Charge transport in doped organic semiconductors
Yulong Shen,1Kenneth Diest,1Man Hoi Wong,1Bing R. Hsieh,2David H. Dunlap,3and George G. Malliaras1,* 1Materials Science and Engineering, Cornell University, Ithaca, New York 14853, USA
2Applied Chemistry, National Chiao Tung University, Hsinchu, Taiwan
3Physics and Astronomy, University of New Mexico, Albuquerque, New Mexico 87131, USA 共Received 19 May 2003; published 20 August 2003兲
We report an unusual transition in the conductivity of an organic semiconductor upon doping: For low doping levels, the conductivity of N,N,N⬘,N⬘-tetra-p-tolyl-4-4⬘-biphenyldiamine dispersed polycarbonate in-creases with doping in a nearly linear fashion, and shows an activation energy of 0.2 eV. At high doping levels, a superlinear increase of conductivity with doping is observed, and the activation energy decreases, reaching a low of 0.12 eV. This behavior is understood in terms of broadening of the transport manifold due to enhanced disorder coming from the dopants.
DOI: 10.1103/PhysRevB.68.081204 PACS number共s兲: 72.80.Le, 85.60.Jb
Doped organic semiconductors were originally studied in the 1960s.1 Recently, renewed interest has been spurred by their utilization as injection and transport layers in organic light emitting diodes.2– 8 Despite over 25 years of develop-ment, some rather ubiquitous features of charge transport in these materials are not understood. One primary example is the fact that the conductivity of conjugated polymers and small molecules is often found to increase in a superlinear fashion with doping.4 –5,9–12There is little understanding as to the microscopic origins of this behavior, and a physical description of the doping process remains challenging.4This is mainly due to the fact that the morphology of organics is complex and often changes upon doping.
Molecularly dispersed polymers 共MDP’s兲,13 which are solid solutions of aromatic molecules in an inert polymer matrix, can serve as model systems for studies of doping. One characteristic example is tri-p-tolylamine 共TTA兲 dis-persed in polycarbonate 共PC兲. Mort et al. described p-type doping in this material using the electron acceptor SbCl5.14,15 MDP’s offer several distinct advantages that make them model systems for doping studies: The hopping sites in MDP’s are well defined, which has motivated numer-ous studies of transport in these materials and enabled a rea-sonable understanding of their transport properties.16 They are available at high purity, and often exhibit trap free transport.16Doping can be performed in such a way that the average distance between the hopping sites is kept constant, avoiding dilution effects that take place in other organic semiconductors. Finally, up to 100% of the transport sites can be doped without causing any changes in the morphol-ogy of the film.
In this paper we report a transition that takes place in the conductivity of an organic semiconductor upon doping. It is manifested by a change in the slope of the conductivity vs doping and the activation vs doping curves. We interpret this behavior in terms of broadening of the transport manifold due to enhanced disorder coming from the dopants.
The prototypical organic semiconductor PC:TMTPD was used for the doping studies, where TMTPD is
N,N,N
⬘
,N⬘
-tetra-p-tolyl-4-4⬘
-biphenyldiamine. This is a hole transport MDP similar to PC:TTA, but exhibiting a higher mobility and a more stable conductivity upondoping.17Doping was performed by replacing a fraction x of the TMTPD molecules with their salt TMTPD⫹:SbF6⫺,17in such a way as to maintain the number density of TMTPD molecules, therefore keeping the number density of hopping
sites p0 constant and equal to 6.7⫻1020cm⫺3 共see Fig. 1兲. To the first approximation, the degree of positional disorder should be the same in all samples. Films were cast from a dichloromethane solution on quartz plates with photolitho-graphically defined interdigitated Pt electrodes. The current was found to be proportional to the voltage, and the conduc-tivity was determined from the slope of the I-V curves.
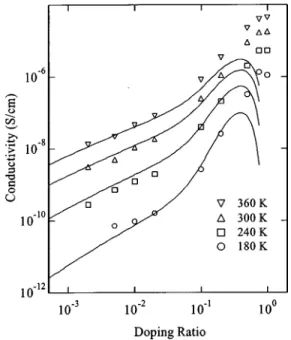
The conductivity was found to exhibit Arrhenius behavior for T⭓200 K 共Ref. 18兲 throughout the doping range 共see Fig. 2兲. Two regimes were found as a function of doping: For low doping (x⬍0.01) the conductivity was found to increase in a nearly linear fashion 共logarithmic slope 0.9兲. This is shown in the inset of Figs. 2 and 3. At the same time, the activation energy EA, shown in Fig. 4, remains approximately constant around 0.2 eV. In the high doping regime (x⬎0.01), the conductivity increases in a superlinear fashion 共logarithmic slope 2.3兲 until x approaches 0.5, after which it levels off at 10⫺5 S/cm; EA decreases dramatically to 0.12 eV.
The change in the slope of the conductivity and EA above
x⫽0.01 cannot be explained with a simple picture of doping,
where holes are generated in a transport level by thermal excitation of electrons to a well-defined acceptor level. This suggests that a transition is taking place in the rate of gen-eration or transport of carriers in the material. We propose a simple model that considers the influence of three mecha-nisms on conductivity and provides an explanation for the origin of this behavior.
共i兲 Filling of the transport manifold. Hole transport in MDP’s takes place via hopping in the manifold of highest occupied molecular orbitals共HOMO兲.19–21The HOMO den-sity of states共DOS兲 approximates a Gaussian due to numer-ous independent contributions to the site energies coming from long-range electrostatic interactions with the surround-ing disordered matrix.22,23Excess holes thermalize in the tail at an average energy2/kT above the mean, where2is the variance of the Gaussian. Consequently, the共zero-field兲 hole mobility should be independent of hole concentration as long as the number of holes is lower than the critical hole density
RAPID COMMUNICATIONS
PHYSICAL REVIEW B 68, 081204共R兲 共2003兲
pC required to fill all states above 2/kT. Deviations are expected when the concentration exceeds pC, so that the average hole energy dips further into the DOS. Time-of-flight measurements in TMTPD estimate to be between 0.102 eV共TMTPD dispersed in polystyrene兲24and 0.078 eV 共TMTPD glass兲.25For a 1 to 1 correspondence between holes and dopants, this would imply pC between 8.0⫻1016 and 2.6⫻1018cm⫺3, respectively. However, the superlinear in-crease in the conductivity 共inset of Fig. 2兲 is not observed until x⬎0.01, which corresponds to pC⬎6.7⫻1018 cm⫺3. This is on the high side of the expected range, indicating that manifold filling alone is not responsible for the observed transition.
共ii兲 Coulombic trapping of carriers. Holes introduced in the HOMO of the TMTPD molecules upon doping are trapped due to the Coulombic attraction with the SbF6⫺ ion. The TMTPD⫹:SbF6⫺ complex resembles a charge transfer exciton with a binding energy ⌬ that depends on the electron-hole distance. In order to model the influence of the Coulombic traps in a simple manner, we will assume that a hole becomes free when it hops to an uncomplexed TMTPD molecule, i.e., the spatial extent of a trap is equal to the intermolecular distance. The overall DOS is a superposition of the Gaussian energy densities for the TMTPD and TMTPD⫹:SbF6⫺ sites, respectively, DOS共E兲⫽共1⫺x兲
冑
2 e ⫺共E/&兲2 ⫹ x冑
2e ⫺关共E⫹⌬兲/&兴2 . 共1兲Assuming that these have the same width, which should be the case for long range electrostatic interactions, what distin-guishes one from the other is the Coulomb trap energy ⌬. The doping fraction determines their relative contribution to the overall DOS.
共iii兲 Broadening of the transport manifold. Introduction of dipoles in an MDP is known to broaden the HOMO manifold.26 Approximating this additional contribution as Gaussian, the width of the DOS should then increase with x as27
⫽
冑
02⫹x冉
7.04 Pa2
冊
2
, 共2兲
where0 is the intrinsic width共sample with x⫽0), P is the dipole moment of the TMTPD⫹:SbF6⫺complex, a is the
av-FIG. 1. Doping in PC:TMTPD. Left: The pristine state, where TMTPD molecules共ovals兲 are dispersed in the polycarbonate ma-trix. Right: A fraction (x⫽0.2) of the TMTPD molecules is re-placed with their salt. The filled circle represents SbF6.
FIG. 2. Temperature dependence of conductivity for films with different doping ratios. The lines are fits to the Arrhenius equation. Inset: Conductivity共at room temperature兲 as a function of the dop-ing ratio. The lines are fits with slopes 0.9 and 2.3.
FIG. 3. Conductivity共at various temperatures兲 as a function of the doping ratio. The lines are fits to Eq.共3兲.
FIG. 4. Activation energy extracted from Arrhenius fits of the experimental data共circles兲 and the calculated values of conductivity
共line兲 as a function of the doping ratio.
RAPID COMMUNICATIONS
SHEN, DIEST, WONG, HSIEH, DUNLAP, AND MALLIARAS PHYSICAL REVIEW B 68, 081204共R兲 共2003兲
erage distance between TMTPD sites 共in Å兲, and the di-electric constant.
In order to quantify the effects described above, we have applied the theory of Ambegaokar, Halperin, and Langer28 for conductivity in the presence of strong spatial and ener-getic disorder. In MDP systems having a high concentration of transport sites, energetic disorder is large enough to make the conductances exponentially disparate at temperatures of interest, but not so large as to induce significant preferential hopping beyond the immediate nearest neighbors.29In such a case, the conductivity g is to be estimated by the critical conductance28
g⫽ep00e⫺共EC⫺EF兲/kT, 共3兲 where0 is a prefactor mobility and EF is the Fermi energy. The critical energy EC determines the half width ␦C⫽EC
⫺EF of an energetic window in the DOS, centered about the Fermi energy, which provides the minimum fraction of nearest neighbor connections required for a percolating net-work of the highest conductances:
⫽
冕
⫺␦C
␦C
DOS共E⫺EF兲dE. 共4兲
Equations共3兲 and 共4兲 have been obtained assuming a Miller-Abrahams form for the underlying hopping rate, ignoring charge-charge interactions except by imposing a maximum of one charge per hopping site, ignoring complications which arise due to spatial correlations in the energetic disorder and, as noted above, the extended nature of the Coulomb traps. For large x, however, we must keep in mind that a number of other effects are expected to become important, such as over-lap of the Coulomb traps, screening of electrostatic disorder, and possible alignment of the TMTPD⫹:SbF6⫺ dipoles. For this reason we have limited the application of the model to the regime of low x.
The solid curves in Fig. 3 were generated from a four-parameter fit to Eq. 共3兲, up to a maximum concentration x ⬵0.2 共hole density 1.3⫻1020 cm⫺3). The percolation frac-tion was taken to be ⫽0.25 共the fit was found not to be particularly sensitive in the value of 兲. The model gives a reasonably good accounting of conductivity versus x in this range, describing the transition between the low and the high doping regimes, while at the same time giving a dependence on T which is in agreement with the experiment. The pre-dicted EA, superimposed with the experimental data in Fig. 4, also shows a transition between the two doping regimes.
The values of the four fitted parameters are 0⫽0.87
⫻10⫺6 cm2/V sec,
0⫽0.086 eV, P⫽28 D, and ⌬
⫽0.36 eV. With the exception of 0, these parameters are within the range expected for the PC:TMTPD system. The value of0 is in the range of values measured with time of flight.24 –25The values of P and⌬ imply a distance between the TMTPD⫹ and the SbF6⫺ ions of 5.8 and 4.3 Å, respec-tively. Although not identical, these values are close to each other and reasonable for a salt. However, the mobility pref-actor0 is approximately 4 orders of magnitude lower than what was measured in TMTPD in polystyrene using time of flight.24 A similar discrepancy of two orders of magnitude
was found by Mort et al. in PC:TTA.14 This is not entirely unexpected since one underestimates the propensity for trap-ping by ignoring the volume of the Coulomb traps, which extends not only to the complexed TMTPD molecule, but to the full extent of the Coulomb radius. Another source of discrepancy comes from the distinction between the true prefactor, and the zero-field ‘‘extrapolated’’ prefactor which is determined from Poole-Frenkel plots.30
Close examination of the fits shows that the increase in dipolar disorder with doping is the primary reason for the transition from the low to the high doping regime, which is manifested by the abrupt reduction of the EA, as well as the superlinear increase of the conductivity. Namely, the width of the DOS 关Eq. 共2兲兴 begins to increase markedly above x ⫽0.01. The resulting broadening of the manifolds of com-plexed and uncomcom-plexed TMTPD molecules increases the density of states in the neighborhood of the Fermi energy, which decreases␦C. This implies an increase in the number of isoenergetic sites which participate in conduction, increas-ing the critical conductance while simultaneously decreasincreas-ing
EA. In such a case, it is not surprising that the exact shape of the conductivity vs doping curve is rather subtle, and de-pends on the detailed manner in which the DOS correspond-ing to the complexed and uncomplexed TMTPD molecules overlap in the neighborhood of the Fermi energy.
It should be noticed that the model proposed here predicts a maximum in the conductivity and a subsequent decrease at high doping ratios. This is a combined effect of dipolar broadening of the DOS and manifold filling. At x⬵0.5, ⫽0.36 eV, the two manifolds are broadened so that they overlap into one, which is half filled. Any further increase of carrier concentration causes a decrease of the conductivity. Such behavior has been experimentally observed in PC:TTA,14,15 where the conductivity abruptly decreases above x⫽0.5. In contrast, the conductivity of PC:TMTPD does not show such a decrease. The reason for this likely resides in the fact that holes are able to access an additional lower lying manifold that corresponds to accommodating two holes per TMTPD molecule. Indeed, such a double oxi-dation of TMTPD has been experimentally observed in elec-trochemical studies.31
In conclusion, we observed a transition in the conductiv-ity of an organic semiconductor upon doping. This was manifested by a change in the slope of the conductivity vs doping curve, which changed from nearly linear 共slope of 0.9兲 for x⬍0.01, to superlinear 共slope of 2.3兲 for x⬎0.01. At the same time, the activation energy changed from being independent of doping for x⬍0.01, to decreasing with dop-ing for x⬎0.01. This behavior was understood in terms of broadening of the transport manifold due to enhanced disor-der coming from the dopants.
Thanks are due to Martin Abkowitz, Jack Blakely, J. Campbell Scott, Steve Barlow, and Seth Marder for fruitful discussions. This work was supported by the National Sci-ence Foundation 共Grant No. 0094047 and No. DMR-0097204兲 and by the Cornell Center for Materials Research 共CCMR兲, a Materials Research Science and Engineering Center of the National Science Foundation共Grant No. DMR-9632275兲.
RAPID COMMUNICATIONS
CHARGE TRANSPORT IN DOPED ORGANIC SEMICONDUCTORS PHYSICAL REVIEW B 68, 081204共R兲 共2003兲
*Author to whom correspondence should be addressed. Email ad-dress: george@ccmr.cornell.edu
1For example, see, M. Pope and C. E. Swenberg, Electronic Pro-cesses in Organic Crystals and Polymers 共Oxford University
Press, Oxford, 1999兲.
2M. Pfeiffer, A. Beyer, T. Fritz, and K. Leo, Appl. Phys. Lett. 73,
729共1998兲.
3M. Pfeiffer, A. Beyer, T. Fritz, and K. Leo, Appl. Phys. Lett. 73,
3202共1998兲.
4M. Pfeiffer, T. Fritz, J. Blochwitz, A. Nollau, B. Plo¨nnigs, A.
Beyer, and K. Leo, Adv. Solid State Phys. 39, 77共1999兲.
5C. Ganzorig and M. Fujihira, Appl. Phys. Lett. 77, 4211共2000兲. 6X. Zhou, M. Pfeiffer, J. Blochwitz, A. Werner, A. Nollau, T. Fritz,
and K. Leo, Appl. Phys. Lett. 78, 410共2001兲.
7W. Gao and A. Kahn, Appl. Phys. Lett. 79, 4040共2001兲. 8W. Gao and A. Kahn, Organic Electron. 3, 53共2002兲. 9D. M. de Leeuw, Synth. Met. 55, 3597共1993兲. 10
A. R. Brown, D. M. de Leeuw, E. E. Havinga, and A. Pomp, Synth. Met. 68, 65共1994兲.
11C. P. Jarrett, R. H. Friend, A. R. Brown, and D. M. de Leeuw, J.
Appl. Phys. 77, 6289共1995兲.
12B. A. Gregg and R. A. Cormier, J. Am. Chem. Soc. 123, 7959 共2001兲.
13This class of organic semiconductors is also referred to as
mo-lecularly doped polymers. The use of the term ‘‘doped’’ in this sense is unfortunate, as the introduction of the small molecule increases the mobility and not the free carrier density. By doping we shall refer to the addition of a species共such as SbF6) that increases the free carrier density.
14J. Mort, S. Grammatica, D. J. Sandman, and A. Troup, J. Electron.
Mater. 9, 411共1980兲.
15A. Troup, J. Mort, S. Grammatica, and D. J. Sandman, J.
Non-Cryst. Solids 35, 151共1980兲.
16P. M. Borsenberger and D. S. Weiss, Organic Photoreceptors for Xerography共Marcel Decker, New York, 1998兲.
17B. R. Hsieh共unpublished兲.
18According to the Gaussian disorder model, one might be tempted
to plot the logarithm of conductivity as T⫺2. The data did not fall into a straight line on this plot, nor did they on a plot of the logarithm of conductivity vs the logarithm of T. The Arrhenius plot gave the best correlation. Moreover, using the Arrhenius plot facilitates comparison with literature data关see Refs. 4, 5, 16, 17, and 25兴.
19
Y. N. Gartstein and E. M. Conwell, Chem. Phys. Lett. 245, 351
共1995兲.
20D. H. Dunlap, P. E. Parris, and V. M. Kenkre, Phys. Rev. Lett. 77,
542共1996兲.
21S. V. Novikov, D. H. Dunlap, V. M. Kenkre, and P. E. Parris,
Phys. Rev. Lett. 81, 4472共1998兲.
22A. Dieckman, H. Ba¨ssler, and P. M. Borsenberger, J. Chem. Phys.
99, 8136共1993兲.
23S. V. Novikov and A. V. Vannikov, J. Phys. Chem. 99, 14573 共1995兲.
24S. Heun and P. M. Borsenberger, Physica B 216, 43共1995兲. 25S. Heun and P. M. Borsenberger, Chem. Phys. 200, 245共1995兲. 26P. M. Borsenberger and J. J. Fitzgerald, J. Phys. Chem. 97, 4815
共1993兲.
27R. H. Young, Philos. Mag. B 72, 435共1995兲.
28V. Ambegaokar, B. I. Halperin, and J. S. Langer, Phys. Rev. B 4,
2612共1971兲.
29P. E. Parris, J. Chem. Phys. 108, 218共1998兲.
30P. E. Parris, V. M. Kenkre, and D. H. Dunlap, Phys. Rev. Lett. 87,
126601共2001兲.
31E. T. Seo, R. F. Nelson, J. M. Fritsch, L. S. Marcoux, D. W.
Leedy, and R. N. Adams, J. Am. Chem. Soc. 88, 3498共1966兲.
RAPID COMMUNICATIONS
SHEN, DIEST, WONG, HSIEH, DUNLAP, AND MALLIARAS PHYSICAL REVIEW B 68, 081204共R兲 共2003兲