Advances in Brief
Glutathione S-Transferase P1 and NADPH Quinone Oxidoreductase Polymorphisms
Are Associated with Aberrant Promoter Methylation of P16
INK4aand
O
6-Methylguanine-DNA Methyltransferase in Sputum
1Frank D. Gilliland, Heidi J. Harms, Richard E. Crowell, Yu-Fen Li, Randy Willink, and Steven A. Belinsky
2Keck School of Medicine, Department of Preventive Medicine and Norris Comprehensive Cancer Center, University of Southern California, Los Angeles, California 90089-9021 [F. D. G., Y-F. L.]; University of New Mexico School of Medicine [H. J. H.], and Division of Pulmonary, Critical Care and Allergy [R. E. C.], University of New Mexico, Albuquerque, New Mexico 87106; Pulmonary and Critical Care, New Mexico Veterans Healthcare System, Albuquerque, New Mexico 87108-5153 [R. E. C.]; and Lovelace Respiratory Research Institute, Albuquerque, New Mexico 87108 [R. W., S. A. B.]
Abstract
Inactivation of the p16INK4atumor suppressor gene and O6
-methylgua-nine-DNA methyltransferase (MGMT) DNA repair gene by aberrant pro-moter methylation appears to be an important step in respiratory carci-nogenesis after exposure to tobacco smoke and radon progeny. The determinants of aberrant promoter methylation are not well character-ized. Polymorphic variants of genes of which the products are involved in pathways that modulate and repair DNA damage after carcinogen expo-sure may affect the occurrence of de novo promoter methylation. On the basis of their associations with risk of lung cancer, we hypothesized that functional polymorphic variants of the NADPH quinone oxidoreductase, glutathione S-transferases P1 and M1, myeloperoxidase, and XRCC1 genes are associated with p16 and/or MGMT promoter methylation in sputum from cancer-free subjects at high risk for developing lung cancer. This hypothesis was tested by conducting a cross-sectional study of 70 former uranium miners from the Uranium Epidemiological Study cohort who were at high risk for lung cancer. The polymorphic variant genotypes were characterized through PCR-RFLP on DNA isolated from peripheral lymphocytes, and the methylation status of the p16 and MGMT promoters was determined by methylation-specific PCR on DNA isolated from spu-tum. Subjects who had at least one GSTP1 polymorphic allele (A-to-G at bp 104) had an increased risk for MGMT methylation [odds ratio (OR), 4.8; 95% confidence interval (CI), 1.2–18.6] or for either p16 or MGMT methylation (OR, 4.4; 95% CI, 1.3–14.2). Lack of a wild-type NADPH quinone oxidoreductase allele (C at bp 609) was also associated with methylation of either p16 or MGMT (OR, 3.1; 95% CI, 1.0 –9.2). These results provide the first link between germ-line functional deficits in pathways that protect the cell from tobacco- and radon-induced DNA damage, and the development of aberrant promoter methylation of the
p16 and MGMT genes in the respiratory epithelium of individuals at high
risk for lung cancer. Introduction
Lung cancer is the leading cause of cancer-related death for both men and women in the United States. A substantial body of scientific evidence shows that inhalation of tobacco smoke and/or radon prog-eny is the leading cause of lung cancer in the United States (1, 2). Although the decreasing mortality rates from lung cancer in men illustrate the success of preventive efforts, the increasing mortality rates among women and the increase in smoking around the world
indicate that a substantial population of exposed individuals will remain at high risk for lung cancer for the near future. Furthermore, exposure to residential radon progeny is common, and a large popu-lation is at elevated risk of lung cancer from lifelong exposure. Improved secondary preventive methods, including early detection and risk-reduction strategies such as chemoprevention are urgently needed. The development of a molecular-based marker approach would greatly advance the early detection of lung cancer.
Emerging in the field of cancer biology is the recognition that epigenetically mediated gene silencing through promoter hypermeth-ylation is a common event critical to the initiation and progression of cancer (1). Inactivation of the p163
tumor suppressor gene and MGMT DNA repair gene by aberrant promoter methylation occurs frequently in non-small cell lung cancer. A recent study by our group has demonstrated that methylation of these genes can also be detected in DNA from sputum in 100% of squamous cell carcinomas up to 3 years before clinical diagnosis (2). These methylation changes can also be detected in cancer-free smokers and former uranium miners exposed to radon progeny at a prevalence that approximates lifetime risk for lung cancer (3). Progress in developing aberrant gene meth-ylation as a molecular marker system for lung cancer risk would be facilitated by the identification of the determinants for methylation. Epidemiological studies continue to support the premise that genetic variation in individual response to carcinogens in tobacco smoke is important for defining inherent susceptibility to this disease (4). Polymorphic variants of genes of which the products are involved in pathways that modulate and repair DNA damage after carcinogen exposure may affect the occurrence of de novo promoter methylation. On the basis of their associations with risk for lung cancer, we hypothesized that functional polymorphic variants of the NQO1, GSTP1, GSTM1, MPO, and XRCC1 genes are associated with p16 and/or MGMT promoter methylation in sputum from cancer-free subjects at high risk for developing lung cancer. This hypothesis was tested by conducting a cross-sectional genetic epidemiological study of 70 former uranium miners from the UES cohort who were at high risk for lung cancer.
Materials and Methods
Study Population. The study population consisted of 70 former under-ground uranium miners who were recruited for a cancer surveillance study from the most highly exposed miners in the UES cohort of 3469 Grants, New Mexico, Mineral Belt miners established by Samet et al. (5). Cumulative exposure to radon progeny in WLMs was estimated as part of the UES (6, 7).
Received 11/15/01; accepted 2/26/02.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
1Supported by NIH Grant R01 CA70190 under Department of Energy Cooperative
Agreement DE-FC04-96AL76406; by a Special Emphasis Research Career Award, Na-tional Institute of OccupaNa-tional Health and Safety, FG02-90ER60939; and by the NaNa-tional Institute of Environmental Health Sciences, NIH, Grant 5P30 ES07048-06.
2To whom requests for reprints should be addressed, at Lovelace Respiratory Research
Institute, 2425 Ridgecrest Drive SE, Albuquerque, NM 87108. Phone: (505) 348-9465; Fax: (505) 348-4980; E-mail: [email protected].
3The abbreviations used are: p16, p16INK4a; MGMT, O6-methylguanine-DNA
meth-yltransferase MSP, methylation-specific PCR; MPO, myeloperoxidase; NQO1, NADPH quinone oxidoreductase; GSTP1, glutathione transferase P1; GSTM1, glutathione S-transferase M1; GST, glutathione S-S-transferase; UES, Uranium Epidemiological Study; WLM, working level month, OR, odds ratio; CI, confidence interval.
The smoking histories were collected from interviews between 1993 and 1995 from mining company records, and from records of New Mexico uranium miners who had mining-related physical examinations during 1957–1976 (7). Selection of participants was based on cumulative exposure to radon through uranium mining or a combined history of tobacco smoking and uranium mining. All of the patients gave written informed consent for this study.
Human Tissue Samples. Induced sputum and venous blood samples were collected during clinic visits. Sputum was induced by nebulized normal saline solution. Expectorated sputum was collected and stored in Saccamanno’s solution until DNA was extracted.
Nucleic Acid Isolation. Leukocytes from venous blood samples were isolated by standard procedures. DNA was then extracted from both sputum and leukocyte samples by digestion with Pronase in 1% SDS followed by standard phenol-chloroform extraction and ethanol precipitation. DNA was stored at 4C until additional analysis.
MSP. The methylation status of the p16 and MGMT gene promoters in sputum samples was determined by a nested, two-stage MSP assay (2). Genomic DNA isolated from sputum was modified by treatment with sodium bisulfite, which converts only unmethylated cytosines to uracil. PCR primers specific to both methylated and unmethylated template were used. The con-ditions for the nested, two-stage PCR approach have been described (2). Products were visualized on 2% agarose gels. Normal human tissue collected at autopsy of never-smokers, and cell lines positive for p16 (Calu6) and MGMT (SkuLu1) methylation served as negative and positive controls, respectively. The two-stage MSP assay requires only nanogram quantities of DNA and can detect methylated alleles in the presence of unmethylated alleles (as is the case with sputum) at a sensitivity of 1 in⬎50,000 copies.
Polymorphism Assays. The presence of polymorphic variants of the NQO1, GSTP1, XRCC1, and MPO genes was determined by PCR-RFLP
analysis of genomic DNA isolated from leukocytes. Briefly, a region surround-ing the polymorphic site of each gene was generated through PCR amplifica-tion. Primer sequences for each polymorphism have been described (8 –12). Each PCR was performed with⬃200 ng of genomic DNA in a 50-l reaction volume using Taq Gold polymerase (Perkin-Elmer). Each polymorphism either destroys or creates a specific restriction site within its amplification fragment. Subsequent digestion of the PCR products with appropriate restriction en-zymes specific for each variant yields distinct banding patterns that correspond either to the wild-type or polymorphic allele of each gene.
PCR conditions for analysis of the MPO gene are as follows: 95°C for 11.5 min, denature at 95°C for 60 s, anneal at 58°C for 60 s, and extension at 72°C for 60 s for 40 cycles, followed by a 5-min final extension. The 350-bp PCR product was then digested with AciI. The presence of the polymorphic variant (G to A at position⫺463 in the promoter region of the MPO gene) destroys an
AscI restriction site. An invariant AciI restriction site exists within the
ampli-fication fragment. The three possible genotypes are defined by the following banding patterns: G/G (169-, 120-, and 61-bp fragments), G/A (289-, 169-, 120-, and 61-bp fragments), and A/A (289- and 61-bp fragments).
Conditions for analysis of GSTP1 were as follows: 95°C for 11.5 min, denature at 95°C for 30 s, anneal at 55°C for 30 s, and extension at 72°C for 30 s for 40 cycles, followed by a 5-min final extension. The 176-bp PCR product was then digested with BsmA1. The presence of the polymorphic variant (A to G at bp 1578 of exon 5) creates a BsmA1 restriction site. The three possible genotypes are defined by the following banding patterns: A/A (uncut 176-bp fragment), A/G (176-, 91-, and 85-bp fragments), and G/G (91- and 85-bp fragments).
Because the GSTM1 genotype is either wild type or null, a multiplex PCR was performed to amplify the GSTM1 and-IFN genes (positive control) as described (10). The PCR amplification of GSTM1 was as follows: 95°C for 10 min, denature at 95°C for 1 min, anneal at 60°C for 1 min, and extension at 72°C for 1 min for 40 cycles, followed by a 5-min final extension.
The PCR amplification of NQO1 was as follows: 95°C for 11.5 min, denature at 95°C for 30 s, anneal at 56°C for 30 s, and extension at 72°C for 30 s for 40 cycles, followed by a 5-min final extension. The 211-bp PCR product was then digested with HinfI. The presence of the polymorphic variant (C to T at bp 609 of exon 6) creates a HinfI restriction site. The three possible genotypes were defined by the following banding patterns: C/C (uncut 211-bp fragment), C/T (211-, 181-, and 30-bp fragments), and T/T (181- and 30-bp fragments).
The PCR conditions for amplification of the XRCC1 gene was as follows:
95°C for 11.5 min, denature at 95°C for 30 s, anneal at 62°C for 30 s, and extension at 72°C for 30 s for 40 cycles, followed by a 5-min final extension. The Arg allele at codon 399 contains a MspI site that is lost on conversion to the glutamine amino acid. The 198-bp PCR product was digested overnight with 20 units of MspI (New England BioLabs Inc., Beverly, MA) at 60°C. The homozygous Gln allele produced a single 198-bp product, the homozygous Arg allele produced 145-bp and 53-bp products (the 53-bp product produced was too small to accurately resolve), and the heterozygous Arg/Gln allele produced three products of 198-bp, 145-bp, and 53 bp.
Data Analysis. Logistic regression models were used to assess the effect of genotype on the occurrence of methylation in sputum samples using additive and dominant models for MPO, XRCC1, GSTP1, and NQO1. The number of subjects who were homozygous for the variant alleles was insufficient to fit codominant models for alleles of these genes. The GSTM1 genotype was defined as null or present. In the additive model, the number of polymorphic variants was used as an independent variable, i.e., wild-type (0), heterozygous (1), and homozygous (2). The polymorphic variant genotypes for MPO,
XRCC1, NQO1, and GSTP1 were compared with the homozygous wild-type
genotype (the reference group). Data were analyzed using SAS software version 8e (13).
Results
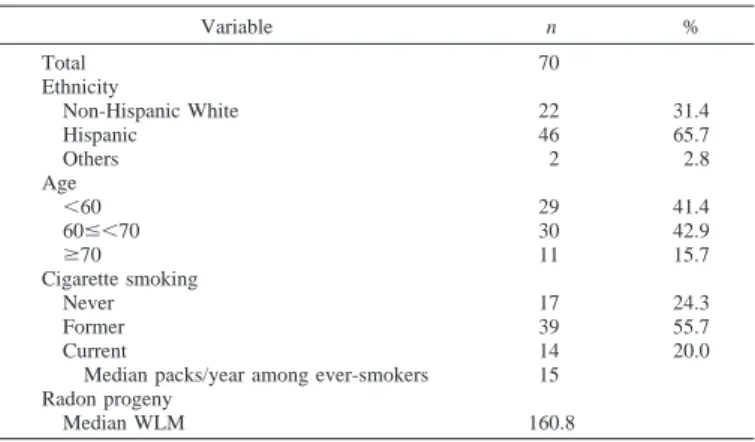
Demographics. Demographic information and smoking histories
for study subjects are presented in Table 1. The percentage of His-panics (65.7%) was twice that of Anglo subjects (31.4%). Two sub-jects were Native American. All of the subsub-jects had a history of radon exposure through uranium mining, and⬃76% had a history of smok-ing (mean pack-years, 15). Radon exposure ranged from 4 to 608 WLMs with a mean of 161 WLMs; 75% of the subjects had⬎100 WLMs. The median age was 62 (range 39 – 83 years).
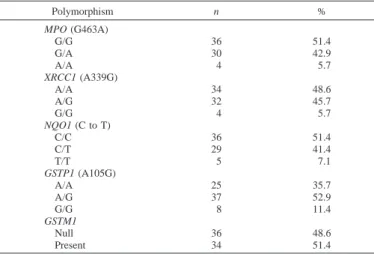
Distribution of Polymorphisms in the MPO, XRCC1, NQO1, GSTP1, and GSTM1 Genes. The genotype distribution of the five
genes among the study population is shown in Table 2, and an example of genotyping by RFLP is depicted in Fig. 1. The MPO genotype was distributed as 51.4% wild-type (G/G), 42.9% heterozy-gous (G/A), and 5.7% homozyheterozy-gous (A/A). The frequency for the wild-type MPO risk genotype was 59.1% and 47.8% in Anglo and Hispanic subjects, respectively. Smoking status of subjects with the wild-type genotype was similar (52.9% never-smokers, 51.3% for-mer-smokers, and 50% current-smokers, respectively).
The XRCC1 genotype was distributed as 48.6% wild-type (A/A), 45.7% heterozygous (G/A), and 5.7% homozygous (G/G). The fre-quency for the wild-type genotype differed among non-Hispanic white and Hispanic subjects, 63.6% and 39.1%, respectively. The percentages of the wild-type genotype showed little variation by smoking status (47.1% never-smokers, 48.7% former-smokers, and 50% current-smokers, respectively).
Analysis of the NQO1 polymorphism revealed a distribution of
Table 1 Demographics of subjects
Variable n % Total 70 Ethnicity Non-Hispanic White 22 31.4 Hispanic 46 65.7 Others 2 2.8 Age ⬍60 29 41.4 60ⱕ⬍70 30 42.9 ⱖ70 11 15.7 Cigarette smoking Never 17 24.3 Former 39 55.7 Current 14 20.0
Median packs/year among ever-smokers 15 Radon progeny
Median WLM 160.8
51.4% wild-type (C/C), 41.4% heterozygous (C/T), and 7.1% ho-mozygous (T/T). The frequency of the wild-type NQO1 genotype among non-Hispanic white subjects was 68.2%, whereas that in Hispanic subjects was 41.3%. The distribution of homozygous wild-type NQO1 genowild-type varied by smoking status (58.8% of never-smokers, 56.4% of former-never-smokers, and 28.6% of current-never-smokers, respectively).
The distribution of the three GSTP1 genotypes was 35.7% wild-type (Arg/Arg), 52.9% heterozygous (Arg/Gly), and 11.4% homozy-gous (Gly/Gly). The frequencies of the wild-type GSTP1 genotype among non-Hispanic white and Hispanic subjects were 36.4% and 32.6%, respectively. The homozygous wild-type genotype occurred in 35.3% of never-smokers, 43.6% of former-smokers, and 14.3% of current-smokers.
Lastly, the distribution of the GSTM1 genotype was 48.6% null and 51.4% present. The frequencies of the present genotype among non-Hispanic white and non-Hispanic subjects were 63.6% and 41.3%, respec-tively. The percentages of the GSTM1 genotype were 58.8% of never-smokers, 41.3% of former-smokers, and 57.1% of current-smokers, respectively.
Methylation of p16 and MGMT in Sputum. The frequency of p16 and MGMT promoter gene methylation was determined previ-ously in sputum samples (2). Abnormal p16 and MGMT gene meth-ylation was present in sputum from 10 (14.3%) and 21 (30.0%) of 70 subjects, respectively. The frequency of methylation of these two genes was similar in tobacco-plus-radon versus radon exposure alone. Methylation of either p16 or MGMT was detected in 40% of subjects, and of p16 and MGMT in 3 subjects. Methylation among ethnic groups was not statistically different.
Relationship between Genetic Polymorphism and Gene-specific Promoter Methylation. Polymorphic variants of GSTP1 and NQO1
were significantly associated with p16 and/or MGMT promoter meth-ylation (Table 3). Interestingly, although there was not a significant association between the variant NQO1 genotype and methylation of the p16 or MGMT promoters individually, the presence of p16 or MGMT was significantly associated (OR, 3.1; 95% CI, 1.0 –9.2). The absence of one or both wild-type alleles for the GSTP1 gene was significantly associated with MGMT methylation alone (OR, 4.8; 95% CI, 1.2–18.6) and for either p16 or MGMT methylation in sputum (OR, 4.4; 95% CI, 1.3–14.2). These significant associations were still observed in the additive model as well (Table 3). No association was apparent among the MPO, XRCC1, or GSTM1 polymorphisms and p16 or MGMT promoter methylation. Although ORs were adjusted for ethnicity, no evidence of confounding was found in our data set.
Ethnicity or exposure to tobacco smoke or radon progeny did not modify the relationships between the genotypes and promoter meth-ylation in the sputum.
Discussion
These studies provide the first potential link between germ-line polymorphisms in genes involved in the protection of the cell from heritable DNA damage stemming from tobacco exposure and the epigenetic-mediated silencing of cancer genes by promoter hyperm-ethylation. Although there were small ethnic differences, the overall distribution of genotypes for all five of the genes was similar to those reported previously in cancer-free subjects (11, 12, 14 –18). More-over, the additive model substantiated that the variant alleles for the NQO1 and GSTP1 genes were associated with methylation.
The GSTP1 genotype was significantly associated with MGMT methylation alone, and this association was increased when either p16 or MGMT methylation was present in cells in the sputum sample. Although there was not a significant association between the NQO1 genotype and methylation of the p16 or MGMT promoters individu-ally, the presence of p16 or MGMT methylation was significantly associated. It is plausible that reduced defenses for oxidant damage from radiation and tobacco smoke exposure as a result of variant NQO1 and GSTP1 enzymes may work in concert to produce DNA damage severe enough to affect the fidelity for maintenance of CpGs in the p16 and MGMT promoters.
The GSTs form a superfamily of four distinct genes important for defense against oxidative damage to DNA (14, 19). These cytosolic enzymes play an important role in protecting DNA against damage because of adduct formation through the detoxification of reactive oxygen species and electrophiles formed during carcinogen metabo-lism and absorption of radiation. The major GST protein in the human lung is GSTP1. A polymorphic site at codon 105 of the P1 gene (an A-to-G substitution) leads to an isoleucine-to-valine substitution in the hydrophobic binding region of the protein. This substitution alters the catalytic properties of the enzyme and is associated with a signif-icantly higher level of hydrophobic DNA adducts (12). Lung cancer
Fig. 1. Ethidium bromide gels show the possible genotypes from the polymorphisms of the MPO (G-to-A), GSTP1 (A-to-G), and NQO1 (C-to-T) genes. A, MPO gene, the 350-bp amplification product contains an invariant AscI restriction site yielding a constant 61-bp fragment seen in all lanes. Individuals homozygous for the A allele have no additional recognition sites and show only two bands at 289- and 61-bp. The G allele creates an additional restriction site such that homozygous G individuals have three bands at 169, 120, and 61-bp. Heterozygotes show all four bands. B, GSTP1 gene, as a result of the A-to-G polymorphism, a BsmA1 restriction site is created. Homozygous A individuals show an uncut 176-bp fragment, whereas homozygous G individuals have two bands at 91- and 85-bp. Heterozygotes show all three bands. C, NQO1 gene, as a result of the C-to-T polymorphism, a HinfI restriction site is created. The homozygous C individuals show an uncut 211-bp fragment, whereas homozygous T individuals show a 181-bp fragment (the resulting 30-bp fragment is not shown on the gel photo). Heterozygotes show the 211- and 181-bp fragments (again, with the 30-bp fragment not shown). Table 2 Distribution of the wild-type and variant alleles of the MPO, XRCC1, NQO1,
GSTP1, and GSTM1 polymorphisms Polymorphism n % MPO (G463A) G/G 36 51.4 G/A 30 42.9 A/A 4 5.7 XRCC1 (A339G) A/A 34 48.6 A/G 32 45.7 G/G 4 5.7 NQO1 (C to T) C/C 36 51.4 C/T 29 41.4 T/T 5 7.1 GSTP1 (A105G) A/A 25 35.7 A/G 37 52.9 G/G 8 11.4 GSTM1 Null 36 48.6 Present 34 51.4 2250
patients have also been shown to have a higher frequency of the GG phenotype than controls (12). GSTM1 is involved in Phase II detox-ification of activated xenobiotic metabolites and has been weakly associated with lung cancer risk (15, 16). A substantial portion of the population has a null genotype for GSTM1. Our finding of more methylation associated with the G allele of GSTP1 but not the GSTM1 null genotype is consistent with high expression of GSTP1 in the lung and the protective role of wild-type GSTP1 in lung cancer risk.
NQO1 is a cytosolic enzyme that carries out the two-electron reduction of a variety of quinone substrates present in the environment as combus-tion by-products, including that of automobile exhaust and cigarette smoke, and endogenously as antioxidants (17, 18). In addition, nitroaro-matic compounds, heterocyclic amines, and cigarette smoke condensate are metabolized by NQO1. However, NQO1 is also involved in antiox-idant defenses through metabolism of quinone antioxantiox-idants. A polymor-phic variant of the gene (a C-to-T transition at bp 609 of exon 6) is associated with an ⬃3-fold decrease in NQO1 activity; however, the association of the wild-type allele with lung cancer risk has been incon-sistent (9, 20, 21). Interestingly, Lafuente et al. (22) found an association between the NQO1 polymorphic variant and an increased risk of colon cancer. In addition, the polymorphic variant genotype was associated with colon tumors that exhibited mutation in the K-ras gene. The mutated ras gene functions as an oncogene, leading to a prolonged mitogenic signal in affected cells. Consistent with this study, we found that the variant NQO1 allele confers an increased risk of p16 suppression through hypermethylation. However, studies suggest that, depending on the sub-strate, the wild-type NQO1 allele may confer either an increased or decreased risk for various malignancies. Our results support the hypoth-esis that NQO1 is an important enzyme involved in modulating lung cancer risk, but the effects of the variant allele may depend on specific exposures.
The connection between DNA methylation and gene expression has been known for almost 20 years. The predominant consequence of gene promoter methylation is transcriptional repression, which can be mediated either directly, by blocking the binding of transcription factors, or indirectly by proteins that specifically bind to methylated DNA (23–25). However, the genetic or environmental factors that underlie the targeting of specific gene promoters for hypermethylation are unknown. DNA damage by tobacco carcinogens could be one
factor leading to regional disruption of normal chromatin structure. This change in genomic integrity could lead to inappropriate methy-lation by the cytosine DNA methyltransferases of CpG sites in the normally protected promoter region of genes such as p16 and MGMT. A limitation of our study was the low power to detect ORs⬍2.5. Population stratification may be a source of bias in multiethnic pop-ulations. We adjusted for ethnicity but did not have information to adjust for admixture within ethnic groups. Furthermore, ethnic differ-ences in genetic effects could not be adequately assessed. In the present study, all of the subjects had substantial radon progeny expo-sure through uranium mining. The frequency of methylation of the p16 and MGMT genes was similar in tobacco-plus-radon versus radon exposure alone. Additional investigations with an expanded study population will more precisely define the influence of these and other germ-line polymorphisms working together or alone on the propensity to acquire multiple epigenetic changes in sputum. This information could greatly aid in the ultimate development of a genetic and epige-netic risk model for lung cancer.
References
1. Baylin, S. B., Herman, J. G., Graff, J. R., Vertino, P. M., and Issa, J. P. Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv. Cancer Res., 72: 141–196, 1998.
2. Palmisano, W. A., Divine, K. K., Saccomanno, G., Gilliland, F. D., Baylin, S. B., Herman, J. G., and Belinsky, S. A. Predicting lung cancer by detecting aberrant promoter methylation in sputum. Cancer Res., 60: 5954 –5958, 2000.
3. Law, M. R., Morris, J. K., Watt, H. C., and Wald, N. J. The dose-response relationship between cigarette consumption, biochemical markers and risk of lung cancer. Br. J. Cancer, 75: 1690 –1693, 1997.
4. Anonymous. Human genetic variation in response to medical and environmental agents: pharmacogenetics and ecogenetics. International Titisee Conference Schwar-zwald-Hotel, Titisee, Black Forest, Federal Republic of Germany, October 13th–15th, 1977. Hum. Gen., 1 (Suppl.): 1–192, 1997.
5. Samet, J. M., Pathak, D. R., Morgan, M. V., Key, C. R., Valdivia, A. A., and Lubin, J. H. Lung cancer mortality and exposure to radon progeny in a cohort of New Mexico underground uranium miners. Health Phys., 61: 745–752, 1991.
6. Lundin, F. E., Jr., Lloyd, J., Smith, E., Archer, V., and Holaday, D. Mortality of uranium miners in relation to radiation exposure, hard-rock mining and cigarette smoking–1950 through September 1967. Health Phys., 16: 571–578, 1969. 7. Morgan, M. V., and Samet, J. M. Radon daughter exposures of New Mexico U
miners, 1967–1982. Health Phys., 50: 656 – 662, 1986.
8. London, S. J., Lehman, T. A., and Taylor, J. A. Myeloperoxidase genetic polymor-phism and lung cancer risk. Cancer Res., 57: 5001–5003, 1997.
9. Wiencke, J. K., Spitz, M. R., McMillan, A., and Kelsey, K. T. Lung cancer in Mexican-Americans and African-Americans is associated with the wild-type geno-Table 3 The effects of the wild-type and variant alleles of the MPO, XRCC1, NQO1, and GSTP1 polymorphisms on MGMT and p16 gene promoter methylation
Polymorphism modelsa MGMT p16 MGMT/p16 OR 95% CI OR 95% CI OR 95% CI MPO Dominant model GG 1 — 1 — 1 — GA/AA 0.8 0.3–2.2 1.2 0.3–4.5 0.8 0.3–2.0 Additive modelb 0.8 0.3–2.0 1.3 0.4–4.0 0.9 0.4–2.1 XRCC1 Dominant model AA 1 — 1 — 1 — AG/GG 0.8 0.3–2.4 1.6 0.4–6.7 0.8 0.3–2.2 Additive modelb 0.9 0.3–2.1 1.2 0.4–3.8 0.8 0.4–1.9 NQO1 Dominant model CC 1 — 1 — 1 — CT/CC 1.8 0.6–5.4 1.9 0.5–8.0 3.1 1.0–9.2 Additive modelb 1.9 0.8–4.5 1.6 0.6–4.7 2.5 1.0–5.9 GSTP1 Dominant model Ile/Ile 1 — 1 — 1 — Ile/Val, Val/Val 4.8 1.2–18.6 2.4 0.5–12.5 4.4 1.3–14.2 Additive modelb 2.3 1.0–5.4 1.9 0.7–5.5 2.4 1.0–5.5 GSTM1 Present 1 — 1 — 1 — Null 2.0 0.7–6.0 0.6 0.1–2.4 1.3 0.5–3.7
aModels are adjusted for ethnicity.
bAdditive: wild-type (0), heterozygous (1), and homozygous (2).
type of the NAD(P)H: quinone oxidoreductase polymorphism. Cancer Epidemiol. Biomark. Prev., 6: 87–92, 1997.
10. Brockmoller, J., Kerb, R., Drakoulis, N., Staffeldt, B., and Roots, I. Glutathione S-transferase M1, and its variants A, and B as host factors of bladder cancer susceptibility: a case control study. Cancer Res., 54: 4103– 4111, 1994.
11. Divine, K. K., Gilliland, F. D., Crowell, R. E., Stidley, C. A., Bocklage, T. J., Cook, D. L., and Belinsky, S. A. The XRCC1 399 glutamine allele is a risk factor for adenocarcinoma of the lung. Mutat. Res., 461: 273–278, 2001.
12. Ryberg, D., Skaug, V., Hewer, A., Phillips, D. H., Harries, L. W., Wolf, C. R., Ogreid, D., Ulvik, A., Vu, P., and Haugen, A. Genotypes of glutathione transferase M1 and P1 and their significance for lung DNA adduct levels and cancer risk. Carcinogenesis (Lond.), 18: 1285–1289, 1997.
13. SAS Institute Inc. SAS user’s guide: statistics, version 8e. Cary, NC: SAS Institute, Inc., 2001.
14. Hayes, J. D., and Strange, R. C. Glutathione S-transferase polymorphisms and their biological consequences. Pharmacology (Basel), 61: 154 –166, 2000.
15. Reszka, E, and Wasowicz, W. Significance of genetic polymorphisms in glutathione S-transferase multigene family and lung cancer risk. Int. J. Occup. Med. Environ. Health, 14: 99 –113, 2001.
16. Houlston, R. S. Glutathione S-transferase M1 status and lung cancer risk: a meta-analysis. Cancer Epidemiol. Biomark. Prev., 8: 675– 682, 1999.
17. Dinkova-Kostova, A. T., and Talalay, P. Persuasive evidence that quinone reductase type 1 (DT diaphorase) protects cells against the toxicity of electrophiles and reactive forms of oxygen. Free Radic. Biol. Med., 29: 231–240, 2000.
18. Ross, D., Kepa, J. K., Winski, S. L., Beall, H. D., Anwar, A., and Siegel, D. NAD(P)H. quinone oxidoreductase 1 (NQO1): chemoprotection, bioactivation, gene regulation and genetic polymorphisms. Chem. Biol. Interact., 129: 77–97, 2000.
19. Hayes, J. D., and Strange, R. C. Potential contribution of the glutathione S-transferase supergene family to resistance to oxidative stress. Free Radic. Res., 22: 193–207, 1995.
20. Chen, H., Lum, A., Seifried, A., Wilkens, L. R., and Le Marchand, L. Association of the NAD(P)H:quinone oxidoreductase 609C–⬎T polymorphism with a decreased lung cancer risk. Cancer Res., 59: 3045–3048, 1999.
21. Rosvold, E. A., McGlynn, K. A., Lustbader, E. D., and Buetow, K. H. Identification of an NAD(P)H:quinone oxidoreductase polymorphism and its association with lung cancer and smoking. Pharmacogenetics, 5: 199 –206, 1995.
22. Lafuente, M. J., Casterad, X., Trias, M., Ascaso, C., Molina, R., Ballesta, A., Zheng, S., Wiencke, J. K., and Lafuente, A. NAD(P)H:quinone oxidoreductase-dependent risk for colorectal cancer and its association with the presence of K-ras mutations in tumors. Carcinogenesis (Lond.), 21: 1813–1819, 2000.
23. Wade, P. A. Methyl CpG binding proteins: coupling chromatin architecture to gene regulation. Oncogene, 20: 3166 –3173, 2001.
24. Rountree, M. R., Bachman, K. E., Herman, J. G., and Baylin, S. B. DNA methylation, chromatin inheritance, and cancer. Oncogene, 20: 3156 –3165, 2001.
25. Robertson, K. D. DNA methylation, methyltransferases, and cancer. Oncogene, 20: 3139 –3155, 2001.