ALTERATION OF PROTEIN KINASE C ISOFORMS IN THE LIVER OF
SEPTIC RAT
Chin Hsu, Ya-Ching Hsieh, Hseng-Kuang Hsu, Shiao-Ching Jao, and
Rei-Cheng Yang
Department of Physiology, Kaohsiung Medical University, Kaohsiung, Taiwan, R.O.C.
Received 24 October 2000; first review completed 1 December 2000; accepted in final form 27 February 2001 ABSTRACT—The present study investigated the alteration of protein kinase C (PKC) isoforms in rat liver during the progression of sepsis. Cecal ligation and puncture (CLP) model of polymicrobial sepsis was used, with early and late sepsis referring to those animals sacrificed at 9 and 18 h, respectively, after CLP. The protein contents of various PKC isoforms were quantified by Western blot and densitometric analysis. PKC␣activity was performed after immunoprecipi-tation and assayed based on the incorporation rate of32p from [␥-32p] adenosine triphosphate (ATP) into histone. The distribution of PKC␣was evaluated by immunohistochemical staining. The steady state expression of PKC␣mRNA was estimated by reverse transcriptase-polymerase chain reaction (RT-PCR). The results indicated that 1) five isoforms (␣,, ␦,,) could be detected in normal rat liver. PKC␣andwere predominantly present in the cytosolic fraction, while membrane-associated PKC␦was more prominent than that of cytosolic fraction; 2) the protein content of membrane-associated PKC␣was significantly decreased at early (P < 0.05) and late (P < 0.01) sepsis; 3) there was no significant difference of protein contents of PKC-␦, -and -between sham-operated and septic rat liver; 4) the activity of membrane-associated PKC␣was significantly declined under detection level during sepsis; 5) at both early and late sepsis, the immunohistochemical staining of PKC␣was significantly diminished, especially in the nucleus; 6) the RT-PCR product of PKC␣mRNA of septic liver was significantly less than the sham-operated liver. These results suggest that inactivation and the suppression of PKC-␣gene transcription might be involved in modulating hepatic failure during sepsis.KEYWORDS—PKC␣, hepatic failure, sepsis
INTRODUCTION
Protein kinase C (PKC) mediates phosphorylation of
proteins and modulates the function of signal transduction
pathways leading to gene expression (1), cell proliferation (2,
3), and cell survival (4, 5). In metabolic homeostasis, PKC
plays an important role in the regulation of hepatic glucose
metabolism (6). Our previous result has shown that PKC
activ-ity of rat liver is inactivated during the late the hypoglycemic
phase of sepsis (7). Similar correlation has also been
docu-mented between PKC and numerous pathological situations,
such as Alzheimer disease, pancreatic cancer, myocardium
cardiomyopathy, or human proliferative glomerulonephritis.
Currently, at least 12 mammalian PKC isoforms have been
identified (8). Each individual PKC isoform possesses distinct
mode of activation, kinetic property, substrate specificity,
diverse patterns of tissue expression, and intracellular
distribu-tion (9, 10). PKC isoforms are mainly located in the cytoplasm.
Once activated, they translocate from the cytoplasm to the
plasma membrane and also to the nucleus, termed as
membrane-associated form (11). The differential subcellular
localization is associated with regulation of cell function in situ
(12). Specific PKC isoform exerts spatially defined effects by
virtue of their directed translocation to distinct intracellular
sites (13).
In mammalian liver, Stravitz et al. (14) have
immunochemi-cally shown that there are at least 5 PKC isoforms,
␣
,

,
␦
,
,
and
, expressed constitutively. Further functional studies
revealed that PKC-
␣
,
␦
, and
isoforms are involved in cell
growth events of human hepatoma Hep 3B cells (15).
Inhibi-tion of PKC

II was found to lead to cell cycle arrest in the
G
2/M phase (16). The expressions of PKC-
␣
, and -

II in
hepa-tocytes were selectively increased in diabetes (17).
Strepto-zotocin (STZ)-induced diabetes also induces the expression of
a biologically inactive form of PKC
␣
in liver by an undefined
post-translational modification (18). PKC
␣
and PKC
␦
are
suggested to have different roles in liver regeneration and cell
proliferation (19). Moreover, PKC-
is abundant in fetal liver
and plays a role in cell proliferation (20). These data indicate
that specific PKC isoform plays a different role in the hepatic
survival and metabolism. Therefore, it is interesting and
essen-tial to investigate which isoform(s) is (are) major involved in
modulating hepatic failure during sepsis, for both the
pathoge-netic research and clinical therapeutic intervention. In this
study, we analyzed the expression and distribution of various
PKC isoforms by immunochemical, immunohistochemical
studies, and enzyme activity measurement to explore the
molecular metabolic alternation leading to the liver
dysfunc-tion during sepsis.
MATERIALS AND METHODS
Materials
Monoclonal antibodies against PKC-␣, -, ␦, and - and positive control were purchased from Transduction Laboratories (Lexington, KY). Polyclonal anti-PKC- rabbit polyclonal antibody IgG was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Pre-stained protein molecular weight standard was obtained from Bio-Rad Laboratories (Hercules, CA). Biotin and streptoavidin-peroxidase were purchased from DAKO (Carpinteria, CA). Diaminobenzidine (DAB) was purchased from Vector Laboratories (Burlingame, CA). Other chemicals and reagents were of analytical grade.
Animal model
In conducting the research described in this study, the authors adhered to the guidelines of the National Institutes of Health for the use of the experimental animals and all experiments were approved by the Committee for the Use of Experi-Address reprint requests to Rei-Cheng Yang, MD, Department of Physiology,
Kaohsiung Medical University, Kaohsiung City, Taiwan
normal saline at the completion of surgery and also at 7 h post-surgery. Animals were fasted but had free access to water after operative procedures. Early and late sepsis refers to those animals sacrificed at 9 and 18 h, respectively, after CLP. The livers were sampled from rats of each group under pentobarbital anesthesia. The tissues were then chilled immediately in ice-cold buffer A [(2 mM EDTA, 10 mM EGTA, 10 mM dithiothreitol, 0.15 M sucrose, 25 mM Tris-HC1, pH 7.4), and protease inhibitor (0.1 mM PMSF, 1 mM pepstatin A, 0.1 mM benzamide, 1 mM leupeptin, 1 mg/mL aprotinin, 1 mg/mL trypsin-inhibitor)] for further experiments.
Subcellular fraction preparation of rat liver homogenate
Different fraction of tissue proteins was extracted and partially purified by the method of Wise B.C. et al., (22) with modification. Approximately 2–3 mg of minced liver, trimmed free of visible fatty and vascular tissues, was homogenized in 2 mL of buffer A with a Tekmar tissumizer (Cincinnati, OH; Model SDT). The homogenate was centrifuged at 100,000 g for 60 min, and the resultant supernatant was filtered through glass wool and was used as a crude cytosolic PKC-contained extract. To prepare membrane-associated PKC-contained fraction, the above pellet was rehomogenized in 2 mL of buffer A containing 0.25% TritonX-100. The mixture was shaken for 2 h followed by centrifugation at 30, 000 g for 20 min. The resulting supernatant was filtered through glass wool and was used as crude membrane-associated PKC-contained extract.Western blot analysis
The protein content was estimated using the Bio-Rad protein microassay proce-dure. Optimal amount (20 g) of cytosolic extract and the same volume of membrane-associated extract were loaded and separated by 10% SDS-PAGE simul-taneously. The gel was transferred to polyvinylidene fluoride (PVDF) membrane by a Bio-Rad transblot apparatus at constant 100 V for 1 h. The membranes were blocked with 5% nonfat dry milk/TBS-T (20 mM Tris-HCl, pH7.4, 150 mM NaCl, 0.1% Tween-20) for 1 h and then incubated with the monoclonal anti-PKC-␣, -, -␦, - Transduction Laboratories; Cat #P16520, P17720, P36520, P14820) or poly-clonal anti-PKC- (Santa Cruz; Cat #SC-216-G) antibody, respectively, in 5% nonfat dry milk/TBS-T for 1 h at room temperature. Membranes were washed completely with TBS-T buffer and incubated with anti-mouse IgG-HRP (Transduc-tion Laboratories; Cat #M15345, for isoforms␣, , -␦, and -) or anti-rabbit IgG-HRP (Transduction Laboratories; Cat #R14745, for isoform ) for 1 h at room temperature. Target isoforms were developed using the ECL (Amersham Life Science; Piscataway, NJ) detection system and autoradiography. The bands were scanned and quantified.
Immunoprecipitation of PKC-
␣
Samples were incubated with 5 g of monoclonal anti-PKC␣ (Transductional Lab.) by rotation for 2 h at 4° C. Then, protein G plus/protein A-agarose (CALBIO-CHEM; Cat #IP05) was added to each sample, followed by agitation at 4°C for 4 h to precipitate the antigen-antibody complex. The suspension was subjected three times successively to a 5-min spin in a microcentrifuge followed by washing of the pellet. Immune complexes were recovered from the beads by suspension of the pellet in 100l of activity assay buffer.
Assay of PKC-
␣ activity
PKC activity was assayed according to the method of Wise et al. with modifi-cation. The standard reaction mixture in a final volume of 0.25 mL contained 40 mM Tris-HCl (pH 7.4), 5 mM MgCl2, 10 mM free Ca
2+, 0.25 mg histone, 0.2 m M
[␥-32
p] ATP with a radioactivity of approximately 2.5 × 106
cpm. The reaction was performed with or without 70 mg of phosphatidylserine plus 40 mM of diacylglyc-erol. The reaction was initiated by the addition of partially purified PKC enzyme preparation and was allowed to proceed for 2 min at 37°C. At the end of incubation, the reaction was terminated by the addition of 1 mL of 15% trichloroacetic acid solution containing 1 mg of bovine serum albumin as a carrier protein. The mixture was centrifuged at 3,000 × g for 10 min. The resultant pellet was dissolved in 0.2 mL of 1 N NaOH, and the protein was reprecipitated. This procedure was repeated one more time. The final pellet was dissolved in 0.4 mL of 1 N NaOH, and the radioactivity was determined by a liquid scintillation counter. The enzymatic
activ-then were incubated with blocking solution for 1 h at room temperature. The sections were incubated 2 h at 37°C in a humid chamber with PKC antibody. After washing, the sections were incubated with Biotin labeled secondary antibody for 20 min and then incubated with streptoavidin-peroxidase for another 20 min. The sections were finally developed with a DAB containing 0.01% hydrogen peroxidase for 5 min (Vector Laboratories).
TUNEL staining
The specimens were de-paraffinized and then subjected to proteinase K (20 g/mL) digestion (25°C, 20 min). Three percent hydrogen peroxide was used to quench endogenous peroxidase activity. DNA strand breaks were identified by TUNEL assay using the KLENOW FragEL™ DNAfragmentation detection kit (Calbiochem, Cat #QIA21) following the manufacturer’s protocol. The Signal of TdT-mediated dNTP nick end labeling (TUNEL) was then detected by streptavidin conjugated with HRP, a reporter enzyme that catalytically generates a brown-colored product from the chromogenic substrate diaminobenzidine (DAB). The cells were counter stained with methyl green. Negative controls were processed identi-cally except that TdT was not added. The incidence of apoptosis was derived from the quotient of TUNEL positive cell mumber divided by the sum of total cell munbers in each section. All measurements were made by a single person.
RT-PCR of PKC-
␣ mRNA
Total RNA was isolated from liver tissue according to a method described by Chomczynski P. & Sacchi N (23). First-strand cDNA was synthesized using oligo (dT) primers and reverse transcriptase using an RT-PCR kit (Titan ™ Boehringer Mannheim). The sense primer used was 5⬘-TGAACCCTCAGTGGAATGAGT-3⬘ and the antisense primer was 5⬘-GGCTGCTTCCTGTCTTCTGAA-3⬘. The cDNA of-actin was also amplified as an internal standard. The primers used were 5⬘ primer: 5⬘-CTACAATGAGCTGCGTGTGG-3⬘. 3⬘ primer: 5⬘-TAGCTCTTCTC-CAGGGAGGA-3⬘. The amplification profile involved denaturation at 95°C for 1 min, primer annealing at 59°C for 45 s, and extension at 72°C for 4 min. This cycle was repeated 35 times using a Perkin-Elmer thermocycle (model 2400) with 1U of Taq DNA polymerase.
Statistical analysis
The statistical analysis of the data was performed using one-way analysis of variance followed by Newman-Keuls test. A 95% confidence limit was accepted as statistically significant.
RESULTS
Expression of PKC isoforms in normal rat liver
The expression of PKC isoforms in normal rat liver was
investigated by Western bolt and immunochemical staining by
antibodies recognizing PKC-
␣
, -

, -
␦
, -
, -
. The identification
of each isozyme was confirmed by characterizing
immunore-active proteins purchased from Transduction Laboratories. As
shown in Figure 1, all five detected PKC isoforms were
visu-alized in cytosolic and membrane-associated fractions of the
liver extract.
Alterations in expression of PKC isoforms of rat liver
during CLP-induced sepsis
After quantification of the band of protein by densitometer,
the content of membrane-associated PKC-
␣
was decreased by
25% (P < 0.05) and 35% (P < 0.01) during early and late
sepsis, respectively. However, the content of cytosolic PKC-
␣
was not altered significantly between the early or late stage of
sepsis (Fig. 2). As shown in Figure 3, the content of cytosolic
PKC-

was increased by 46% (P < 0.05) during late sepsis,
while no significant difference of membrane-associated PKC

was found between sham and septic rats. No significant
differ-ence was observed PKC
␦
(Fig. 4), PKC-
(Fig. 5), and PKC-
(Fig. 6), both in cytosolic and membrane-associated fractions,
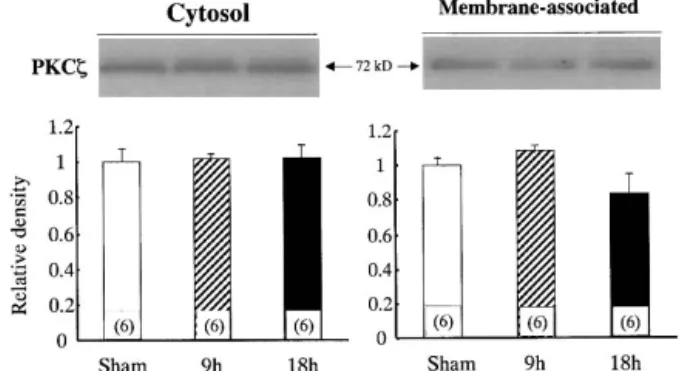
between sham and septic rat liver.
Changes of PKC-
␣ activities in rat liver during
CLP-induced sepsis
Owing to the significant change observed in the quantitative
study, we concentrated the focus on the PKC-
␣
in the
follow-ing investigations. Befollow-ing similar with the result of protein
content, the in vitro activities of cytosolic PKC-
␣
showed no
significant difference between sham- operated and septic rat
livers. As expected, the membrane-associated PKC-
␣
activities
were undetectable by the current method in both early and late
septic rat liver (Table 1).
Immunohistochemical study of PKC-
␣ in rat liver during
CLP-induced sepsis
To understand the definite cellular redistribution of PKC-
␣
in liver cells during sepsis, we performed the
immunohisto-chemical study. As shown in Figure 7A, immunoimmunohisto-chemical
activity was homogenous stained in cytoplasm, with condensed
FIG. 1. Protein kinase C isoform distribution in rat liver. Proteinextracts from rat liver were prepared as described in Materials and Method. Liver cytosolic and membrane-associated and nuclear protein (20 µ g protein/lane) extract were separated in an 10% SDS-polyacrylamide gel, transferred to PVDF membrane, and subjected to Western immunoblotting. PKC isoforms were detected by enhanced chemiluminescence (ECL, 5–20 min exposure). For all isoforms, rat brain was extracted as a positive control. A molecular mass maker for each isozyme is shown at the left. Specificity of immunoreactive bands was demonstrated for each antibody by preincubating the respective primary antibody with the PKC isoform-specific peptide against which it was raised. P: positive control, C: Cytosolic, M: membrane-associated.
FIG. 2. The cytosolic and membrane-associated PKC␣ protein expressions in rat liver derived from sham-operated rats or at 9 and 18 h after CLP. The molecular weight of PKC␣ is 82KD. The data shown indi-cates mean ± SD of six samples in each group. *Indiindi-cates P < 0.05, **indi-cates P < 0.01.
FIG. 3. The cytosolic and membrane-associated PKC protein expressions in rat liver derived from sham-operated rats or at 9 and 18 h after CLP. The molecular weight of PKC is 80KD. The data shown indi-cates mean ± SD of six samples in each group. *Indiindi-cates P < 0.05.
FIG. 4. The cytosolic and membrane-associated PKC␦ protein expressions in rat liver derived from sham-operated rats or at 9 and 18 h after CLP. The molecular weight of PKC␦ is 78KD. The data shown indi-cates mean ± SD of six samples in each group.
FIG. 5. The cytosolic and membrane-associated PKC protein expressions in rat liver derived from sham-operated rats or at 9 and 18 h after CLP. The molecular weight of PKC is 90KD. The data shown indi-cates mean ± SD of six samples in each group.
stain in the nucleus, in the specimen of sham rat. During the
early stage of sepsis, the staining of nuclei was fading away
while the cytosolic staining was not significantly changed (Fig.
7B). As the sepsis progressed, the characteristics of condensed
staining in nuclei nearly disappeared from the specimen of late
sepsis, leading to an ill-defined picture through the whole field
(Fig. 7C).
The increase of apoptotic incidence in rat liver during
CLP-induced sepsis
The incidence of hepatic apoptosis per 1,000 cells in the
liver of sham-operated rat (2.96% ± 0.97%) was significantly
lower than that in early (10.45% ± 1.24%) (P < 0.01) and late
phases of sepsis (13.87% ± 1.76%) (P < 0.01) (Fig. 8). The
number of rats in each group was six.
The RT-PCR product of PKC
␣-mRNA in rat liver
during sepsis
Figure 9 showed the changes of the RT-PCR product of the
steady-state level of PKC-
␣
mRNA in rat liver during sepsis.
The reverse transcription and amplification of total RNA
isolated from liver tissues resulted in a single band of 325 bp
using PKC-
␣
specific primer and a single band of 450 bp using

-actin specific primer. Analysis of the densitometric signals
revealed that the RT-PCR product of PKC-
␣
mRNA was
significantly decreased by 59% (P < 0.01) and 69% (P < 0.01)
during early and late sepsis, respectively.
DISCUSSION
It is well known that inactivation of PKC causes cellular
dysfunction resulting in various organ failures. With the
advance in biochemical characterization, at least 12 isoforms
have been reported (8). Each isoform was regulated by its own
way and exerted its physiological function independently. In a
pathological situation, specific isoform was revealed to be
responsible for specific diseases. In our previous study, we
have shown that the hepatic PKC was inactivated during late
sepsis and suggested to be the factor leading to the
hypo-metabolic phase during severe infection (7). However, which
isoform is the major one is still obscure.
In the present study, we demonstrated that 5 PKC isoforms,
-
␣
, -

, -
␦
, -
, and -
are expressed in rat liver. During sepsis,
only isoforms -
␣
, and -

altered in specific distribution. The
protein content of membrane-associated PKC-
␣
was
signifi-cantly decreased, which was further confirmed by decline of
enzyme activity. Although PKC-
␣
protein content was only
decreased by 25–35% (Fig. 2), there was a near complete loss
of kinase activity. Since post-translational modification may
occur during septic condition, the PKC-
␣
activity might not
FIG. 6. The cytosolic and membrane-associated PKC proteinexpressions in rat liver derived from sham-operated rats or at 9 and 18 h after CLP. The molecular weight of PKC is 72KD. The data shown indi-cates mean ± SD of six samples in each group.
TABLE1. Activities of cytosolic and membrane-associated PKC␣ of the liver from sham and CLP-induced septic rats
Fraction
Activity (p moles/mg.min)
Sham 9 h after CLP 18 h after CLP Cytosol 32.36 ± 4.33 (3) 39.14 ± 4.18 (3) 38.75 ± 5.66 (3)
Membrane-associated 28.24 ± 4.24 (3) U.D. (3) U.D. (3) Activities are expressed as p moles/mg of protein/min (m ± SEM). Numbers in parenthesis indicates number of rats.
U.D.: under detection.
FIG. 7. Immunohistochemical staining of the PKC␣-isoform of liver tissues in sham-operated or septic rats. (A) liver tissue of sham operated rat, (B) 9 h after CLP, and (C) 18 h after CLP (original magnification ×200).
absolutely correlate with protein content. However, we can’t
exclude the possibility that the activity assay might not be
sensitive enough to detect the very low protein content of
membrane-associated PKC-
␣
, which is significantly less than
that of cytosolic fraction, especially during sepsis. Our
previ-ous report (7) showed that, during early sepsis, both
membrane-associated and cytosolic PKC activity remained
relatively unaltered. However, the membrane-associated
PKC-
␣
activity and protein expression were decreased (P <
0.05) during early sepsis in the present result. The PKC activity
showed in the previous report represented the total activity of
all PKC isoforms (at least all calcium-dependent isoforms).
Whereas, the PKC-
␣
activity showed in the current data is the
activity of PKC-
␣
only, because immunoprecipitation of
PKC-
␣
isoform was performed before activity assay. A
decrease of specific isoform could be present, while the total
activity unchanged. The present observation indicated that
PKC-
␣
might be crucial in hepatic failure during sepsis.
PKC modulates the function of a variety of signal
transduc-tion pathways leading to gene expression and each isoform
may perform distinct functions via its translocation to discrete
regions within the cell (13). Recent studies indicate that PKC
itself may translocate to the nucleus and directly phosphorylate
nuclear regulatory proteins (24, 25, 26). Our data of both
West-ern blot study and activity assay showed that the membrane
fraction decreased progressively and activity became
undetect-able during the progression of sepsis. In addition, the result of
immunohistochemical staining showed that the decrease of
PKC
␣
during sepsis seems more prominent in nucleus than in
cytosol or plasma membrane. Recently, we further found that
the nuclear PKC
␣
was significantly decreased at early and late
phases of sepsis (data not shown). Moreover, the RT-PCR
product of PKC
␣
mRNA in the liver of septic rat was
signifi-cantly less than that of sham-operated rats. These observations
provided strong evidence that suppressed expression and
inac-tivation of PKC
␣
might be involved in mediating liver
func-tional failure during sepsis.
One possible cause of progressive organ failure in sepsis
could be cell apoptosis (27, 28). PKC
␣
plays an important role
in cell proliferation and tumorogenecity (15, 19, 29). Although
PKC
␣
inactivation is concerned in apoptosis and subsequent
functional failure of different cell types, it remains unclear how
PKC
␣
regulates nuclear events or modulates hepatic apoptosis
during sepsis. Previous report indicated that a decrease of PKC
activity is associated with apoptosis in freshly isolated rat
hepatocytes (4). Furthermore, we identified the level of hepatic
apoptosis during sepsis by many methods, such as annexin-V
& PI stainings followed by flow-cytometry, DNA ladder
elec-trophoresis and PARP-cleavage (30). Herein, the TUNEL
posi-tive cells were significantly increased at early and late stages of
sepis (Fig. 8). Additionally, our recent results indicated that
inactivation of PKC
␣
may play an important role in
modulat-ing hepatic apoptosis durmodulat-ing sepsis and this apoptosis is closely
associated with the alterations of Bcl-2 family proteins (30). It
has been reported that PKC
␣
phosphorylates the lamin B,
which participates in the activation of DNA fragmentation and
nuclear apoptosis in HL60 cells (31). More interestingly,
PKC
␣
phosphorylates the CREB and up-regulate the bcl-2
expression (32) or cause a conformational change in the CREB
FIG. 8. TUNEL stain positive cells in rat liver of sham-operated (A), 9h (B) and 18 h (C) after CLP. Strongly labeled nuclei are observed in the cells of septic rat liver tissue.
FIG. 9. RT-PCR product of PKC␣ mRNA in rat liver of sham-operated, 9 and 18 h after CLP. (A) Alteration in relative density of PKC␣ mRNA. (B) RT-PCR product of PKC␣ mRNA in liver derived from sham-operated rats or septic rats. The RT-PCR product of PKC␣ mRNA was 325 bp. The -actin (450 bp) was used as a housekeeper. DNA crude preparations were prepared and separated by 2% agarose gel. M: 100 bp DNA ladder. Data are representative of four independents.
In conclusion, we present fundamental evidence of specific
PKC
␣
inactivation in rat liver during the progression of
experi-mental sepsis. We suggest that PKC
␣
might be the major
candidate causing the hypometabolic phase through the failure
in expression and/or translocation of the PKC
␣
during sepsis.
This finding provides certain insights into the molecular
patho-genesis of liver dysfunction and denotes the necessity for
searching for the possible therapeutic intervention of PKC
␣
activator in treating such a disease entity.
ACKNOWLEDGMENT
This work was supported by NSC-89-2320-B-037-019 (Taiwan).REFERENCES
1. Han Y, Meng T, Murray NR, Fields AP, Brasier AR: Interleukin-1-induced nuclear factor-kB-IkB␣ autoregulatory feedback loop in hepatocytes. J Biol
Chem 274:939–942, 1999.
2. Leszczynski D, Joenvaara S, Foegh ML: Protein kinase C-␣ regulates prolif-eration but not apoptosis in rat coronary vascular smooth muscle cells. Life Sci 58:599–606, 1996.
3. Mischak H, Goodnight J, Kolch W, Martiny-Baron G, Schaechtle C, Kazanietz MG, Blumberg PM, Pierce JH, Mushinski JF: Overexpression of protein kinase C-␦ and - in NIH3T3cells induces opposite effects on growth, morphology,
anchorage dependence, and tumorigenecity. J Biol Chem 268:6090–6096, 1993. 4. Sanchez V, Lucas M, Sanz A, Goberna R: Decreased protein kinase C activity is associated with programmed cell death (apoptosis) in freshly isolated rat hepatocytes. Bioscience Report 12:199–206, 1992.
5. Shen L, Glazer RI: Induction of apoptosis in glioblastoma cells by inhibition of protein kinase C and its association with the rapid accumulation of p53 and induction of the insulin-like growth factor-1-binding protein-3. Biochem
Phar-macol 55:1711–1719, 1998.
6. Inaba H, Filkins JP: Augmentation of endotoxin lethality and glucose dysho-meostasis by phobol ester. Am J Physiol 260:R494–R502, 1991.
7. Hsu C, Jao HC, Yang SL, Hsu HK, Liu MS: Inactivation of protein kinase C in rat liver during late hypoglycemic phase of sepsis. Mol Cell Biochem 181:181– 189, 1998.
8. Mellor H, Parker PJ: The extended protein kinase C superfamily. Biochem J 332:281–292, 1998.
9. Mochly-Rosen D, Kauvar LM: Modulating protein kinase C signal transduc-tion. Adv Pharmacol 44:91–145, 1998.
10. de Moel MP, van Emst-de Vries SE, Willems PH, de Pont JJ: Purification and isotype analysis of protein kinase C from rat liver nuclei. Int J Biochem Cell
Biol 30:185–195, 1998.
11. James G, Olson EN: Deletion of the regulatory domain of protein kinase C␣ exposes regions in the hinge and catalytic domains that mediated nuclear target-ing. J Cell Biol 116:863–874, 1992.
transition of cell cycle. J Biol Chem 171:15045–15053, 1996.
17. Tang EY, Parker PJ, Beattie J, Houslay MD: Diabetes induces selective alter-ations in the expression of protein kinase C isoforms in hepatocyte. FEBS Lett 326:117–123, 1993.
18. Nivet V, Antoine PJ, Amessou M, Descamps G, Desbuquois B, Clot JP, Durand D: Increased expression of liver PKC alpha in hypoinsulinemic diabetic rats: a post-translational effect. Mol Cell Endocrinol 146:177–185, 1998.
19. Alessenko A, Khan WA, Wetsel WC, Hannun YA: Selective changes in protein kinase C isozymes in rat liver nuclei during liver regeneration. Biochem
Biophys Res Commun 182:1333–1339, 1992.
20. Goldberg M, Steinberg SF: Tissue-specific developmental regulation of protein kinase C isoforms. Biochem Parmacol 51:1089–1093, 1996.
21. Wichterman KA, Baue AE, Chaudry IH: Sepsis and septic shock—a review of laboratory models and a proposal. J Surg Res 29:189–201, 1980.
22. Wise BC: Phospholipid-sensitive Ca2+-dependent protein kinase from heart. I.
Purification and general properties. J Biol Chem 257:8481–8488, 1982. 23. Chomczynski P, and Sacchi N: Single-step method of RNA isolated by acid
guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162: 156–159, 1987.
24. Boyle WJ, Smeal T, Defize LHK, Angel P, Woodgett JR, Karin M, Hunter T: Activation of protein kinase C decreases phosphorylation of c-jun at sites that negatively regulates its DNA-binding activity. Cell 64: 573–584, 1991. 25. Bsckmann R, Buchner K, Jungblut PR, Eckerskorn C, Weise C, Hilbert R,
Hucho F: Nuclear substrates of protein kinase C. Eur J Biochem 210:45–51, 1992.
26. Olson EN, Burgess R, Staudinger J: Protein kinase C as a transducer of nuclear signal. Cell Growth Differ 4:699–705, 1993.
27. Hotchkiss RS, Swanson PE, Cobb JP, Jacobson A, Buchman TG, Karl IE: Apoptosis in lymphoid and parenchymal cells during sepsis: findings in normal and T- and B-cell-deficient mice. Crit Care Med 25:1298–1307, 1997. 28. Hiramatsu M, Hotchkiss RS, Karl IE, Buckman TG: Cecal ligation and
punc-ture (CLP) induced apoptosis in thymus, spleen, lung and gut by endotoxin and TNF-independent pathway. Shock 7:247–253, 1997.
29. Wang XY, Repasky E, Liu HT: Antisense inhibition of protein kinase C␣ reverses the transformed phenotype in human lung carcinoma cells. Exp Cell
Res 250:253–263, 1999.
30. Jao HC, Yang RC, Hsu HK, Hsu C: The decrease of PKC␣ is associated with hepatic apoptosis at early and late phases of polymicrobial sepsis. Shock 15: 130–134, 2001.
31. Shimizu T, Chun-Kia C, Shao RG, Pommier Y: Lamin B phosphorylation by protein kinase␣ and proteolysis during apoptosis in human leukemia HL-60 cells. J Biol Chem 273:8669–8674, 1998.
32. Wilson BE, Mochon E, Boxer LM: Induction of bcl-2 expression by phosphor-ylated CREB proteins during B-cell activation and rescue from apoptosis. Mol
Cell Biol 16:5546–5556, 1996.
33. Kreisberg JI, Radnik RA, Kreisberg SH: Phosphorylation of cAMP responsive element binding protein after treatment of mesangial cells with high glucose plus TGF or PMA. Kidney International 50:805–810, 1996.
34. Leirdal M, Sioud M: Ribozyme inhibition of the protein kinase C␣ triggers apoptosis in glioma cells. Br J Cancer 80:1558–1564, 1999.
35. Sioud M, Leirdal M: Design of nuclease resistant protein kinase C␣ DNA enzymes with potential therapeutic application. J Mol Biol 296:937–947, 2000.