-ray structural determinations of the intermediates are not in agreement. RhCl,(COCH,CH,Ph)(PPh,), 151 is reported to have a square pyramidal geometry, whereas RhCl,(COCH,Ph)(PPh,), [6] is reported to have a trigonal bipyramidal structure, The structure assignments were made in the pre- liminary states of refinement and as such are subject to significant uncertainty. In this study, we explore the initial step of the decarbonylation of acid chlorides in the presence of the Wilkinson catalyst. A kinetically labile cis-RhCl,(PPh,),(COC,H,) complex was isolated at low temperature and fully characterized by single crystal X-ray diffraction study.
Experimental General
The ‘H and 31P NMR spectra were recorded on a Bruker AM-300WB FT NMR spectrometer using 5-mm NMR tubes. 31P NMR spectra of Rh-phosphine com- plexes were recorded with broadband proton decoupling in the composite pulse decoupling mode. Infrared spectra were recorded on a Perkin-Elmer 9836 Infrared spectrometer in solution cells equipped with calcium fluoride windows and 0.5 mm path length. All air-sensitive compounds were manipulated in a nitrogen-filled dry glovebox (VAC HE-63-P) or handled by use of Schlenk techniques. Chloroacetyl chloride, acetyl chloride, propionyl chloride and 2-chloropropionyl chloride were obtained from Merck. THF was dried by refluxing with Na/benzophenone under nitrogen before use. CH,Cl, and n-hexane were freshly distilled from calcium hydride under nitrogen. PPh, was recrystallized from ethanol.
Reactions
Wilkinson’s catalyst, RhCl(PPh,), (1) [7] was prepared by a published method. 31P NMR: 52.9 ppm (dt, JRh_p = 189 Hz, Jp_p = 37.5 Hz), 35.9 ppm (dd, JRh_p =
385
139 Hz, Jr-p = 37.5 Hz). The phosphine truns to the chloride gives a peak at lower field than the mutually truns phosphine ligands. The values of JRh_p for the phosphine truns to the chloride are much greater than for the mutually truns ones. Reactions of 1 with acid halides. Typically, a solution of ca. 30 mg complex 1 (0.03 mmol) was mixed with 5-10 ~1 of acid chloride to make a total volume of ca. 0.6 ml CDCl, solution in an NMR tube at 0°C and the reaction was then monitored by lH and 31P NMR spectroscopy at room temperature as a function of time.
NMR spectroscopy of the reactions of acetyl and propionyl chloride with 1 gives the same results as those obtained by Wilkinson et al.; namely, at room tempera- ture, the reaction of CH,COCl with 1 proceeds with the formation of the 5-coordi- nate cis-acyl isomer, ‘H NMR: 3.37 (s); 31P NMR: 29.8 (d, JRh_r = 145 Hz), then the truns-acyl isomer, ‘H NMR: 2.49 (s), “P NMR: 23.6 (d, JRh_P = 108 Hz), and finally an equilibrium mixture of the truns-isomer with the decarbonylation product RhCl,(PPh3),(CH,), ‘H NMR: 0.08 (m, JRH_H = 1.9 Hz, JP_” = 4.8 Hz); 31P NMR: 18.5 (d, JRh_r = 90 Hz). For CH,CH,COCl, no alkyl complex was observed at room temperature, i.e. after 7 h, an equilibrium mixture of cis-isomer, ‘H NMR: 4.03 (q, JH_H = 7.2 Hz), 1.17 (t); 31P NMR: 30.3 (d, JIul_P = 146 Hz), and truns-isomers, ‘H NMR: 2.98 (q, JH_H = 7.2 Hz, CH,), 0.08 (t, CH,); 31P NMR: 23.6 (d, JIuI_P = 111 Hz), was obtained. This solution was dried under vacuum and redissolved in CH,Cl, for growing single crystals of the cis-isomer. Since our interest is in the c&isomer, no heating was applied to cause decarbonylation.
For chloroacetyl chloride, the signals from the cis-complex RhCl,(PPh,),(CO- CH,Cl), ‘H NMR: 5.35 (s); 31P NMR: 29.9 (d, JRh_P = 139 Hz), are observed in the NMR spectrum as soon as the two components are mixed. NMR monitoring of the initial stage of the reaction, reveals the formation of a six-coordinate complex RhCl,(CO)(PPh,),(CH,Cl), ‘H NMR: 3.71 (dt, JP_H = 6.3 Hz, JRh_” = 2.7 Hz); 31P NMR: 17.7 ppm (d, JRh_P = 90 Hz), without detection of the corresponding truns-complex. Only once the reaction had gone to about 75% completion, was a trace amount of the truns-complex detected. 31P NMR: 22.4 ppm (d, JRh_P = 102 Hz).
When CH,CHClCOCl was mixed with 1, oxidative addition of the acid chloride immediately gave the 5-coordinated cis-Rh acyl complex which exhibits two doub- lets of doublet pairs in its 31P NMR spectrum, ‘H NMR: 5.94 (q, JH_H = 7.2 Hz), 1.40 (d); 31P NMR: 28.7 (dd, JRh_P = 139 Hz, JP_P = 15 Hz), 25.5 (dd, JRh_P = 139 Hz, Jp-p = 15 Hz). The truns-isomer, lH NMR: 4.63 (q, JH_H = 7.3 Hz), 0.76 (d);
31P NMR: 22.7 (d, JRh_P = 106 Hz), began to form after about an hour and after 12 h the cis- and truns-isomers were present in a ratio of 1 : 9. No virtual P-P coupling was detected for the truns-isomer. At room temperature, no decarbonylation prod- uct was detected in the NMR spectrum as was the the case for CH,CH,COCl. No heat was applied to cause decarbonylation.
Gystul structure determination
Crystals suitable for single crystal X-ray analysis were obtained by slow evapora- tion from a CH,Cl, solution containing an equilibrium mixture of cis- and truns-isomers of RhCl,(PPh,),(COEt) in a freezer at -20°C. It seems that it is easier for the &s-isomer to form single crystals. Crystals of cis- RhCl,(COC,H,)(PPh,), are monoclinic with a = 13.329(3), b = 14.644(3) c =
Crystal and intensity collection data for cis-Rh(PPh,),(COCH2CHs)CI, (at room temperature)
mol formula C,,H,,OCl,PzRb
space group a,A b, A 0 z, :eg v, K radiation MC+K, 28 range
scan speed, deg/min scan type
scan width
total no. of reflections
no. of unique reflections with 1) 2.50(I) standard reflections R RW P&/c 13.329(3) l&644(3) 19.712(3) 9952(l) 3794.72 A = 0.7107 A 20-50° 20/2-20/13 26/o 2(0.8 +0.35 tan 0) 6969 2954 (-2,2,-7) (2,2,7) (2, - 2,7) 0.045 0.038
19.712(3) A and /3 = 99.52(1)O as determined from a least-squares refinement of the angular settings of 25 reflections centered accurately on an Enraf-Nonius CAD4 diffractometer. Successful solution and refinement was achieved when the centric space group P2,/c (2 = 4) was used. A total of 6969 unique reflections were
Table 2
‘H and 31P NMR data for RhCl,L,(COR) and RhCl,L,(CO)R
Complex ‘H NMR 31P NMR cis-2a tram-2a 3a cis-2b #runs-2b 3b cis-242 WlWW2C cis-2d trams-2d 3.37 (s) 2.49 (s) 0.08 (m) (JahH =1.9 Hz) (Jr_ H = 4.8 Hz) 5.35 (s) - 3.71 (dt) (Jr.” = 6.3 Hz) (Jm_r = 2.7 Hz) 4.03(q), 1.17(t) (Jrr_u = 7.2 Hz) 2.98(q), 0.08(t) (JH_H = 7.2 Hz) 5.94(q), 1.40(d) (JH_u = 7.2 Hz) 4.63 (9). 0.76 (d) (JH_u = 7.2 Hz) 29.8 (d, JRh_,, =145 Hz) 23.6 (d, &,_r =108 Hz) 18.5 (d, Ja,,_p = 90 Hz) 29.9 (d, Ja,_p = 139 Hz) 22.4 (d, Jm,_p = 102 Hz) 17.7 (d, Ja,,_p = 90 Hz) 30.3 (d, &,,_r = 146 Hz) 23.6 (d, &,_r = 111 Hz) 28.7 (dd, JRh_r = 139 Hz) (Jr-p = 15 He) 25.5 (dd, JRh_r = 139 Hz) (Jr-p =15 Hz) 22.7 (d, &,_ r = 106 Hz)
387
measured in the scan range 28 = 2-50 o using graphite monochromatized MO-K, radiation (MO-K,, X = 0.7107 A) and employing a variable rate o-28 scan tech- nique. No decay was noted in the intensities of three standard reflections recorded after every 7300 sec. After correction for Lorentz, polarization, absorption and
Table 3
Atomic coordinate and isotropic thermal parameters for nonhydrogen atoms of cis-2c
Atom x Y z Bisn Rh Cl(l) CW) C(1) C(2) C(3) o(l) P(l) P(2) C(ll) C(l2) C(l3) C(l4) C(l5) C(l6) C(21) c(22) ~(23) c(24) c(25) c(26) C(31) ~(32) C(33) C(34) C(35) C(36) C(4U c(42) C(43) C(44) C(45) C(W C(51) c(52) c(53) C(54) C(55) C(56) C(61) C(62) C(63) C(64) C(65) C(66) 0.27555(6) 0.43056(20) 0.24469(23) 0.1841(8) 0.1955(8) 0.1244(12) 0.1233(5) 0.34369(20) 0.14730(20) 0.3779(7) 0.3883(g) 0.4159(10) 0.4345(9) 0.4255(9) 0.3978(7) O&27(7) 0.4607(S) 0.5502(10) 0.6404(g) 0.6433(8) 0.5551(8) 0.2811(7) 0.3276(8) 0.2863(g) 0.1995(10) 0.152q9) 0.1909(8) 0.1996(8) 0.1503(8) 0.1943(9) 0.2850(10) 0.3351(9) 0.2933(8) 0.0876(8) -0.0003(8) -0.0507(9) -0.0119(10) 0.0752(9) 0.1252(8) 0.0323(7) -0.0374(9) -0.1294(9) -0.1473(9) -0.0774(9) 0.0111(S) 0.11217(6) 0.05806(19) 0.17897(20) 0.0074(7) -0.0439(8) -0.1136(11) -0.0157(S) 0.05937(18) 0.20950(18) -0.0631(6) -0.1183(7) -0.2100(7) -0.2450(7) -0.1907(7) -0.1000(7) 0.1215(7) 0.2144(7) 0.2649(8) 0.2235(X) 0.1307(9) 0.0786(7) 0.0736(7) 0.1203(8) 0.1176(9) 0.0688(9) 0.0219(8) 0.0250(7) 0.3252(7) 0.3999(8) 0.4857(X) 0.5005(8) 0.4301(8) 0.3425(7) 0.1995(7) 0.1485(7) 0.1427(8) 0.1893(S) 0.2382(8) O-2433(7) 0.2142(7) 0.2818(X) 0.2830(g) 0.2174(9) 0.1511(X) 0.1482(8) 0.23281(4) 0.20257(14) 0.12181(14) 0.2168(5) 0.152q6) 0.1312(7) 0.2519(3) 0.34167(13) 0.25313(14) 0.3509(5) 0.2963(5) 0.3073(6) 0.371q6) 0.426q5) 0.4165(5) 0.3639(5) 0.3588(5) 0.3721(5) 0.3913(6) 0.3976(6) 0.3832(5) 0.4172(5) 0.4758(5) 0.5360(5) 0.5375(6) 0.4793(6) 0.4189(5) 0.2575(5) 0.2837(6) 0.2873(7) 0.2678(7) 0.2442(6) 0.2383(5) 0.3286(5) 0.3264(5) 0.3818(7) 0.442q6) 0.4456(6) 0.3908(5) 0.186q5) 0.1879(6) 0.1431(7) 0.0929(6) 0.0904(6) 0.1370(6) 2.34(3) 3.50(12) 3.98(14) 3.0(5) 4.3(6) 9.8(11) 4.1(4) 2.37(12) 2.55(12) 2.5(5) 3.9(6) 4.9(7) 4.5(6) 4.1(6) 3.2(5) 2.8(5) 3.6(5) 4.7(6) 4.8(6) 4.8(7) 3.7(5) 2.8(5) 3.8(5) 4.8(6) 5.6(7) 4.6(6) 3.5(5) 3.2(5) 4.3(6) 5.4(7) 5.6(7) 4.7(6) 3.3(5) 3.0(5) 3.9(6) 5.6(7) 5.6(7) 4.8(6) 3.5(5) 3.0(5) S-4(7) 5.8(7) 5.4(7) 5.3(6) 4.q6)
used in all the subsequent calculations. A three dimensional Patterson function revealed the positions of the rhodium atom. Fourier and difference Fourier analysis revealed the positions of all the remaining nonhydrogen atoms. Full matrix least- squares refinement, with all nonhydrogen atoms being refined anistropically, con- verged to final R and R, values of 0.045 and 0.038 respectively. Data collection parameters are summarized in Table 1, NMR data are listed in Table 2, and the final values of the positional and the isotropic thermal parameters are given in Table 3. Scattering factors and anomalous dispersion terms were taken from the literature [S].
Results and discussion
DecarbonyJation in the presence of Wilkinson’s catalyst
Acid halides, including CH ,COCl, CH,CH ,COCl, ClCH ,COCl and CH,CHClCOCl were allowed to react with Wilkinson’s catalyst RhCl(PPh,), (1) in CDCl, at room temperature. These reactions were monitored by 31P and ‘H NMR spectroscopy. A 14-electron species, RhCl(PPh,),, has been proposed as a solvent-stabilized, and very reactive intermediate in reactions involving Wilkinson’s catalyst [9]. Oxidative addition of an acid halide to RhCl(PPh,), initiates the stoichiometric decarbonylation. This very first step probably yields a 5-coordinate acyl complex with two PPh, ligands in cis configuration. Trigonal bipyramidal (TBP) and square pyramidal (SP) are two commonly observed geometries for complexes with 5-coordination. From the structure determination of RhCl,(PPh,),(COEt) described below, it is reasonable to assume that the acetyl complex also possesses SP geometry with the acetyl ligand occupying the apical position. The acyl complex isomerizes from a &-form to a trans-form which shows peaks at different chemical shifts and with different coupling constants in their respective ‘H and 31P NMR spectra. In addition, in the infrared spectra, absorp- tions in the Rh-Cl and carbonyl regions of the cis-isomer are also different from those of the trans-isomer [lo]. The 5-coordinate acyl complex possesses an open coordinate site that facilitates alkyl migration. Finally, the alkyl complex undergoes intramolecular reductive elimination to yield a chloroalkane and RhCl(CO)(PPh,),. An alkyl group bearing a &hydrogen probably undergoes elimination to give olefin and hydrogen chloride.
Interaction of various acid chlorides with 1
For the reaction of acetyl chloride with 1. Baird and co-workers [5] have proposed the reaction pathway mentioned above, namely, the formation of an unstable cis-acetyl complex, RhCl,(PPh,),(COMe) (cis-2a) which is followed by its isomerization at room temperature to another acyl complex, trans-2a. Then a six-coordinate complex RhCl z (PPh 3) 2 (CO)(Me) (3a) forms and establishes an equilibrium with tram-2a. For the reaction of CH,ClCOCl, Baird [lo] had reported the isolation of RhCl 2(CO)(PPh3) ,(CH,Cl) (3b) obtained directly from the reaction of ClCH,COCl with 1 in refluxing CH,Cl,. In our experiment, cis- RhCl,(PPh,),(COCH,Cl) (cis-2b) could be observed in the initial stage provided the solution was not heated. Complex cis-2b exhibits a resonance at 5.34 ppm for CH, group in its ‘H NMR spectrum, and a doublet peak at 29.9 ppm (JRh_p = 140
389
Hz) for PPh, in the 31P NMR spectrum. Alky 1 migration occurred in about 10 min while only a trace amount of tram-2b was detected by 31P NMR. The existence of
complex truns-2 was later confirmed by a weak doublet peak at 22.4 ppm in the 31P NMR spectrum after 2 h. It is reasonable to assume that the decarbonylation step involves the intermediate trans-2b, which is less stable than trans-2a.
Wilkinson and co-workers [ll] have described the reaction of excess CH,CH,COCl with 1 in refhtxing CH,Cl,. After some solvent had been removed rapidly from and diethyl ether added to the concentrated solution, trans-
RhCl,(PPh,),(COEt) (I runs-k) was obtained as a solid which gave a quartet/ triplet pair at 2.98/0.78 ppm in the ‘H NMR spectrum. In our experiment, when EtCOCl was added to a solution of RhCl(PPh,), in CDC13 at room temperature, another quartet/triplet pair at 4.03/1.17 ppm with Ju_u = 7.3 Hz was observed in the initial stage. The corresponding 31P NMR signal appeared as a doublet at 35.9 ppm with Jru_r= 146 Hz. This is assigned to the cis-RhCl,(PPh,),(COEt) (cis-2c). Later, the 2.98/0.78 ppm quartet/triplet pair appeared. These signals can be assigned to the complex tram-2c which shows peaks at 29.24 ppm with .JRh_r = 111 Hz in the 31P NMR spectrum. The peak attributed to the RhCl,(PPh,),(CO)(Et) complex was not detected during the course of reaction at room temperature during about 7 h.
It is expected that when CH,CHClCOCl reacts with 1, the resonances of the cis- and tram-acyl complexes on the 31P NMR spectrum should appear as doublets in the region of 29 and 22 ppm respectively. But upon mixing CH,CHClCOCl with RhCl(PPh,), in CDCI,, two doublets of doublet peaks appeared at 28.7 ( JRh_ r = 139 Hz) and 25.5 ppm (Jru_r = 140 Hz) in the 31P NMR spectra and in the ‘H NMR spectra, a quartet/doublet pair at 5.94/1.40 ppm also appeared. These peaks gradually decreased and a new doublet peak at 28.3 ppm ( JRh_p = 106 Hz) in the 31P NMR spectra grew up and the corresponding peaks on the lH NMR were present at 4.63/O-76 ppm. The presence of two doublets of doublet peaks in the 31P NMR spectrum indicated that two of the PPh, ligands are magnetically inequiv- alent in cis-2d, and the small coupling constant of 15 Hz is attributable to the virtual
coupling of magnetically inequivalent PPh, ligands. This inequivalence arises from the diastereoisomeric nature of cis-2d, the complex having a chiral carbon, C(O)C*HClMe, and a cis arrangement of the phosphines. The cis-2d isomerizes to the corresponding tram-2d with chemically and magnetically equivalent phosphine ligands.
As to the structure of pentacoordinate complex of Rh, a number of papers have been reported [12] and these may have either trigonal bipyramidal (TBP) or square pyramidal (SP) structure. Interestingly for a TBP structure, the coupling constant J Rh_p for the equatorial phosphite is larger than that for the corresponding axial phosphite, for example, [Rh(P(OMe),),]BPh,, JRh_Rej = 206, Hz, JRh_PCaj = 143 Hz [13]. A similar dependence of Jm_ p has been observed in the trigonal bipyra- midal complex, RhCl(Ph,POCH,CH=CH,), and other related complexes in which one phosphorus occupies an equatorial and the other an axial position. A TBP arrangement has been suggested for RhCl ,(PhCH,CH,CO)(PPh, ) 2, on the basis of
preliminary X-ray data; two phosphines occupy the axial positions.
For a 5-coordinated acyl complex the transformation between TBP and TP is probably a fast equilibration process. For acetyl chloride, there is an equilibrium between trans-acyl complex and alkyl complex with the major stable species being
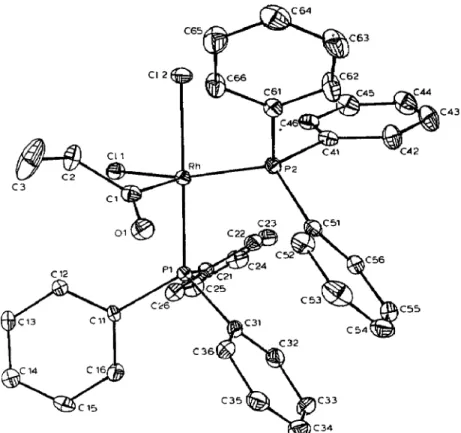
Fig. 1. ORTEP drawing of ci.+RhCl,(PPh,),9COEt), cis-2~. Hydrogen atoms are omitted for clarity.
the trans-acyl complex. However, under similar reaction conditions, the ethyl complex, Cl,Rh(CO)(CH,CH,)(PPh,),, was not observed in our experiment but Cl,Rh(CO)(CH,Cl)(PPh,), was readily formed. Thus electronic effects play an important role in the alkyl migration reaction. The electron-withdrawing group enhances the ability to undergo the acyl-to-ahcyl rearrangement. This conclusion is consistent with a report by Stille, who carried out decarbonylations on a number of benzoyl complexes, (p-YC6H,CO)Cl,Rh(PPh3), (Y = OCH,, H, Cl, NO,) [13]. The first-order rate constant of alkyl migration was found to fall in the order Y=NO,>Cl>H>OCH,.
Solid state structure of cis-Rh(COC, H,)CI,(P(C,H,),),.
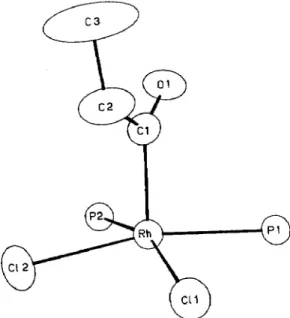
The crystal structure of cis-2c contains discrete monomeric molecules. An ORTEP perspective drawing along with the labeling scheme is shown in Figure 1. The coordination geometry around the metal is square pyramidal with the propionyl group occupying the apical position. A view of the SP coordination geometry is presented in Figure 2. Atomic coordinates and isotropic thermal parameters are given in Table 3 and important intramolecular distances and angles for the structure are given in Table 4. The orientation of the acyl group is such that the acyl oxygen points between the two phosphine ligands which might minimize nonbonded repulsions with the PPh, ligand and thus restricting rotation about the Rh-acyl bond. For a low spin, pentacoordinate d6 complex, square pyramidal geometry is expected, which is precisely what is observed here. Pignolet and co-workers [4] used
391
Fig. 2. Square pyramidal geometry around the Rh atom for cis-2c.
a chelating ligand to elucidate the crystal stpcture of cis-RhCl,(COPh)(dppe) and found that the Rh-C separation is 1.992(3) A. A list of Rh(III)-C u-bond distanc!s has been tabulated by Collman et al. [14]. All values are in the range 2.05-2.26 A, except for the 1.97 A in the chelating carbene complex RhI,(CO)(CPhN(Me)C(Ph)-
Table 4
Selected bond distances and angles for cis-2c Bond distances (2) WI-Cl(l) Rh-Cl(Z) Rh-C(1) Rh-P(1) Fur-P(Z) C(l)-C(2) c(1)--O(1) Bond angles ( “) Cl(l)-Fur-Cl(Z) Cl(l)-Rh-C(1) Cl(l)-Rh-P(1) Cl(l)-Rh-P(2) Cl(Z)-Rh-C(1) C1(2)-Rh-P(1) CI(2)-Rh-P(2) C(l)-Rh-P(1) C(l)-Rh-P(2) P(l)-Rh-P(2) Rh-C(l)-C(2) Rh-C(l)-O(1) (2)-C(l)-o(1) 2.379(3) 2.370(3) 1.953(10) 2.320(3) 2.311(3) 1.504(14) 1.198(12) 86.07(10) 104.0(3) 84.11(10) 161.24(10) 99.6(3) 166.69(11) 83.40(10) 91.5(3) 93.0(3) 103.47(10) 112.4(7) 126.3(8) 121.3(9) wbc(11) P(l)-C(21) P(l)-c(31) P(2)-~(41) P(2)-C(X) P(2)-C(61) C(3)-C(2) Rh-P(l)-C(11) Rh-P(l)-C(21) Rh-P(l)-C(31) C(ll)-P(l)-C(21) C(ll)-P(l)-C(31) C(21)-P(l)-C(31) Rh-P(2)-C(41) EUt-P(2)-C(51) Rh-P(2)-C(61) C(41)-P(2)-C(51) C(41)-P(2)-C(61) C(51)-P(2)-C(61) 1.X52(9) 1.819(10) 1.X35(10) 1.X29(10) 1.X05(10) :.x48(10) 1.409(17) 117.8(3) 104.4(3) 123.5(3) 105.7(5) 99.7(4) 104.1(4) 107.0(3) 122.2(3) 116.7(3) 104.6(S) 105.3(5) 99.4(5)
NMe) where metal --, ligand back bonding undoubtedly occurs. in the present structure, the very short Rh-acyl carbon bond distance is 1.95(l) A, even shorter than that of the Rh-carbene separation. Such a short separation could be rational- ized in terms of two factors, namely (i) the decrease of the covalent radius of carbon on going from the sp3 hybridization of alkyls to the sp* hybridization of an acyl, and (ii) a back-bonding interaction between a filled d, orbital of the Rh(II1) center and a vacant T* orbital of the acyl ligarid. [4]. Other parameters within the structure are more or less as expected. The Rh-P bond lengths average 2.316(3) A and agree with typical Rh(III)-phosphine values reported. Experiments in progress are di- rected at determining the Rh-C separation for the truns-isomer.
Acknowledgements
We thank the National Science Council of the Republic of China for financial support.
References
1 (a) J.P. Collman and L.S. Hegedus, J.R. Norton and R.G. Finke, Principles and Applications of Organotransition Metal Chemistry, publisher(s)?, 1987, p 768. (b) H.M. Walborsky and L.E. Allen, J. Am. Chem. Sot., 93, (1971) 5465. (c) H.M. Walborsky and L.E. Allen, Tetrahedron Lett., (1970) 823. 2 (a) K. Obno and J. Tsuji, J. Am. Chem. Sot., 90 (1968) 99. (b) J. Tsuji and K. Ohno, J. Am. Chem.
Sot., 88 (1966) 3452. (c) J. Tsuji and K. Ohno, Tetrahedron Lett., (1966) 4713.
3 L.H. Pignolet, Homogeneous Catalysis with Phosphine Complexes, Plenum Press, New York, 1983, p 343-375.
4 M.F. McGuiggan, D.H. Doughty, L.H. Pignolet, J. Organomet. Chem., 185 (1980) 241.
5 D.L. Egglestone, MC. Baird, C.J.L. Lock and G. Turner, J. Chem. Sot., Dalton Trans., (1977) 1576. 6 K.S.Y. Lau, Y. Becker, F. Haung, N. Baenziger and J.K. Stille, J. Am. Chem. Sot., 99 (1977) 5664. 7 J.A. Osbom, F.H. Jardine, J.F. Young and G. Wilkinson, J. Chem. Sot. (A), (1966) 1711.
8 International Tables for X-ray Crystallography, Kynoch Press, Birmingham, 1974, Tables 2.2B and 2.3.1.
9 (a) J. Halpem and C.S. Wong., J. Chem. Sot., Chem. Commuu., (1973) 629. (b) J. Halpem, Inorg. Chim. Acta., 50 (1981) 11. (c) F.H. Jardine, Prog. Inorg. Chem. 28, (1982) 64 (d) D.A. Wink and P.C. Ford, J. Am, Chem. Sot., 109 (1987) 436.
10 D.A. Slak D.L. Egglestone and M.C. Baird, J. Grganomet. Chem., 146 (1978) 71. 11 M.C. Baird, J.T. Mague, J.A. Osbom and G. Wilkinson, J. Chem. Sot. (A), (1967) 1347.
12 P.S. Pregosin and R.W. Kunz, P. and C NMR of Transition Metal Phosphine Complexes, Springer- Verlag, New York, 1979, p. 38.
13 J.K. Stille and M.T. Regan, J. Am. Chem. Sot., 96 (1974) 1508.
14 (a) J.A. Evans, D.R. Russell, A. Bright, B.L. Shaw, J. Chem. Sot., Chem. Commun., (1971) 841. (b) P.C. Appleton, H.C. Clark, L.E. Manzer, Coord. Chem. Rev., 10 (1973) 335. (c) J.P. Collman, P.A. Christian, S. Current, P. Denise&h, T.R. Halbert, E.R. Schmittou, K.O. Hodgson, Inorg. Chem., 15 (1976) 223.
15 P.B. Hitchcock, M.F. Lappert, G.M. McLaughlin, A.J. Oliver, J. Chem. Sot., Dalton Trans., (1974) 68.