Molecular Binding in Structure-based Drug Design: a Case Study of
the Population-based Annealing Genetic Algorithms
C'hieii-cheng Clhen
Leuo-hong Miang
Cheng-yan Lao
Ming
Ouhyoung
Wen- c
hi
n
C
hen
Department of Computer Science
and
Information Engineering
National Taiwan University, Taipei, Taiwan,
R.O.C.
Abstract
The molecular banding problem, one of the most irnpoi-tant problems tn structure-based drug deszgiz ccin be formulated as a global energy optimzzatzoii problem by usziag niolecular mechanics In thas pa- per, a noire1 computataonal ulgorathm as proposed to address the molecular bandiiig problem T h e algo-
I ithnz zs dei rued f r o m genetac algorathms(GA) plus simulated annealzng{SA) hybrad techniqt~es, namely populatron-based annealriig genetac algorathms(PAG)
CI.e h a w applzed the algorzthm t o find bandaiig
structures f o i three drug-protean molecular pairs One of the three drugs 2s un antz-cancer drug n r e t h o t r e ~ a t e { M T X ) und the other two are analogue of antibacterral drug trzmethopi-am Moreover, we
l i a i e also studzed two other well-resolved Ezguiid-
itceptor molecular complex whzch are obtazned f r o m the Pi oteari Data B a n k { P D B ) Thermolysan-HONH- ben-ylnialoiiyl-L-Ala-Gly-p-natroanalide coinplex(5tln)
o i i d HI\'- 1 protease-Hydroxyethylene isostere anhrbitoi. complex Hydroxyethylene rsostere aizhzbztor 6s one of n e w potentzal NlL'-I protease inhabitors synthesazed Through o u r experzments, all of the bindang re5tIk5
not only keep the energy a t low levels, but also h a t e
a prorniszng bindrng geometracal structure an terms o f number of hydrogen bonds formed
1
Introduction
Many cliiiica.1 drugs used today are generated in
a lengthy drug discovery cycle. Generally speaking, such a lengthy cycle includes several important steps, including finding good startting molecular st,ructures which are called lead conipounds for optimization, re- fining t~he starting molecular structures to generat,e po- t,ential drugs, biologica.lly testing potential drugs gen- erated from previous steps and testing new drugs clin- i d l y [3]. Each step requires 1-3 years t,o complete. It requires 6-12 years to bring a new drug from discovery
t.o ma.rl;et in 1990[10]. Therefore, many researchers desire to shorten the cycle.
One of t8he effortss in speeding up t,he time required to find lead coinpounds is a strategy extended from Cohen's paradigm[9]. In 1977, Cohen proposed a
genera.1 pa.ra.digm to design drugs for infectious dis- eases. His paper revealed tha.t infectious agents, such as viruses, bind themselves with crucial proteins or nucleic acids. These crucial proteins serve as targets for intervention and can be utilized t80 design anti- cancer or ant.i-virus drugs. In other words, if certain drugs can bind appropriately with targets of infections agents, the diseases will be inhibited. Extending
Co-
hen's paradigm, alternative methods were applied t o drug discovery[a]
[
191. Those met>hods are categorized in structure-based d r u g design[18]. T h e key issue of structure-ba.sed drug design is to find a small molecule which b i d s well wit,h t,he receptor geomet,rically and biologically. Finding a good lead compound with a de- sirable mode of act.ion and acceptable biological prop- erties corresponding to a specific receptor has been termed t,he moleculul- binding problem,.In the past, many researchers have proposed st.rat#e- gies, including simulated anneding-based [l] E121 a,nd genetic algorit,hm-based strategies
[5]
[71[23] t o conquer the molecular binding problem. In this pa.per, we have proposed a novel stra.t,egy t o address the molecular problem. Our strategy is based on genetic algorithms plus simulated annealing hybrids, namely population- based annealing genetic algorithms(PAG) .2
Population-based annealing genetic
algorithm
Genetic algorithins(GA) [14][15] and simulated an- nealing(SA) [17][20] are two kiiids of useful stochastic techniques which can be used to solve optimization problems approximately. T h e research incorporating GA with SA can be roughly considered to be two com-
plenienta.ry categories, one using genetic approach t,o design parallel SA [13][22] and the other considering
SA
a.lgorithm as a, neighborhood operator of the ge- net'ic algorithms [4][2l]. The categories are not, appar- ent for some cases, since these cases can be explained by different ways from different perspectives.Observing the GA/SA hybrids listed, t8here is a
common characteristic. No matter what the combi- i d i o n between GA and SA is, a partial sequence of SA algorithm was performed on each individual of the population. From thse view point of
G A ,
this part,ial sequence of SA algor.ithm takes ca.re of improving in- dividuals before or afim genetic operators are applied. We can treat this sequence of SA algorithm as a iieigh- borhood operator which searches tthe neighbors of in- dividuals under the control of Metropolis condition.Motivated by the clbservation mentioned above, SA algorit,hm being a neighborhood operator seems to be good for GA/SA hybrids. So we fix the nuniber of steps of SA algorithm performed on each individua.1 and define a set of population-based annealing(PA) operators. That. is, a PA operator performs K times of one-step
SA
on the whole population. Following the concept, we can define a set, of population-based annealing operator:Definition 1 A population-based annealing(PA) op- e,rator is a neighborhood operator of G A . P A can be de- scribed ns a three tuples operator: P A = { K ,
N g ,
R.A}. I< i s the nzim,ber of steps of one-step SA algorithm ap- plied t o each i n d i v i d d of current population whereIi
2
1..&/y
is the neighbor generation method that isused by each individual of current population when per-
forming one-step SA algorithm.
R.A
is the acceptance criterion used by one-step S A algorithm f o r competa- tion of the current individual and its neighbors.According to Definition 1, when we incorporate PA wit,h GA, we can derive a new class of GA/SA hy- ]>rids, na.mely population-based annealing genetic al- gorit,hms(PAG)~ [27][28][30]. The pseudo code of the a.lgorithm is given below:
A. Initialize the parameters, i.e., populationsize, To,
and decreasing factor tr(0
< a
<
1 )B. Randomly generate in,itial population.
C. Repeatedly generate new populations as follows until system is frozen :
1. For each individual do
Best_point:=Currentpoint:=Currentindividual ;
Do E; times :
a. Generate nextpoint from currentpoint using the n o r m a l probability d e n s i t y function(nOrma1 P.D.F.) applied on the whole solution space ;
b. Accept next-point as currentpoint by probability:
Prob
-
e z p ( - c C , e z t / T k ) eap(--C,,,,,,t/Tk)tESP(--C,,,tITk) ' 2. 3 . c. if (Ccrrrrent<
C h e s t ) t h e n Bestpoint := Currentpoint ;Pick Best-point into the transient population. G e n e t i c stage.
Apply the genetic operators to transient population;
if (it is the first stage) then
determine the initial temperature 7'1
TI = fifaT,(Ck,,,-c:rn)
populatton-szze/l else
T A + ~ = C Y . T k :
There is somet.hing interesting in the initia.lization of the system temperature. It. is recognized t,hat the execution time of SA depends on the init,ial temper- ature and the decreasing factor of the temperature.
PAG
includes SA as an operator, so t,he efficiency of PAG a.lso depends on the init.ia1 temperature. .4ct8u- ally, adjust,ing the initial temperature is a tradeoff be- t,weeii efficiency arid the mpability of diversit.y ma.in- tenance of the PA operat,or. When tshe init,ial tem- perat.ure is high, PA can accept worse solutions with a larger proba,bilities. That means, much more solu- tion space can be explored. But, on t,he other side, the higher initial temperature is, the more time a system takes to reach to the frozen condit,ion. In contrast, when the init,ial temperature is low, the exploration capability of PA is inhibited.In ['Ll], the aut,liors provided a method deciding a
reasona.ble range of initia.1 temperature to prevent the deficiency. Here we provide another strategy. The strategy is defined as follows. For balancing the ef-
ficiency and the capa.bility of diversity mairiteiiaiice of the PA operat,or, we define the acceptance proha.- bility of detriment,al move t,o be 0.6 when the algo- rit,hni starts. From Met.ropolis criterion Prob(AC) =
exp(-AC/T), we obtain T
=
2.AC where A C is determined by t,he largest possible detrimental niove of current g e n e d i o n . Since we are not sure how largea possible detrinienta.1 niove can be, we try to est,imate tshe possible det,riinent,al move by the values generated from the first cycle of PAG algorithm. In the first cy- cle of PAG, a piecewise Markov chains are generated from each individual of initial population, these indi- vidua,ls inay be located a t very different hills. We have to consider all chains t o estimate the largest possible detrimental move. Therefore, we calculate the initial temperatsure as :
Ma.cc;(c:n,,
-
CLin) T. . -znzt - populat ion_size/2
where C&,,
,C:nz,,
are the largest and lowest cost of the ith sequence of Markov chain generated by ith individual of first generation. We take the maximaldifference of all sequences of Markov chain as AC to det,ermine the initial temperature.
As for the genetic phase, t,here are two important issues. One is the selection mechanism and the other is the genetic operators.
At the genetic phase, we use the ranking algorithm as a selection mechanism. As presented by Whitley [29], the ranking algorit,hm not only prevented GA from premature convergence but also provided a di- rect control on selective pressure which affects the search speed. iVe propose a dynamic ranking proce- dure which adaptively changes the select#ive bias from generatmion t o genemtion. The dyna.mic ra.nking pro- cedure is given as :
Assign initial value of bias. The value biases the ratio of the offspring with expected value between the best and the worst, individual in current pop- ulation.
For each individual, evaluate t.he number of the expected offspring, E,, based on the bias and the following linear function from the best, one to the worst one.
( p o p u l a t z o n - s i z e . b i a s ) - ( 2 . ( b i a s - 1) ‘ r a n k , ) (population-size
+
bias - 1) E , =(2) where rank, is the rank of each individual. The best one is zero, and the worst one is (popula- tionsize-1). When bias equals 1.2 , the ratio be- tween t.he best and the worst is about 1.5. When the bias reaches 1.5 , the ratio becomes 3. Raise the bias by a. factor ct from generation to
generation.
Since we want t o keep tjhe diversity at, the first few generations of
PAG
and after that we need to grad- ually inherit good materials from better individuals, therefore dynamically changing the bias of ranking is a.ppropriate. After a few tests, we use 1.2 as t.he initial bias and multiply t,he bias by 1.005 after ea,ch genera- t,ion.Moreover, the crossover and mutation opera.tors are performed according to the following steps.
1. At first, t,he parents are selected from the popula- tion randomly. The crossover operator is applied with a predefined crossover rate. After t,hat, two offspring are produced.
2. T he offspring survive only when the costs of these two offspring are both less than average cost of the previous generation. Otherwise, give up the offspring and continue t o apply the mutation
opera.t,or t,o the parents. The mut,ation opera- tor is an annealing-like mut,ation opera.tor which mimics t.he exploration capability of SA. It ex- plores the neighborhood of the parents. Mu- tation occurs with a logistic probability given as e z p ( - E $ e w ) / ( e z p ( w ) + e z p (
-*pew)),
whereE,,, and Eold are the evaluation functions of a
problem, and are binding energies in our problem domain.
3 . Finally, the offspring or the mutated parents are copied into the ilew popula.tion.
Finally, for the frozen condition, PAG will stop when 80% chromosomes’ genes are all the same. Choosing 80% as the stopping criterion is only for the sake of efficiency for our problem domain.
3
The
Problem
The key issue of the molecular binding problem is to find the binding structure of the drug-receptor complex. To judge whether a binding structure of molecules is stable or not, scientists use the potential energy between binding pairs as a reference.
Generally speaking, the interaction pqtential en- ergy of a specific binding structure can be approxi- mately evaluated by molecular mechanics[6]. In gen- eral, the potential energy of a given system is deter- mined by the summation of all kinds of force fields. The following equation is a canonical form of the total potential energy function:
where
Vb
is the bond strekhing force field,1$
the bond angle bending force field, V+ the bond torsion force field,V,
t8he out-of-plane bending force field, Vnb the non-bonded interaction force field, Ve the electrostatic interaction energy and is the hydrogen bonding force field.Fortunately, not all of these terms are crucial t o solve the molecular binding problem. Actually, only three terms of them are dominant when calculating
the intermolecular potential energy. These three terms are non-bonded interaction, bond torsion(steric) and electrostatic force fields. Therefore, without loss gen- erality, we can simplify Equation 3 as follows:
Since we start with a simplified case based on pre- vious work of [24]. Therefore, about the scoring func- tion, we follow authors in [24] to use the Lennard- Jones 6-12 pot,ential function to represent non-bonded
interaction !& and electrostatic interaction
V,.
The Lennard-Jones equation is defined as follows[26] :-’
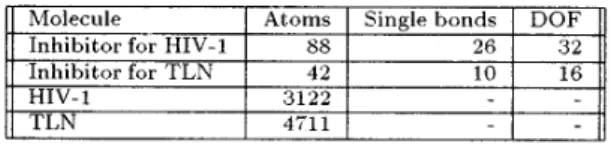
(5) Molecule Inhibitor f i r HIV-1 Atoms SS Single bonds 26 DOF 32 Inhibitor for TLN 42 10 16
where
Kat
is the total binding energy. qr andqd
are the partial charges of atoms b_elongin$to the receptor and the drug respectively.R,
and Rd represent the three dimensional coordinates of atom in the-receptor and the drug molecule. Therefore,IZ,.
- RdJ is the distance between atoms of the receptor and drug dif- ferently. E , Ardl B p d are the dielectric and non-bond const,ants. In this function, the first summation simu- lat,es the electrostatic interaction between each pair of atoms, and the second and third summation simulate the repulsive and attractive terms in Van der Waals interaction energy respectively.Following the work in
[24],
we will first bind three ligands t o dihydrofolate reductase(DHFR) t o demon- strate our algorithms. Three kinds of ligands are used to be “docked” t o DHFR: methotrexate, inhibitor 91 and inhibitor309.
Methotrexate is an anti-cancer drug which is used clinically t o cure patients. Inhibitor 91 and 309 are analogues of trimethoprim which is a n anti-bacterial drug. To reduce computational com- plexity in this pilot research] we only fetch the active site of DHFR as ourt
srget molecule. The constitution of all molecules including the receptor and the number of degrees of freedom are listed on Table 1.Table 1: The data set of our simulation model.
In addition, we also study two other well-resolved ligand-receptor molecular complex which are obtained from the Protein Data Bank(PDB): Thermolysin- HONH-benzylmalonyl-L-Ala-
Gly-p-nitroanilide coinplex(5tln) and HIV-1 protease- Hydroxyethylene isostere inhibitor complex. Ther- molysin is one of the important enzymes tha t ex- ist widely and participate in a number of impor- t,ant biological and physiological processes. HONH- benzylmalonyl-L-Ala-Gly-p-nitroanilide is one of the well-known inhibitors which bind well t o thermolysin
[16]. Hydroxyethylene isostere inhibit,or is one of new potential HIV-1 prot,ease inhibitors synt,liesized[l1]. The DOF and numbers of at80m of receptors an d lig- ands are listed on Table 2 .
I _.
HIV-1
I
3122I
TLN
I
4711I
Table
2.
T h e data set of our sirnulataon model. whereDOF
as the number of sangle bonds plus sax degrees of freedom an translatzon and rotataon. T L Nas
ther-molysan,
HI\‘-1
as H u m a n Immunodeficaency Varus-1 Protease.All test molecules listed above can be viewed as a number of atoms defined by their three-dimensional coordinates. Using these d a t a sets as well as scoring functions mentioned previously, we will use PAG t o regenerate binding structures of these molecular com- plex.
Int,uitively, the three-dimensional location of the drug and its three rotational angles relative t,o
3
axes are all adjustable. Moreover, the molecules have anumber of deformable single bonds. Each single bond is a degree of freedom. Based on the description, the energy minimization between a molecular system be- comes:
1. Fix the location of the receptor. Initialize the structure of the drug molecule. Evaluate the in- teraction energy based on the scoring function.
2.
Repeatedly adjust different degrees of freedom, including translating and rotating the drug molecule and twisting single bonds inside the drug, to fit the receptor. Evaluate the interac- tion energy of each new configura.tion.3.
Find the best configuration wit,li t,he lowest inter- actmion energy from these configurations.By adjusting the value of each degree of freedom, a new configuration will be generated. Th e whole search space is the combimtion of possible values of all de- grees of freedom. Therefore we encode all degrees of freedom as a chromosome:
where
t , ,
t , and t , represent the positmion of the drug molecule relative to the centroid of the receptor, T ,,
Table 3 . T h e results evaluated by
PAG,
where the bandang energy need t o be globally manamazed. Ewaluataon means the count of ewaleiataoii about scorang functaonry ~ r, are the rotational angles of the drug, and s, are
the twisting angles about the single bonds inside the drug molecule.
For convenience, we use real-coded scheme to ea- code each parameter. T h a t is, all parameters are real numbers. In addition, we use one-point crossover and blend crossover as genetic operators.
4
Experimental results and discussion
4.1Molecular binding results
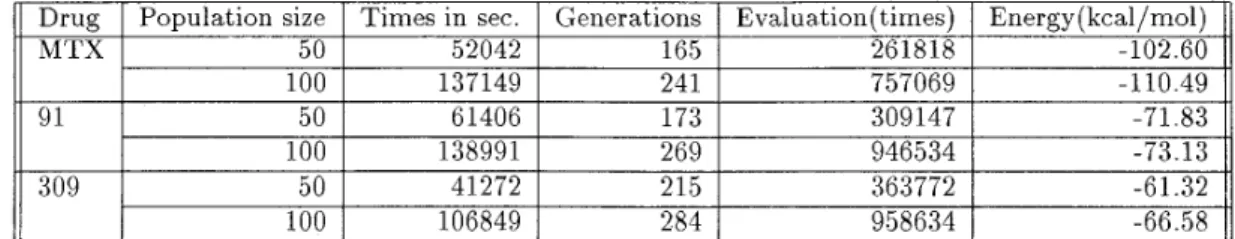
We have implemented PAG algorithms on a Sun UltraSparc workstation.(hloreover, we also have im- plemented another P C version of PAG algorithms.) Three pairs of receptor-ligand: DHFR-MTX, DHFR- Inhibitor 309 and DHFR-Inhibitor 91 are evaluated by using the algorithm listed in previous section. Th e preliminary results in this pilot research are given in Ta.ble 3 .
In the table, PAGs are executed 10 times for each case. All of the parameter settings are the same. Th e decreasing factor of temperature is 0.9. K , the number of steps performing PA operator in each generation is equal t o degrees of freedom in each problem. We use one-point crossover and blend crossover as the genetic operators. The probability of crossover and blend crossover are both 0.5. Based on the design of genetic stage explained in the previous section, the mutation rate is dependent on the success of the crossover opera- t,ors. Moreover, the final results of PAG is improved by a steepest-descent local minimizer. Since we use the real number encoding, each DOF is a 64-bit real num- ber, the total number of possible candidates for each DOF are 264. Therefore, if a drug molecule possesses 1 2 single bonds, the total number of possible position
a i d conformation will be 2G4*18 = 6.07*1034G. We also compare our results with similar results from Oshiro, Kuntz and Dixon [23]. The following is a table of com- parison (Table 4). Please note t ha t Kuntz’s method is
a pure genetic algorit,hni, and uses binary coding for the solution space, while we use real number coding; furthermore, Kuntz’s algorithm needs t o know the ac- tive sitme first, but ours can use random position for the starting points. For initial population, we use a much smaller size (100) as compared to 12167 from Kuntz. The result,s show that our P,4G is 24% better in terms of number of energy function evaluations, even if our solution space is much bigger.
Drug
I
PopulationI
EvaluationI
RmsdI
SolutionspaceI
KuntzI
12167I
940332I
0.8 AI
active sitePAG
I
100 I 757069 I 0.8 A I whole maceTable 4: Table of compcarzson wath the bandang of an anhabator M T X t o a receptor
DHFR,
where R m s das
root- mean-squa re dew a at aon .
4.2
Comparison of experimental results
There are many criteria to judge whether the bind- ing structures generat,ed by the program are reason- able. The first criterion is the binding energy. Since we use the sa.me d a t a set and energy functions as the ex- periment done in [24][25], the results can be compared with the d a t a listed in [24]. In [24], the best binding energy of t.hese three drugs are from -4OKcal/mol t o -120Kcal/mol. PAGs have obtained the minimal en- ergy at the range from -40I<cal/mol t o -1lOKcal/mol. The binding energy of the pairs MTX-DHFR, 91- DHFR and 309-DHFR are in the range of -7OKcal/mol t o -1 lOKcal/mol, -4OKcal/mol to -8OKcal/mol and -SOKcal/mol to -70Kcal/mol respectively.
The results listed above roughly verify that PAGs can find out the binding structures witah reasonable low binding energy. However, the corresponding drug structures of approximative binding energy may be different in orientation and conformation.
In addition t o t,he binding energy, the existence of hydrogen-bonds is the other stronger criterion that de- termines the goodness of fitting between ligands and proteins. The hydrogen bond is formed when the hy- drogen atom is close enough (2.4,a
-
2.8A in distance) to another at om such as oxygen. Since the hydrogen bond involves a strong “binding energy”, two to fivepairs of such hydrogen bonds becomes dominant in the total binding energy. There are 4 primary hydrogen bonds formed between MTX and DHFR. These 4 pri- mary hydrogen bonds construct the elementary con- formational structure of MTX-DHFR complex. The same situation happens to the 309-DHFR and 91- D H FR complex.
Table 5 shows our experimental results obtained from the PAG. From the analysis of hydrogen bond-
Table
5:
T h e hIydrogen bonds found byPAG.
aThe largest distance of forming a hydrogen bond bForms 4 hydrogen bonds
‘Forms at least 3 ]hydrogen bonds
ing between drugs and DHFR, we can claim the pre- sented PACT have found some good fitting molecular structures between drugs-DHFR complex, since most of t he resulting drug structures getting from PAG are buried deeply into the correct pocket of DHFR.
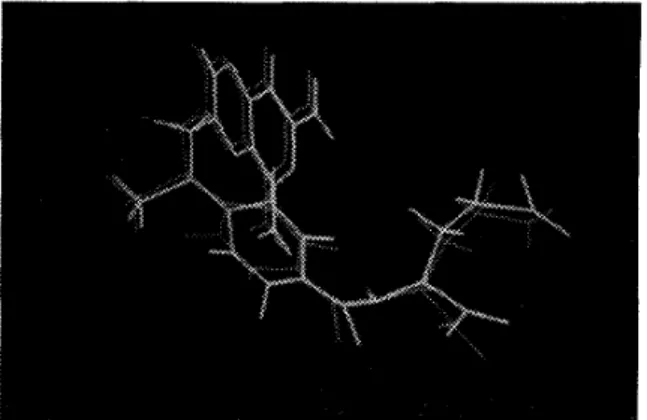
Figure 1 illustrates the superimposing of MTX crystal structure with the best of our results. The rinsd(root-mean-square deviation) value of this struc- ture is 0.81k
4.3
Experimental results
of more general
cases
We have applied PAG t o solve more compli- cated cases. Thes8e cases are molecular complex obtained from PDB, including Thermolysin-HONH- benz ylma1onyl-L- Ala-
Gly-p-nitroanilide complex( 5 t h ) and HIV-1 protease- Hydroxyethylene isostere inhibitor complex. Since the problem size is bigger than the cases of DHFR, we use larger population siz,e for PAG. The experimental re-
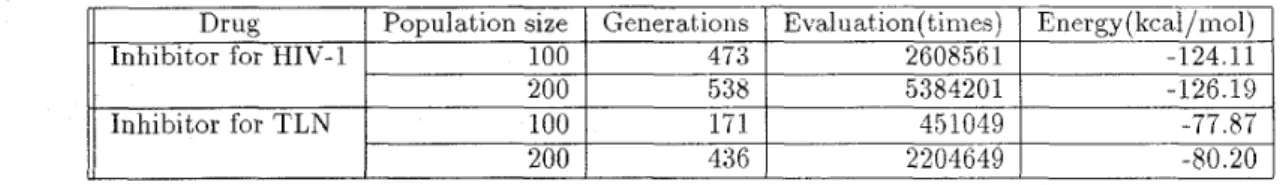
sults of more general cases are listed in Table 6. All of the cases listed above are the best cases. The parameter settings and genetic operators are all the
Figure 1. T h e superimposzng of our resultzng structure with the crystal structure of hfT.ri wath rnisd value of 0.8-4. T h e brzghter lzne segments alp the crystal struc- tures of M T X , and the darker lzne scgments are oui-
resultzng structure.
same with the ca.ses of DHFR. We will discuss the results with the following issues:
Binding energy
When the population size is 200, the binding energy of HIV-1 protease with it.s ,in- hibitor obtained from PAG are in the range of -110 Kcal/mol t,o -130 Kcal/mol . As to the cases of Thermolysin, the binding energy are in the range of -40 Kcal/mol to -80 Kcal/mol. Com- parison with the binding energy of crystal struc- tures which are -145 Kcal/mol(HIV-1) and -88 Kcal/mol(TLN) respectively, our results are near enough. Moreover, we also try another exper- iments of parameter settings of PAG. We have focused on the initial temperatureTo
and tem- perature decreasing factor a respectively. Since raising the initial temperature and the tempera- ture decreasing factor could strengthen the effects of simulated annealing to prevent from trapping into local optimal. Due to the stochastic property of PAC, the results aren’t, always much better. We will try more experiments about parameter settings of PAG in the future.’
Hydrogen bonding
According to the results pre- sented in [ll] and[16],
there are a few primary hydrogen bonds formed between HIV- 1 protease and Thermolysin with their inhibitors. There are5
primary hydrogen bonds between HIV-1 pro- tease with its inhibitor. In one experiment done by Wang [30] has showed 5 out of 10 runs could lo- ca,te these5
hydrogen bonds. In the case of Ther- molysin, there are 4 primary hydrogen bonds be-Drug Population size
Inhibitor for HIV-1 100
200
Inhibitor for TLN 100
200
Table 6: T h e results eualuuted by
PAG.
Eualuatzon nzeuns the count of euulziatzon about scorzng functzon.Generations Evaluation( times) Energy (kcal/mol)
473 2608561 -124.11
538 5384201 -126.19
171
451049
-77.87
436 2204649
-80.20
tween Thermolysin with its inhibitor. 6 out of 10 runs in his results locate these 4 hydrogen bonds. From the a.nalysis of hydrogen bonds, PAG still have found good fit.ting molecular st r uch re s over 50% even when the problem size is much larger than the cases of
DHFR.
From the discussion list,ed above, we can claim
PAG
still have found the reasonable fitting struct,ures over 50% even when the problem size is getting much larger. The following figure(Figure 2) shows the illus- tration of the binding structures.Figure 2: T h e bandang structure of Inhzbztor wzth HIV-
i,
where the central small molecule as the anhabator, and the outsade rabbon model as theHIV-1.
5
Conclusion
T he importance of the molecular binding prob- lem is without questioning. In this paper, we have developped a ciass of neighborhood operators called populat.ion-based annealing operators for genetic al- gorit.hms. Incorporating a different PA operator with genet,ic algorithms will form a new
GA/SA
hybrid. Pire design t,hePAGs
to solve the level-2[S]
molecular binding problem. Experimental studies not only indi- cate t,he usefulness of PAGs but also the properties of different PA operators.The near-optimal solutions obtained by PAG can be used for the work of drug design. We have verified that the genetic algorithms incorporating with SA are suitable to solve the molecular binding problem and believe these results are very useful for the biotechnol- ogy community. In summary, the contribution of this paper are:
1. We have discussed the GA/SA hybrids
in
de- tails. Aftfer some elementary analysis, we design the GA/SA hybrids. T h e algorithm can be used to solve the level-2 molecular binding problem. Furthermore, in the binding problem, we don’t need to know the binding pocket site a t first, where most researchers have to know for their algorithms to reduce the problem size. We also hied the case of HIV-1 protease with its inhibitor, where the size of the problem have kept others from automatically finding its binding position. 2. Our PACT algorithms are extensible since we havefocused on ba.lancing the efficiency a.nd enhanc- ing the capabilit,y of diversity maintenance of the algorithms. Based on the extensibilit,y, we ha.ve a
great chance to examine the level 3 binding prob- lem.
References
[I] R. Abagyan, M. Totrov, D. Kuznetsov, ICM-A New
Method for Protein Modeling und Design: Applica- tions to Docking und Structure Prediction from the Distorted Native Conjormution, Journal of Computa-
tional Chemistry, Vol. 15, No. 5, pp. 485-506, 1994.
[2] K . Appelt, et, al., Design of Enzyme Inhibitors Using iterative Protein Crystallographic Anulysis, Journal of
Medicinal Chemistry, vo1.34, No, 7, pp. 1925-1934, 1991.
[3] L. NI. Balbes, S. W. Mascarella, D. B. Boyd, A Per-
spective of Modern Methods in Computer-Aided Drug Design, Reviews in Computational Chemistry, Vol- ume V, K . B. Lipkowitz and D. B. Boyd, Ed. VCH Publishers. Inc. New York, 1994.
[4] D. E. Brown, C. L. Hunt.ley, A. R. Spillane,..4 parcrllel genetic heuristic: f o r the quadratic assignment proh-
lem, Proceeding of the Third International Conference i n Genetic Algorithms, pp. 406-415, 1989.
[5] T . Brodmeier, E . Pretsch, Application of Genetic 4 1 -
gorithms in hfoleculer Modeling Journal of Comput,a- tional Chemistry, Vol. 15, No. G , pp. 588-595, 1994.
[6] Lf. Burkert, N. L. Allinger, hfolecular Mechanics, American Chemical Society, Washington D.C., 1983. [7] I<. P. Clark, Ajay Flexible Ligand Docking without Parameter Adju’ustment across Four Ligand-Receptor Complexes, Journal of Computational Chemist.ry, Vol. 16, No. 10: pp. 1210-1226, 1995.
[8] D. E. Clark, D. R. Westhead, Evolutionary adgo- rithms in computer-aided molecular design, Journal of Computer-Aided Molecular Design, Vol. 10, pp. 337- 358, 1996.
[9] S. S. Cohen, A Strategyfor the Chemotheraphy of ln- fectious Disease, Science, Vol. 197, No. 4303, pp. 431-
432, 1977.
[lo] J . A. DiMasi, R. W. Hansen, H. G. Grabowski, L.
Lasagna Cost of Innovation in, the Pharmaceutical In- dustry, Journal of Health Economics.
[ll] G. B. Dreyer, et al. Hydroxyethylene Isostere In- hibitors of Human 1mmunodeficien.cy Virus-1 Pro- tease: Structure-Activity Analysis Using Enzyme Iii- netics, X-ray Crystallography, and Infected T-cell Assays, Biochemist.ry, vol. 31, pp. 6646-6659, 1993. [I31 D. I<. Gehlhaar, IC. E. Moerder, D. Zichi, C. J . Sher-
man, R. C . Ogden,
S.
T. Freer, De Novo Design of Enzyme Inhibitors b y Monte Carlo Ligand Genera- tion, Journal of Medicinal Chemistry. Vol. 38, No.3, pp. 466-472, 1995.[13] D. E. Goldberg, .4 Note on Boltzmann Tournament Selection for Genetic Algorithms and Population- Oriented Simulated Annealing, Complex Systems, 4,
pp. 445-460, 1990.
[14] D. E. Goldberg, Genetic algorithms : In Search, Optimization and Machine Learning, Addison-Wesley Publishing Company, 1989.
[15] J . H. Holland, Adcrptation in natural and artificialsys-
[16] NI. A. Holmes, B. W. Matthews, Binding of Hydrox- amic Acid Inhibitors to Crystalline Thermolysin Sug- gests a Pentacoordinate Zinc Intermediate in Cataly- sis, Biochemistry, vol. 20, pp. 6912-6920, 1981.
tems, T h e universi.ty of Michigan Press, 1975.
[17]
S.
Kirkpatrick, C. D. Gelatt Jr., M. P. Vecchi, Op- timization b y simulated annealing, Science, Vol. 220,No. 4598, pp. 671-680, 1983.
[lS] I. D. Kuntz, Structure-Based Strategies f o r Drug De- sign and Discover:y, Science, Vol. 357, pp. 1078-1083, 1993.
[19] L. F. Kuyper. et al., Receptor-based Design of Dihy- drofolnte Rediictase Inhibitor$: Comparison of Crys- tallographically Determined Enzyme Binding with Emyme .4finity in a Series of Carboxy-Subsituted Trimethoprim Analogues, Journal of Medicinal Chem- istry.. 1’01 23, pp.303-311, 1985.
[20] P. J. M. van Laarhoven, E. H. L. Aarts, Simulated i l n n d i n y : theory and applications, D. Reidel Pub- lishing Company, Dordrecht, Holland, 1987.
[all
F. T. Lin, C. Y. Kao, C:. C. Hsu, Applying theGenetic Approach to simulated .Inizealing in Solving S o m e NP-Hard Problems. IEEE Transaction on Sys- tem, Man, Cybernetics, Vol. 2 3 No. G, pp. 1752-1767> 1993.
[ 2 2 ]
S.
W. Mahfoud, D. E. Goldberg, Pnrallel Recombina- tive Simulated Annealing: .4 Gen,etic Alyorithm, Par- allel Chnput.ing, Vol. 21, No. 1, pp. 1-25, 1995. [23] C. hl. Oshiro. I . D. I<unt,z, J. S. Dixon, Flexible Lig-and Dockir2y Using o Genetic Algorithm Journal of Computer-Aided hiIolecular Design, Vol 9, pp. 113- 130, 1995.
[24] M. Ouhyoung, Force Display in Molecular Binding, Ph. D dissertation, CS dept.., University of North Car- olina a t Chapel Hill, 1990.
[ 2 5 ] F. P. Brooks? Jr., M. Ouhyoung. J. J . Batter, P. J . Kilpat.rick, Project Grope: Haptic Display for Sciera- tific Visualization, ACM SIGGRAPH’SO, pp. 177-186, (also Computer Graphics Vol. 24, No.4, 1990).
[%GI N. P a t h b i r a m a n et al., Computer’Graphics and Drug Design: Real Time Docking, Energy Culcrtlation and Minimization, Journal of Computational Chemistry, [?7] L. H. Wang, C. Y . b o , M. Ouhyoung, W. C. Chen, Using an Annealing Genetic Algorithm to Solve Global Energy Minimization Problem in Molecular Binding, IEEE International Conference on Tools wit.h Artifi- cial Intelligence, pp. 404-410, New Orlean. USA, 1994. [38] L. H. Wang, C. Y. Kao, hf. Ouhyoung, W. C. Chen, Molecular Binding: a Cast. Stzidy of the Population-
based A n n e a h g Genetic Algorithms, IEEE Interna- tional Conference on Evolutionary Computing, Perth, Australia, 1995.
[29] D. Whit,ley, The Gentitor Algorithm and Selection Pressure : Why Rank-Based allocation of Reproduc- tive Trials is Best, Proceedings of the Third Inter- nat,ional Conference on Genetic Algorithms, pp. 116- 121, 1989.
[30] L. H. Wang, Molecular Binding in Structure-based Drug Design: a Case Study 0.f the Population-based dept., National Taiwan University, 1997.
Vol. 6, pp. 433-436, 1985.