Synthesis and characterization of H-bonded side-chain and crosslinking LC
polymers containing donor/acceptor homopolymers and copolymers
Hong-Cheu Lin
a,b,*, Jemmy Hendrianto
baDepartment of Materials Science and Engineering, National Chiao Tung University, Hsinchu, Taiwan, ROC bInstitute of Chemistry, Academia Sinica, Taipei, Taiwan, ROC
Received 12 September 2005; received in revised form 20 October 2005; accepted 31 October 2005 Available online 14 November 2005
Abstract
Side-chain copolymers consisting of proton acceptors (stilbazoles) and proton donors (benzoic acids) connected to polyacrylate backbones with different methylene spacer lengths are prepared by tuning their donor/acceptor molar ratios. The H-bonded copolymer networks are formed once they were synthesized, and showed more homogenous phase behavior than the physical blending supramolecular networks composed of donor and acceptor homopolymers. Analogous monomer–monomer and polymer–monomer (H-bonded side-chain polymer) complexes of similar structures are also investigated. According to DSC, POM, and XRD studies, it reveals that the copolymers with various spacer lengths and donor/acceptor molar ratios show the smectic A (SA) phase. The d spacing values of XRD results suggest that the H-bonded copolymers may be
constructed by either a monolayer or an interdigitated bilayer structure. Generally, the d spacing values of copolymers (in the SAphase) increase
with higher H-bonded crosslinking density between benzoic acids and stilbazoles, i.e. decrease with higher molar ratios of benzoic acids, which may be owing to the formation of hydrogen bonds between benzoic acids from different backbones. The isotropization temperatures of the H-bonded blends and copolymers increase as the molar ratios of benzoic acids increase, while the higher H-H-bonded crosslinking density between benzoic acids and stilbazoles stabilizes the liquid crystalline phase.
q2005 Published by Elsevier Ltd.
Keywords: H-bonded LC polymer networks; Donor and acceptor copolymers; Crosslinking density
1. Introduction
Recently, self-assembled phenomena through the molecular recognition between individual constituents have been explored in various areas [1–4]. Hydrogen bonding is one of the most important key factors for the molecular recognition in nature. Intensive investigation has been concentrated on the usage of non-covalent interactions, such as ionic force [5,6]
and hydrogen bonding [7–17], to generate liquid crystalline polymers. Among these approaches, intermolecular hydrogen bonding is easily obtained by complexation of carboxylic (or benzoic) acid and pyridyl moieties[17,18]. Several series of H-bonded complexes and side-chain liquid crystalline polymers through intermolecular hydrogen bonding (between
benzoic acid and pyridyl interactions) have been reported previously[8–12].
In our previous research [4], a series of supramolecular polymers with different molecular weights have been developed, and the concept of the angular (bending) structures formed by hydrogen bonding has been introduced to the side-chain supramolecular liquid crystalline (LC) polymers. The proton donors containing 4-oxybenzoic acid units are connected to the side chains of the polysiloxane polymers in order to serve as donor polymers in our past survey. Since novel properties are generated in the side-chain supramolecular polymers, the backbones of the acid donor polymers can be modified by the replacement of the polysiloxane backbones with the polyacrylate structures. In this investigation, similar approaches are applied to the reverse form of the previous supramolecular polymers by connecting proton acceptors to the polymer backbones to become acceptor polymers, i.e. poly-p-stilbazoles, to which the proton donors (benzoic acids) are H-bonded. Besides, it is also interesting to evaluate the LC properties by switching the H-bonded pendant groups of the side-chain supramolecular polymers from previous proton acceptors (stilbazoles) to the proton donors (benzoic acids).
www.elsevier.com/locate/polymer
0032-3861/$ - see front matter q 2005 Published by Elsevier Ltd. doi:10.1016/j.polymer.2005.10.136
* Corresponding author. Address: Department of Materials Science and Engineering, National Chiao Tung University, Hsinchu, Taiwan, ROC. Tel.: C886 3 5712121x55305; fax: C886 3 5724727.
So far, some publications related to supramolecular polymer networks with/without LC phase[19,20], including hydrogen-and metal-bonding[21], have been explored. For instance, as poly(N-vinyl pyrrolidone) (PVP) macromolecules are blended with short chain poly(ethylene glycol)s (PEGs) bearing reactive hydroxyl groups at both chain ends, which develop two H-bonds through both terminal OH-groups and act as reversible H-bonded crosslinkers, the PVP–PEG complexes will form non-mesogenic network supramolecular structures
[20]. With regard to H-bonded LC polymers, one of the H-bonded LC networks was achieved by self-assembly of bis-imidazolyl-terminated mesogenic compounds and poly(acrylic acid)s, where imidazolyl groups excluded from the cores were only used as the H-bonded crosslinkers [10]. Furthermore, H-bonded liquid crystalline polymer networks under mechan-ical stress also can be stabilized by these dynamic H-bonds
[16]. The formation of intermolecular H-bonds between carboxylic groups of functionalized LC copolymers and the pyridine fragments of the dopants also leads to stable, nonseparating mixtures in a broad interval of contents (up to 30 mol% dopant molecules) [17]. Another H-bonded LC copolymer networks were composed of various amounts of 40-(11-acryloylundecyloxy)biphenyl-4-carboxylic acids and acrylic acids, whose monomers have carboxylic acids on each terminus [22]. Dynamically crosslinked structures can also be obtained by blending a carboxyl-functionalized polyacrylate with a mixture of a stilbazole (with a single H-bonded acceptor) and a bipyridine (with two H-bonded acceptors) [23]. In addition, supramolecular mesogenic net-works were prepared by self-assembly involving multifunc-tional low-molecular-weight components, including trifunctional H-bonded donors and bifunctional H-bonded acceptors (maintaining a 1:1 molar ratio of acid and pyridyl groups) [24]. Similar H-bonded LC networks have been prepared by self-assembly of a chiral bifunctional H-bonded acceptor and achiral trifunctional H-bonded donors. These H-bonded complexes could exhibit the cholesteric phase and glass transition behaviour [25]. Furthermore, interesting polymerization occurred in the cholesteric phase induced by addition of chiral molecules in H-bonded nematic monomers
[26]. Another anisotropic networks consist of donor groups connected to a poly(methacrylate) backbone, H-bonded side-chain stilbazoles, and bipyridyl crosslinking units. The anisotropic networks are smectogenic (SA and SB), and the
glass transition temperature (Tg) increases with the
concen-tration of the bipyridyl crosslinking unit[27]. Though there are some results regarding supramolecular mesogenic networks, to our knowledge, a copolymer consisting of proton donor and acceptor pendants on the same backbone (thus to form a H-bonded crosslinking polymer) has not been published yet.
Accordingly, in this report, novel H-bonded LC polymer networks are formed by the copolymers, where the H-bonded interactions of donor and acceptor moieties on the same or different polymer backbones take place once they are synthesized. This kind of H-bonded donor/acceptor copoly-mers can avoid the process of blending (by heating blends or evaporation of the polymer solution) two complementary
donor/acceptor homopolymers, which may have the immisib-lity problem to cause the phase separation. With the purpose of examining the above influence, two analogous donor/acceptor homopolymers are also synthesized and blended to compare with the H-bonded donor/acceptor copolymers. As a whole,
Fig. 1displays various types of idealized H-bonded side-chain and crosslinking liquid crystalline structures demonstrated in this report, i.e. (a) donor monomers and acceptor polymers, (b) donor polymers and acceptor monomers, (c) physical blends of donor and acceptor homopolymers, and (d) donor and acceptor copolymers. In order to study the effect of the crosslinking density of H-bonds on mesogenic properties, acceptor monomers (stilbazoles) and donor monomers (benzoic acids) are copolymerized by different feeding donor/acceptor molar ratios and corresponding copolymers with similar output donor/acceptor molar ratios can be generated. Thus, H-bonds of interchains and intrachains are formed between acceptor (stilbazole) and donor (benzoic acid) moieties as soon as the copolymers are made, and the crosslinking density can be controlled by the donor and acceptor ratios in the copolymers. Therefore, different H-bonded LC polymers, i.e. side-chain LC polymers in Fig. 1(a) and (b) or crosslinking LC polymers in
Fig. 1(c) and (d), are easy to be acquired and controlled in order to adjust their supramolecular properties. Besides, different lengths of methylene spacer groups are tuned to investigate their effect on the mesogenic properties of the H-bonded polymer networks.
2. Experimental 2.1. Measurements
1H NMR or 13C NMR spectra were recorded at ambient
temperature for monomers and at 40 8C for polymers (concerning the incomplete solubility of the polymers at ambient temperature) using 300 MHz Bruker MSL spec-trometer. Chemical shifts are given in ppm with respect to the solvent residual resonances fixed at 7.27, 2.49 ppm for CDCl3and DMSO, respectively. The actual compositions of
the copolymers were determined by 1H NMR spectroscopy. The elemental analyses were carried out by Perkin–Elmer 2400 CHN type.
The mesogenic behavior was measured on a differential scanning calorimeter (DSC), model Perkin–Elmer DSC-7. The sample chamber was flushed with nitrogen gas. Measurements were performed on 2–4 mg of samples (less for monomers, more for polymers) sealed in aluminium pans. The samples were all previously dried in vacuo for 2 days at room temperature (for monomers) and at 40 8C (for polymers), and scanned at the rate of 10 8C/min. The thermograms used were obtained from the second heating and the first cooling data, as well as the transition temperatures are given by the maximum peaks or minimum peaks.
X-ray diffraction (XRD) patterns were obtained from an X-ray diffractometer Siemens D-5000 (40 kV, 30 mA) fitted with a temperature controller TTK450. Nickel-filtered Cu Ka radiation was used as an incident X-ray beam.
Polarizing optical microscopy (POM) observations of the film samples were made using a Leitz Laborlux S equipped with a THMS-600 heating stage. The film samples were prepared by casting samples between two plates of glass in the isotropic state.
Number-average and weight-average molecular weights, Mn and Mw, respectively, were determined by
polystyrene-calibrated gel permeation chromatography (GPC; Jasco PU-1580 and Jasco RI-930) using THF as the eluent.
2.2. Materials
Unless otherwise specified, the reagents were obtained from Acros. All solvents used (dimethylformamide, THF, benzene,
dichloromethane, acetone, ethyl alcohol) were analytical grade and were distilled before use. 4-Picoline, triethylamine were distilled over calcium hydride. Hexane, ethyl acetate, petroleum ether, acetic anhydride, sodium hydroxide (NaOH), cesium carbonate, acryloyl chloride, acrylic acid, potassium hydroxide (KOH), hydroquinone, PTSA (all of the best reagent grade available) were used as received. Benzoyl peroxide (BPO; Acros), used as a radical initiator, was recrystallized before use. Column chromatography was performed using silica gel [Merck, Kiesegel 60 (70– 230 mesh), and thin-layer chromatography (TLC) was carried out on precoated silica gel plates (Merck, F-254). Water used for reaction and extraction was purified by a Milli Q water purification system (resistance 18 U).
Fig. 1. Schematic illustration of idealized H-bonded side-chain and crosslinking LC polymers: (a) donor monomers and acceptor polymers; (b) donor polymers and acceptor monomers; (c) donor polymers and acceptor polymers; (d) donor and acceptor copolymers.
2.3. Synthesis
2.3.1. 4-((6-(Acryloyloxy)alkyl)oxy)benzoic acid monomers (D1-2)
D1-2 were prepared according to the procedure reported by Portugal et al.[28]. The NMR results of the monomers are as follows.
2.3.1.1. 4-((6-(Acryloyloxy)hexyl)oxy)benzoic acid monomer (D1).1H NMR (300 MHz, CDCl3)d: 1.45–1.53 (m, 4H, CH2), 1.69–1.74 (m, 2H, CH2), 1.80–1.85 (m, 2H, CH2), 4.02 (t, 2H, JZ6.3 Hz, ArOCH2), 4.17 (t, 2H, JZ6.6 Hz, OCH2), 5.81 (dd, 1H, JZ9.3, 1.1 Hz, cis-COCHCH2), 6.13 (dd, 1H, JZ17.3, 10.4 Hz, COCHCH2), 6.37 (dd, 1H, JZ16.1, 1.2 Hz, trans-COCHCH2), 6.92 (d, 2H, JZ8.8 Hz, Ar–H), 8.05 (d, 2H, JZ 8.8 Hz, Ar–H). 13C NMR (300 MHz, CDCl3)d: 25.7, 28.5, 28.9, 64.5, 68.0, 114.1, 121.5, 128.5, 130.5, 132.3, 163.5, 166.3, 171.8. Anal. Calcd for C16H20O5: C, 65.74; H, 6.9.
Found: C, 65.31; H, 6.92.
2.3.1.2. 4-((6-(Acryloyloxy)decyl)oxy)benzoic acid monomer (D2).1H NMR (300 MHz, CDCl3)d: 1.32–1.46 (m, 12H, CH2), 1.65–1.69 (m, 2H, CH2), 1.76–1.83 (m, 2H, CH2), 4.02 (t, 2H, JZ6.4 Hz, ArOCH2), 4.15 (t, 2H, JZ6.6 Hz, OCH2), 5.83 (dd, 1H, JZ9.2, 1.2 Hz, cis-COCHCH2), 6.14 (dd, 1H, JZ10.4, 6.9 Hz, COCHCH2), 6.38 (dd, 1H, JZ16.1, 1.2 Hz, trans-COCHCH2), 6.94 (d, 2H, JZ8.8 Hz, Ar–H), 8.06 (d, 2H, JZ8.8 Hz, Ar–H).13C NMR (300 MHz, CDCl3)d: 23.5, 25.9, 27.5, 28.6, 29.1, 29.2, 29.3, 29.4, 64.7, 68.2, 114.2, 121.4, 128.6, 130.5, 132.3, 163.6, 166.4, 171.7. Anal. Calcd for C20H28O5: C, 68.91; H, 8.1. Found: C, 68.8; H, 7.96.
2.3.2. 4-((6-(Acryloyloxy)alkyl)oxy)stilbazole monomers
(A1-2)
A1-2 were synthesized using the procedure shown in
Scheme 1. The detailed syntheses of intermediates and final products are described as follows.
2.3.2.1. Synthesis of acetic acid stilbazole ester (1). First, 20 g of 4-hydroxy-benzaldehyde was dissolved in 80 mL of acetic acid anhydride, then 24 mL of 4-picoline was added into the solution. The reaction medium was stirred and refluxed for 3 days. The acetic anhydride was removed by rotatory evaporation, and the residue was then extracted by dichloro-methane and aqueous sodium bicarbonate solution. The organic layer was collected and dried with magnesium sulfate. The purification was performed by recrystallization from ethyl alcohol. Yield: 60%.1H NMR (300 MHz, d-DMSO)d: 2.31 (s, 3H, CH3), 6.91 (s, 1H, CH), 7.12 (m, 2H, phenyl-H), 7.25 (s, N CH3 C OH O H N C H CH O C O CH3 N C H CH OH THF / H2O NaOH (1.25 eq) N C H CH N C H CH O O O DMF DCM + Acetic anhydride Reflux 3 days 1.5 eq m Acryloyl acid CH2 (1) (2) (3a) m = 6 (3b) m = 10 (A1) m = 6 (A2) m = 10 Cs2CO3 Et3N O OH m CH2 HO Br m CH2
1H, CH), 7.36 (m, 2H, pyridyl-H), 7.53 (d, 2H, JZ6.9 Hz, phenyl-H), 8.56 (d, 2H, JZ6.0 Hz, pyridyl-H).
2.3.2.2. Synthesis of 4-(2-pyridin-4-yl-vinyl)-phenol (2). 44.42 g of intermediate (1) was dissolved in 180 mL of THF in a reactor, then a solution containing 9.28 g of NaOH in 20 mL of water was added into the reactants. The solution was reacted for 24 h, while reacting, a solution of 3 g of NaOH along with 10 mL of water was added into the reactor. The THF was then removed by a rotary evaporator, then the residue was neutralized with dilute hydrochloric acid to yield in precipi-tation of a yellowish solid. The solid was filtered and recrystallized from ethyl alcohol. Yield: 78%. 1H NMR (300 MHz, DMSO)d: 6.78 (s, 2H, phenyl-H), 6.99 (d, 1H, JZ16.5 Hz, CH), 7.42 (s, 1H, CH), 7.46–7.49 (m, 4H, pyridyl-H and phenyl-pyridyl-H), 8.48 (d, 2pyridyl-H, JZ6.0 pyridyl-Hz, pyridyl-pyridyl-H). 2.3.2.3. Synthesis of 4-((2-pyridin-4-yl-vinyl)-phenoxy)decanol (3a). Under inert nitrogen atmosphere, intermediate (2) was dissolved and stirred in 10 mL of anhydrous DMF, then 4.33 g of 10-bromo-decan-1-ol was added dropwisely. After stirred for 24 h at ambient temperature, the solution was extracted using dichloromethane. Upon the removal of dicholoro-methane using rotatory evaporation, the crude product was recrystallized from DMF. Yield: 60.5%.1H NMR (300 MHz, CDCl3)d: 1.32–1.46 (m, 12H, CH2), 1.65–1.69 (m, 2H, CH2), 1.76–1.83 (m, 2H, CH2), 3.64 (t, 2H, JZ9.6 Hz, –CH2OH), 3.98 (t, 2H, JZ9.9 Hz, –OCH2), 6.88 (d, 1H, JZ16.3 Hz, aCH), 6.92 (d, 2H, JZ8.75 Hz, phenyl-H), 7.26 (d, 1H, JZ 16.3 Hz, aCH), 7.35 (s, 2H, pyridyl-H), 7.47 (d, 2H, JZ 12.9 Hz, phenyl-H), 8.52 (d, 2H, JZ6.0 Hz, pyridyl-H). 2.3.2.4. 4-((2-Pyridin-4-yl-vinyl)-phenoxy)hexanol (3b). The synthesis method was the same as (3a), except that the reactant used here was 6-bromo-decan-1-ol. 1H NMR (300 MHz, CDCl3)d: 1.45–1.53 (m, 4H, CH2), 1.69–1.74 (m, 2H, CH2), 1.80–1.85 (m, 2H, CH2), 3.98 (t, 2H, JZ6.4 Hz, ArOCH2), 4.15 (t, 2H, JZ6.6 Hz, OCH2), 6.88 (d, 1H, JZ16.3 Hz, aCH), 6.92 (d, 2H, JZ8.75 Hz, phenyl-H), 7.26 (d, 1H, JZ 16.3 Hz, aCH), 7.34 (s, 2H, pyridyl-H), 7.46 (d, 2H, JZ 12.9 Hz, phenyl-H), 8.53 (d, 2H, JZ6.0 Hz, pyridyl-H). 2.3.2.5. Syntheses of 4-((2-pyridin-4-vinyl)-phenoxy)alkanoxy acrylates (A1-2). Under inert nitrogen atmosphere, 2 g of intermediate (3) was dissolved in anhydrous dichloromethane (30 mL). Then 2 mequiv of triethylamine was added dropwisely into the solution. After stirring for 1 h at room temperature, 0.72 mL of acryloyl chloride was added into the solution at 0 8C. The reacting solution was then stirred for 18 h at room temperature. The crude product was extracted with dicholoromethane and fractionated by column chromatrogra-phy using hexane/ethyl acetate (2/1) to obtain a yellowish solid. Yield: 26%. 2.3.2.6. 4-((2-Pyridin-4-vinyl)-phenoxy)hexanoxy acrylate (A1). 1H NMR (300 MHz, CDCl3)d: 1.45–1.53 (m, 4H, CH2), 1.69–1.74 (m, 2H, CH2), 1.80–1.85 (m, 2H, CH2), 3.98 (t, 2H, JZ6.4 Hz, ArOCH2), 4.15 (t, 2H, JZ6.6 Hz, OCH2), 5.82 (dd, 1H, JZ9.3, 1.1 Hz, cis-COCHCH2), 6.13 (dd, 1H, JZ17.3, 10.4 Hz, COCHCH2), 6.37 (dd, 1H, JZ16.1, 1.2 Hz, trans-COCHCH2), 6.88 (d, 1H, JZ16.3 Hz, CHa), 6.91 (d, 2H, JZ8.7 Hz, phenyl-H), 7.21 (d, 1H, JZ16.3 Hz, CHa), 7.33 (s, 2H, pyridyl-H), 7.47 (d, 2H, JZ8.7 Hz, phenyl-H), 8.53 (d, 2H, JZ6.1 Hz, pyridyl-H). 13C NMR (300 MHz, CDCl3)d: 25.8, 25.9, 28.5, 29.1, 29.3, 29.4, 64.6, 68.1, 76.5, 77.0, 77.4, 114.8, 120.6, 123.5, 128.3, 128.6, 128.7, 130.3, 132.8, 145.1, 149.9, 160.0, 166.2. Anal. Calcd for C22H25O3N:
C, 75.19; H, 7.17; N, 3.99. Found: C, 75.28; H, 7.53; N, 3.75. 2.3.2.7. 4-((2-Pyridin-4-vinyl)-phenoxy)decanoxy acrylate (A2). 1H NMR (300 MHz, CDCl3)d: 1.32–1.46 (m, 12H, CH2), 1.65–1.69 (m, 2H, CH2), 1.76–1.83 (m, 2H, CH2), 3.98 (t, 2H, JZ6.6 Hz, –CH2OCO), 4.13 (t, 2H, JZ6.6 Hz, – OCH2), 5.84 (dd, 1H, JZ9.2, 1.2 Hz, cis-COCHCH2), 6.13 (dd, 1H, JZ10.4, 6.9 Hz, COCHCH2), 6.37 (dd, 1H, JZ16.1, 1.2 Hz, trans-COCHCH2), 6.88 (d, 1H, JZ16.3 Hz, CHa), 6.93 (d, 2H, JZ8.7 Hz, phenyl-H), 7.26 (d, 1H, JZ16.3 Hz, aCH), 7.35 (s, 2H, pyridyl-H), 7.45 (d, 2H, JZ8.7 Hz, phenyl-H), 8.54 (d, 2H, JZ6.0 Hz, pyridyl-H).13C NMR (300 MHz, CDCl3)d: 25.8, 25.9, 28.5, 29.1, 29.2, 29.3, 29.6, 64.6, 68.0, 76.5, 77.0, 77.4, 114.8, 120.6, 123.4, 128.3, 128.6, 130.3, 132.9, 145.2, 149.8, 145.2, 149.8, 159.8, 166.2. Anal. Calcd for C26H33O3N: C, 76.62; H, 8.16; N, 3.44. Found: C, 76.56; H,
8.43; N, 3.32.
2.4. Polymerization
A solution of 1 g of monomers (D1-2 and A1-2) in 5 mL of anhydrous THF was prepared, and then 1 mol% of benzoyl peroxide was added as an initiator and reacted at 70 8C for 40 h under nitrogen atmosphere. The reacted mixture was poured into a large amount of petroleum ether. The polymers and copolymers were purified by repeated dissolution in THF and precipitation from petroleum ether (40–65 8C). The percent yields of these polymers and copolymers are between 40 and 60%, and their molecular weights and compositions are listed inTables 1 and 2. The output compositions of copolymers (F) were determined by NMR. The F values were obtained by integrating the peak areas of the hydrogens connected to the phenyl rings with different functional groups, i.e. benzoic acid and pyridyl groups. These peaks are solely shifted at 7.8 ppm (benzoic acid’s 2H) and 8.5 ppm (pyridyl’s 2H), respectively. Different given molar ratios did not lead to any significant shifts on the NMR peaks. The 1H NMR results of the homopolymers and copolymers (obtained from the most
Table 1
Polymer compositions and molecular weights of poly(D1-co-A1)s f (input) 0 0.17 0.33 0.5 0.67 0.83 1 F (output) 0 0.2 0.42 0.55 0.75 0.89 1 Mn(!10K3) 11.3 12.3 6.3 5.9 3.6 2.6 5.6 Mw(!10K3) 14.5 15.4 7.1 7.1 4 2.9 5.9 PDI 1.28 1.25 1.13 1.21 1.11 1.11 1.06 fZmolar composition in feed, FZmolar composition in copolymer.
representative ones, i.e. FZ0.55 for the spacer length mZ6 and FZ0.5 for the spacer length mZ10) are listed as follows:
2.4.1. P6OBA (PD1)
1H NMR (300 MHz, d-DMSO)d: 1.25 (4H, CH
2), 1.49 (2H,
OCH2CH2), 1.62 (2H, OCH2CH2), 3.91 (4H, OCH2), 6.9 (2H,
Ar–H), 7.82 (2H, Ar–H).
2.4.2. PD10OBA (PD2)
1
H NMR (300 MHz, d-DMSO)d: 1.19 (12H, CH2), 1.47
(2H, OCH2CH2), 1.59 (2H, OCH2CH2), 3.85 (4H, OCH2), 6.85
(2H, Ar–H), 7.81 (2H, Ar–H).
2.4.3. P6Stilb (PA1)
1H NMR (300 MHz, d-DMSO)d: 1.24 (4H, CH
2), 1.46 (2H,
OCH2CH2), 1.73 (2H, OCH2CH2), 3.96 (4H, OCH2), 6.87 (2H,
Ar–H and CHCH), 7.44 (6H, Ar–H and CHCH), 8.52 (2H, pyridyl-H).
2.4.4. PD10Stilb (PA2)
1H NMR (300 MHz, d-DMSO)d: 1.30 (12H, CH 2), 1.43
(2H, OCH2CH2), 1.73 (2H, OCH2CH2), 3.96 (4H, OCH2), 6.88
(2H, Ar–H and CHCH), 7.31 (6H, Ar–H and CHCH), 8.52 (2H, pyridyl-H).
2.4.5. Poly(D1-co-A1), FZ0.55
1H NMR (300 MHz, d-DMSO)d: 1.22 (4H, CH
2), 1.34 (2H,
OCH2CH2), 1.66 (2H, OCH2CH2), 3.95 (4H, OCH2), 6.93 (4H,
Ar–H), 7.47 (6H, Ar–H and CHCH), 7.82 (2H, Ar–H), 8.48 (2H, pyridyl-H).
2.4.6. Poly(D2-co-A2), FZ0.5
1
H NMR (300 MHz, d-DMSO)d: 1.22 (12H, CH2), 1.49
(2H, OCH2CH2), 1.64 (2H, OCH2CH2), 3.93 (4H, OCH2), 6.90
(4H, Ar–H), 7.46 (6H, Ar–H and CHCH), 7.83 (2H, Ar–H), 8.48 (2H, pyridyl-H).
3. Results and discussion
All proton donor and acceptor moieties used in the H-bonded side-chain and crosslinking polymers are illustrated in Fig. 2. The symbols of P, D, A, 1, and 2 represent the polymer (P), donor (D), acceptor (A), and shorter (1) and longer (2) spacer lengths, respectively. Copolymer compo-sitions and molecular weights of poly(D1-co-A1)s and poly(D2-co-A2)s are shown in Tables 1 and 2, where the output compositions of copolymers (F) may have small variations from their input compositions of corresponding monomers (f). According to the results of GPC (shown in
Tables 1 and 2), their molecular weights (Mnand Mw) are about
3–25!103(low MW) and indexes of polydispersity (PDI) are around 1.1–1.6 (narrow polydispersity by free radical polymerization). Table 3 and Fig. 3 demonstrate the phase transition temperatures of the donor/acceptor monomers
Table 2
Polymer compositions and molecular weights of poly(D2-co-A2)s
f (input) 0 0.17 0.33 0.5 0.83 1 F (output) 0 0.2 0.5 0.63 0.86 1 Mn(!10 K3 ) 20.2 13.6 6.3 7.6 2.8 4.4 Mw(!10 K3 ) 25.7 17 7.1 8.6 4.4 4.7 PDI 1.27 1.25 1.17 1.13 1.6 1.1
fZmolar composition in feed, FZmolar composition in copolymer.
O (CH2) O O O OH m O (CH2) O O O OH m n N CH CH O O O (CH2) m N CH CH O O O (CH2) n m N CH CH O O O O (CH2) (CH2) O O O OH n 1-n m m D1 : m = 6 D2 : m = 10 PD1 : m = 6 PD2 : m = 10 A1 : m = 6 A2 : m = 10 PA1 : m = 6 PA2 : m = 10 Poly(D1-co-A1)s Poly(D2-co-A2)s
and polymers before complexation. Donor monomers contain-ing benzoic acids (D1 and D2) show liquid crystalline behavior resulting from H-bonded dimers due to the interaction of carboxylic acids, while donor polymers (PD1 and PD2) lose their mesophases due to the polymer networks formed by H-bonded crosslinks. Since acceptor monomers containing stilbazoles (A1 and A2) are both non-mesogenic compounds, it is conceivable to have non-mesogenic acceptor polymers (PA1 and PA2). As usual, for donor monomers and polymers containing benzoic acids (D1-2 and PD1-2), the isotropization temperatures (Ti) of polymers are higher than those of corresponding monomers. However, the isotropization tem-peratures of the acceptor polymers containing stilbazoles (PA1 and PA2) are lower than those of their acceptor monomers (A1 and A2), individually, which may be originated from the reduced packing efficiency of the acrylate backbones after polymerization. Similar phenomenon has also been reported by
Yanqian et al.[29]. Another evidence of this speculation is that no peak was found on cooling of stilbazole polymers (PA1-2) as obtained from the DSC data, indicating a poor capability of crystallization in acceptor polymers (PA1-2). However, distinct melting and crystallization peaks were found on heating and cooling of benzoic acid polymers (PD1-2) due to the formation of H-bonded interaction between carboxylic acids[22,30]. For the spacer length (mZ6 and 10) effect on a single component of acid donor monomers (D1 and D2), it favors the nematic phase in D1 (with the shorter spacer mZ6) but favors the SC phase in D2 (with the longer spacer length
mZ10). On the other hand, the isotropization temperatures of analogous components with different spacer lengths inTable 3
are quite similar.
The thermal properties of H-bonded monomer (donor monomer/acceptor monomer) complexes or side-chain poly-mer complexes (donor monopoly-mer/acceptor polypoly-mer or donor polymer/acceptor monomer) are exhibited in Table 4 and
Fig. 4. For example, D1/PA1 and D2/PA2 (donor monomer/
Table 3
Transition temperatures of donor/acceptor monomer and polymer moieties Compound Heating/
cooling
Phase transition temperature (8C)
D1 Heating K 55.8 (16.9) K077.0 (92.2) SC100.7 (0.1) SA102a(K) N 109.2 (3.3) I Cooling I 105.6 (3.1) N 98a(K) SA97.3 (0.6) SC92 a (K) K051.7 (67.1) K PD1 Heating K 147.7 (14.7) I Cooling I 144.8 (12.5) K D2 Heating K 79.0 (149.2) SC97a(K) SA110.0 (26.9) I Cooling I 101.7 (27.0) SA95a(K) SC62.0 (143.9) K PD2 Heating K 147.0 (21.5) I Cooling I 142.3 (18.5) K A1 Heating K 84.4 (108.0) I Cooling I 56.9 (109.3) K PA1 Heating K 62.9 (0.2) I Cooling b A2 Heating K 77.2 (101.9) I Cooling I 48.2 (21.8) K0044.9 (5.1) K037.8 (9.7) K PA2 Heating K 69.8 (9.6) I Cooling I 54.2 (0.8) K a
Observed by POM; the values given in parentheses are delta enthalpy values (J/g).
b
No crystallization peak observed by DSC.
25 75 125 175 D1 PD1 D2 PD2 A1 PA1 A2 PA2 T (˚C) K K' SC SA N
Fig. 3. Transition temperatures (heating) of donor and acceptor moieties (including monomers and polymers).
Table 4
Transition temperatures of donor/acceptor monomer/monomer, monomer/po-lymer, and polymer/monomer supramolecular complexes
Compound Heating/ cooling
Phase transition temperature (8C)
D1/A1 Heating K 95.0 (2.6) SA109.6 (6.4) I Cooling I 104.0 (7.0) SA88.3 (2.5) K PD1/A1 Heating K 55.9 (1.1) K0111.1 (3.9) SA149.7 (8.3) I Cooling I 145.5 (10.6) SA107.9 (3.4) K046.1 (1.0) K D1/PA1 Heating G 78.1 (0.01)aSA127.1 (1.3) I Cooling I 115.1 (0.2) SA75b(K) G D2/A2 Heating K 76.4 (62.3) SF101.7 (7.3) SC105b(K) SA 115.9 (10.9) I Cooling I 111.4 (111.4) SA103b(K) SC97.5 (5.6) SF 62.0 (59.2) K PD2/A2 Heating K 112.2 (4.3) SA139.7 (9.6) I Cooling I 131.4 (9.2) SA108.0 (3.6) K D2/PA2 Heating G 74.8 (0.2)aSA109.4 (5.0) I Cooling I 104.5 (5.4) SA74.5 (0.2) a G a
G is the glassy state and the values given in parentheses are delta specific heat capacity DCp(J/g 8C).
b
Observed by POM; the values given in parentheses are delta enthalpy values (J/g). 25 75 125 175 T ( ˚C) K K' SF SA SC
(D1 / A1) (PD1 / A1) (D1 / PA1) (D2 / A2) (PD2 / A2) (D2 / PA2) Fig. 4. Transition temperatures (heating) of donor/acceptor (monomer/mo-nomer, monomer/polymer, and polymer/monomer) supramolecular complexes.
acceptor polymer) are H-bonded side-chain polymers as shown inFig. 1(a), and PD1/A1 and PD2/A2 (donor polymer/acceptor monomer) are H-bonded side-chain polymers as shown in
Fig. 1(b). These H-bonded monomer complexes and side-chain polymer complexes show better mesogenic behavior than those original benzoic acid and stilbazole components. In the case of H-bonded side-chain polymers D1/PA1 and D2/PA2, the glass transition temperatures, i.e. 78.1 and 74.8 8C on heating, are observed and no crystallization occurs upon cooling, which leads to stabilize and freeze the SAphase (focal-conic texture)
to room temperature. Moreover, the SCand SFphases of D2/A2
are suppressed by the polymerization of D2 or A2 in PD2/A2 and D2/PA2, respectively, which may be attributed to the stabilization of the assembly of the SA phase by the
polymerized acrylate backbones and thus to restrict the tilted SCand SFpacking in the supramolecular polymer form. These
H-bonded side-chain polymer complexes possess higher isotropization temperatures than their analogous H-bonded monomer complexes. Only one exception is that D2/PA2 has a lower isotropization temperature than D2/A2 (Fig. 4), which may be due to the lower packing efficiency and lower isotropization temperature of acceptor stilbazole polymer PA2. For the same explanation, the H-bonded side-chain polymer complexes PD1/A1 and PD2/A2 containing acid donor polymers (PD1 and PD2) possess higher isotropization temperatures than their analogous H-bonded side-chain polymer complexes D1/PA1 and D2/PA2 containing acceptor stilbazole polymers (PA1 and PA2), respectively. However, the lower packing efficiency of acceptor stilbazole polymers (PA1 and PA2) causes better mesomorphism in D1/PA1 and D2/PA2 than that in PD1/A1 and PD2/A2. For the spacer length (mZ6 and 10) effect on monomer complexes and side-chain polymer complexes inTable 4, it favors the Sc phase in D2/A2 (with the longer spacer length mZ10) in comparison with D1/A1 (with the shorter spacer length mZ6), because the Sc phase is favored for longer flexible chains. As a result of the higher packing efficiency of the SAphase for medium flexible
chains (mZ6), the isotropization temperatures of PD1/A1 and D1/PA1 (with the shorter spacer length mZ6), i.e. 149.7 8C (147.7/84.4 8C) and 127.1 8C (109.2/62.9 8C), are higher than those of PD2/A2 and D2/PA2 (with the longer spacer length mZ10), i.e. 139.7 8C (147.0/77.2 8C) and 109.4 8C (110/ 69.8 8C), respectively. In addition, PD1/A1 and D1/PA1 have wider ranges of the SA phase than PD2/A2 and D2/
PA2, since the SA phase is more favored in medium flexible
chains (mZ6) in comparison with long flexible chains (mZ 10). Nevertheless, by reason of easier aggregation of unreacted acrylates for longer flexible chains (mZ10) in monomer complexes, D2/A2 (with the longer spacer length mZ10) has a higher isotropization temperature than D1/A1 (with the shorter spacer length mZ6), i.e. 115.9 8C (110/77.2 8C) vs. 109.6 8C (109.2/84.4 8C), in analogous H-bonded complexes with different spacer lengths.
The physical blends of H-bonded polymer networks, i.e. blend(PD1/PA1)s, are obtained from blends in various molar ratios of acceptor and donor polymers with the spacer length mZ6 and their phase transition temperatures are shown in
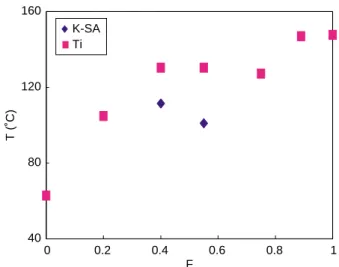
Table 5 and Fig. 5. The schematic structure of H-bonded blending polymer networks of donor and acceptor homo-polymers, i.e. blend(PD1/PA1)s and blend(PD2/PA2)s, is illustrated inFig. 1(c). The value F is the molar ratio of benzoic acid (donor) polymers in the H-bonded physical blends. From these results, it obviously reveals that the incorporation of benzoic acid polymers to the stilbazole polymers does increase the isotropization temperatures of the H-bonded polymers in comparison with those of stilbazole polymers, which are common phenomena in the H-bonded blends. In consideration of stilbazole polymers as semi- semi-crystalline components, the isotropization temperatures of the H-bonded blending polymers[31–33]are in general decreased by adding stilbazole polymers into the H-bonded blends. Liquid crystalline behavior is merely observed in the physical blends of H-bonded polymer networks containing benzoic acid (donor) polymers in the molar fractions around 0.4 and 0.55.
40 80 120 160 0 0.2 0.4 0.6 0.8 1 F T ( ˚C) K-SA Ti
Fig. 5. Transition temperatures (heating) of H-bonded polymer networks with the spacer length mZ6, i.e. blend(PD1/PA1)s, obtained from physical blends of donor and acceptor polymers with various molar ratios of benzoic acid polymers (F).
Table 5
Transition temperatures of H-bonded polymer networks with the spacer length mZ6, i.e. blend(PD1/PA1)s, obtained from physical blends of acceptor and donor polymers with various molar ratios of benzoic acid polymers (F) Composition
FZ(PD1)/(PD1CPA1)
Heating/ cooling
Phase transition temperature (8C)
FZ0 Heating K 62.9 (0.2) I Cooling a FZ0.2 Heating K 104.5 (0.3) I Cooling I 100.7 (0.4) K FZ0.4 Heating K 111.5 (0.5) SA130.4 (1.2) I Cooling I 130.9 (1.6) SA108.5 (0.9) K FZ0.55 Heating K 101.0 (0.5) SA130.4 (1.3) I Cooling I 120.1 (0.3) SA94.6 (0.2) K FZ0.75 Heating K 127.3 (0.7) I Cooling I 117.2 (2.2) K FZ0.89 Heating K 147 (7.1) I Cooling I 139 (8.4) K FZ1 Heating K 147.7 (14.7) I Cooling I 144.8 (12.5) K a No crystallization peak observed by DSC.
The phase transition temperatures of H-bonded copolymers with the spacer length mZ6, i.e. poly(D1-co-A1)s, containing donor and acceptor moieties with various molar ratios of benzoic acids (F) are shown in Table 6 and Fig. 6. The schematic structure of the H-bonded coplymer networks, i.e. poly(D1-co-A1)s and poly(D2-co-A2)s, is demonstrated in
Fig. 1(d). The H-bonded crosslinking density is defined by the ratio of (# of H-bonds between partially H-bonded donors and acceptors) to (# of H-bonds between fully H-bonded donors and acceptors). In terms of the molar ratio of benzoic acids (F), the H-bonded crosslinking density can be simplified to F!2! 100% for F!0.5 and to (1KF)!2!100% for FO0.5. For instance, the H-bonded crosslinking density is 100% in a 1:1 donor/acceptor molar ratio and is 50% in a 3:1 (or 1:3) donor/ acceptor molar ratio. ComparingFigs. 5 and 6, as expected it is proven that H-bonded copolymers containing donor and
acceptor moieties shows enhanced miscibility and better homogeneity comparing to the H-bonded physical blends of analogous homopolymers. As an example, at the molar fraction of benzoic acid (F)Z0.4 (i.e. 80% crosslinking), the SAphase
of H-bonded copolymer is stable at 105.8–133 8C, while it is stable around 111.5–130.4 8C for H-bonded physical blends, which indicates more homogenous phases occur for chemically mixing of donor and acceptor moieties in the H-bonded copolymers. Moreover, the mesogenic properties can be achieved by broader ranges of composition F around 0.42– 0.89 for H-bonded copolymers (in Fig. 6) than those of composition F around 0.4–0.55 for H-bonded physical blends (inFig. 5).
As described above, H-bonded blends and copolymers of stilbazoles and benzoic acids with the spacer length mZ10, i.e. blend(PD2/PA2)s and poly(D2-co-A2)s, show alike tendency as those with the spacer length mZ6, i.e. blend(PD1/PA1)s and poly(D1-co-A1)s. Similar results have been observed in
Tables 7 and 8andFigs. 7 and 8, and more homogenous phases
Table 6
Transition temperatures of copolymers with the spacer length mZ6, i.e. poly(D1-co-A1)s, containing donor and acceptor moieties with various molar ratios of benzoic acids (F)
Composition
FZ(PD1)/(PD1CPA1)
Heating/ cooling
Phase transition temperature (8C)
FZ0 Heating K 62.9 (0.2) I Cooling a FZ0.2 Heating K 98.5 (0.4) I Cooling I 95.1 (0.1) K FZ0.42 Heating K 105.8 (0.6) SA133b(K) I Cooling I 126.4 (0.3) SA103.2 (0.5) K FZ0.55 Heating K 103.7 (0.8) SA132.8 (0.9) I Cooling I 128.9 (0.5) SA101.8 (0.4) K FZ0.75 Heating K 117.4 (0.3) SA134.1 (0.7) I Cooling I 136.7 (1.6) SA115.2 (0.4) K FZ0.89 Heating K 127.1 (0.5) SA143.2 (2.2) I Cooling I 137.2 (1.7) SA124.3 (0.6) K FZ1 Heating K 147.7 (14.7) I Cooling I 144.8 (12.5) K a No crystallization peak observed by DSC.
b Observed by POM; the values given in parentheses are delta enthalpy values (J/g). 40 80 120 160 0 0.2 0.4 0.6 0.8 1 F T ( ˚C) K-SA Ti
Fig. 6. Transition temperatures (heating) of copolymers with the spacer length mZ6, i.e. poly(D1-co-A1)s, containing donor and acceptor moieties with various molar ratios of benzoic acids (F).
Table 7
Transition temperatures of H-bonded polymer networks with the spacer length mZ10, i.e. blend(PD2/PA2)s, obtained from physical blends of acceptor and donor polymers with various molar ratios of benzoic acid polymers (F) Composition
FZ(PD2)/(PD2CPA2)
Heating/ cooling
Phase transition temperature (8C)
FZ0 Heating K 69.8 (9.6) I Cooling I 54.2 (0.8) K FZ0.33 Heating K 105.8 (1.4) I Cooling I 101.8 (1.8) K FZ0.5 Heating K 129.3 (0.9) SA137a(K) I Cooling I 134.3 (0.3) SA123.5 (0.4) K FZ0.67 Heating K 124.4 (0.1) SA131.6 (1.4) I Cooling I 136.1 (0.6) SA117.8 (0.6) K FZ0.83 Heating K 139.7 (6.7) I Cooling I 134.0 (6.8) K FZ1 Heating K 147.0 (21.5) I Cooling I 142.3 (18.5) K
a Observed by POM; the values given in parentheses are delta enthalpy values (J/g).
Table 8
Transition temperatures of copolymers with the spacer length mZ10, i.e. poly(D2-co-A2)s, containing donor and acceptor moieties with various molar ratios of benzoic acids (F)
Composition
FZ(PD2)/(PD2CPA2)
Heating/ cooling
Phase transition temperature (8C)
FZ0 Heating K 69.8 (9.6) I Cooling I 54.2 (0.8) K FZ0.2 Heating K 103.4 (1.2) I Cooling I 98.6 (1.7) K FZ0.5 Heating K 115.6 (1.1) SA129a(K) I Cooling I 122.7 (0.1) SA112.4 (1.1) K FZ0.63 Heating K 117.9 (3.2) SA132.3 (0.6) I Cooling I 129.3 (0.6) SA114.3 (1.3) K FZ0.86 Heating K 120.3 (0.6) SA133.3 (1.5) I Cooling I 131.7 (1.4) SA121.6 (0.7) K FZ1 Heating K 147.0 (21.5) I Cooling I 142.3 (18.5) K a
Observed by POM; the values given in parentheses are delta enthalpy values (J/g).
also occur for chemically mixing of donor and acceptor moieties in H-bonded copolymers with the spacer length mZ 10, i.e. poly(D2-co-A2)s, than those of H-bonded physical blends of two homopolymers, i.e. blend(PD2/PA2)s. AllFigs. 5–8 giving the best mesogenic behavior are around F (acid composition)Z0.5, i.e. 100% crosslinking, which possess a 1:1 molar ratio of acid groups and pyridyl groups. Thus, the highest H-bonded crosslinking density employed by a 1:1 molar ratio of acid donor and stilbazole acceptor can optimally stabilize the liquid crystalline phase. Besides, the higher isotropization temperatures in the higher F values of all H-bonded polymer networks (inFigs. 5–8) is because the contribution of the acid moieties of polymers or copolymers crosslinking themselves to form acid dimmers. Regarding the spacer length effect on the H-bonded polymer networks, Ti does not change significantly as the spacer chain length increases. However, for the same compositions of F, it reveals broader mesogenic phases in
H-bonded physical blends (Fig. 5) and H-bonded copolymers (Fig. 6) with a short spacer length mZ6 than those in H-bonded physical blends (Fig. 7) and H-bonded copolymers (Fig. 8) with a long spacer length mZ10, which may be owing to the SAphase is less favorable in LC polymers with longer spacers
(mZ10). Furthermore, despite of the lower density of H-bonded crosslinks between acids and stilbazoles in poly(D2-co-A2)s of FZ0.86 (i.e. 28% crosslinking plus extra acids) and poly(D1-co-A1) of FZ0.89 (i.e. 22% crosslinking plus extra acids), it is quite interesting to observe that they are still able to form liquid crystalline phases in the low crosslinking density of H-bonded copolymer networks (only in the higher molar ratio of acids, i.e. large F values, but not in the small F regions due to no H-bonded crosslinks between stilbazoles themselves). This consequence might be owing to the stabilization of H-bonded crosslinks between acids in the high F regions of H-bonded copolymers. However, this phenomenon does not occur in the same crosslinking density (large F values with low crosslinking density) of H-bonded physical blending homopolymer networks. The existence and well-dispersity of hydrogen bonds between identical com-ponents (i.e. benzoic acids) help the mesogenic phase to sustain in the H-bonded copolymer networks with the increase of the molar ratios of benzoic acids (F), while this is less easy to occur in the H-bonded blending homopolymer system, which indicates the aggregation of H-bonded crosslinks between benzoic acids in the blending homopolymers is comparably not well dispersed to maintain the mesogenic phase in large F regions.
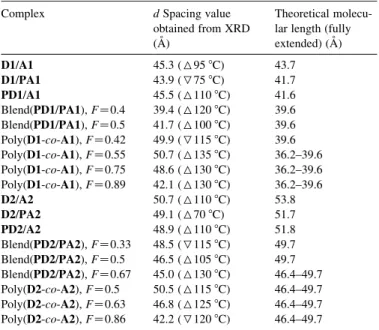
Table 9 shows the d spacing values of all supramolecular monomer complexes, side-chain polymer complexes, polymer blending networks, and copolymer networks that exhibit liquid crystalline structures, where the largest d spacing values are obtained by XRD at the SA phase. In contrast to the
experimental results, the theoretical values of the fully extended molecular lengths for H-bonded complexes and their components are also calculated and listed inTable 9. For D2/A2, PD2/A2, and D2/PA2, their d spacing values are a little shorter than the calculated values from molecular modeling, which might be due to lacking of all-trans conformation in some situations. Generally, the d spacing values of analogous H-bonded systems are in the following order: copolymer networkswmonomer complexesOside-chain polymer com-plexesOpolymer blending networks, e.g. poly(PD2/PA2), FZ 0.5OD2/A2OD2/PA2 (wPD2/A2)Oblend(PD2/PA2), FZ 0.5 (Table 9). It is reasonable to obtain the d spacing values of analogous H-bonded systems in the sequence of monomer complexesOside-chain polymer complexesOpolymer blend-ing networks, which is owblend-ing that the polymerized skeleton of acrylate backbones may confine the flexible part and reduce its possible zig–zag conformation length. Thus, the d spacing values of side-chain polymer complexes decrease as the polymer backbones are introduced into the H-bonded systems, and the d spacing values are further reduced as two homopolymer backbones are involved in the polymer blending networks. According to our recent research, similar results have also been confirmed that the homopolymerization of
40 80 120 160 0 0.2 0.4 0.6 0.8 1 F T ( ˚C) K-SA Ti
Fig. 7. Transition temperatures (heating) of H-bonded polymeric networks with the spacer length mZ10, i.e. blend(PD2/PA2)s, obtained from physical blends of donor and acceptor polymers with various molar ratios of benzoic acid polymers (F). 40 80 120 160 0 0.2 0.4 0.6 0.8 1 F T ( ˚C) K-SA Ti
Fig. 8. Transition temperatures (heating) of copolymers with the spacer length mZ10, i.e. poly(D2-co-A2)s, containing donor and acceptor moieties with various molar ratios of benzoic acids (F).
acrylates does reduce the d spacing values in analogous H-bonded systems, i.e. monomer complexesOside-chain polymer complexesOpolymer blending networks. For most H-bonded physical blending polymer networks and copolymer networks, it shows a maximum in the d spacing value around FZ0.5 and a decrease in d spacing values along with the increase of the molar ratios of benzoic acids (F), indicating the existence of hydrogen bonds between identical components (i.e. benzoic acids) that leads to shorter d spacing values than those between stilbazoles and benzoic acids. However, in poly(D1-co-A1)s, though they have the above trend as mentioned, their d spacing values are much longer than the calculated ones. This discrepancy of the d spacing value from the calculated one (shown inTable 9) is decreased as the molar ratios of benzoic acids (F) is increased, which might be also due to the formation of hydrogen bonds between benzoic acids themselves. In poly(D1-co-A1) of FZ0.75 (i.e. 50% cross-linking plus extra acids), the d spacing shows a value of 44.5 A˚ , indicating that the layer spacing of liquid crystalline domain decrease in length because of the predominance of H-bonded crosslinkings between benzoic acids (from different back-bones). Among them, the d spacing value of the H-bonded copolymer network (FZ0.55, i.e. 90% crosslinking) in poly(D1-co-A1), i.e. 50.7 A˚ measured at 135 8C during heating, has the largest difference from the calculated d spacing value (36.2–39.6 A˚ ). This d spacing value, i.e. 50.7 A˚, is also larger than that of its analogous H-bonded monomer complex D1/A1 (45.3 A˚ as shown inTable 9), which might be reasoned by that the spacer length mZ6 is too short for extension (or interpenetration) of donors and acceptors inside
the copolymers. Therefore, one of the possible layer packing in poly(D1-co-A1) as FZ0.55 is suggested inFig. 9(a), where the interdigitated bilayer structure can induce a larger d spacing value (approximately one benzoic acid, ca. 5 A˚ , longer than that of H-bonded monomer complex D1/A1 if stilbazole units are overlapped). Whereas the d spacing value of H-bonded copolymer network (FZ0.5, i.e. 100% crosslinking) in poly(D2-co-A2), i.e. 50.5 A˚ measured at 115 8C during heating, is similar to the calculated d spacing value (49.7 A˚ ). This d spacing value, i.e. 50.5 A˚ , is also similar to that of its analogous H-bonded monomer complex D2/A2 (50.7 A˚ as shown inTable 9), which might be elucidated by that the length of spacer mZ10 is longer enough for extension (or interpenetration) of donors and acceptors among copolymers. Therefore, another possible layer packing in poly(D2-co-A2) as FZ0.5 is expressed in Fig. 9(b), where the monolayer structure can reveal a similar d spacing value as that of H-bonded monomer complex D2/A2. Due to the predominant crosslinkings between benzoic acids themselves in both copolymer systems, the decrease in layer spacing values of poly(D1-co-A1)s and poly(D2-co-A2)s can be observed as the F values increase. From poly(D2-co-A2)s, the decreasing amount of the d spacing value from FZ0.5 (100% crosslinking) to FZ0.86 (28% crosslinking plus extra acids)
Table 9
Largest d spacing values of the smectic A phase in H-bonded complexes (monomer/monomer, monomer/polymer, polymer/monomer, polymer/poly-mer, and copolymer)
Complex d Spacing value obtained from XRD (A˚ )
Theoretical molecu-lar length (fully extended) (A˚ ) D1/A1 45.3 (695 8C) 43.7 D1/PA1 43.9 (775 8C) 41.7 PD1/A1 45.5 (6110 8C) 41.6 Blend(PD1/PA1), FZ0.4 39.4 (6120 8C) 39.6 Blend(PD1/PA1), FZ0.5 41.7 (6100 8C) 39.6 Poly(D1-co-A1), FZ0.42 49.9 (7115 8C) 39.6 Poly(D1-co-A1), FZ0.55 50.7 (6135 8C) 36.2–39.6 Poly(D1-co-A1), FZ0.75 48.6 (6130 8C) 36.2–39.6 Poly(D1-co-A1), FZ0.89 42.1 (6130 8C) 36.2–39.6 D2/A2 50.7 (6110 8C) 53.8 D2/PA2 49.1 (670 8C) 51.7 PD2/A2 48.9 (6110 8C) 51.8 Blend(PD2/PA2), FZ0.33 48.5 (7115 8C) 49.7 Blend(PD2/PA2), FZ0.5 46.5 (6105 8C) 49.7 Blend(PD2/PA2), FZ0.67 45.0 (6130 8C) 46.4–49.7 Poly(D2-co-A2), FZ0.5 50.5 (6115 8C) 46.4–49.7 Poly(D2-co-A2), FZ0.63 46.8 (6125 8C) 46.4–49.7 Poly(D2-co-A2), FZ0.86 42.2 (7120 8C) 46.4–49.7
Note: theoretical value of fully extended molecular length: D1Z20.2 A˚ , D2Z 25.2 A˚ , A1Z23.5 A˚, A2Z28.6 A˚, PD1Z18.1 A˚, PD2Z23.2 A˚, PA1Z21. 5 A˚ , PA2Z26.5 A˚; (6) data obtained on heating; (7) data obtained on cooling.
Fig. 9. Schematic structures of possible (idealized) H-bonded copolymer liquid crystalline networks: (a) interdigitated bilayer smectic A phase built by copolymers with the spacer length mZ6; (b) monolayer smectic A phase built by copolymers with the spacer length mZ10.
is about 8 A˚ , and this value is acceptable if we adopt the same concept from those of poly(D1-co-A1)s. Comparing the d spacing values of the copolymers with mZ6 and mZ10, it is found that the shorter spacer length tends to improve the liquid crystalline organization, while the spacer length of copolymers with mZ6 is likely to enable an interdigitated bilayer packing to form. Though the H-bonded interdigitated bilayer structure has more stable mesogenic phases than the H-bonded monolayer structure in poly(D2-co-A2)s as FZ0.5 with a longer spacer length mZ10, the origin of this occurrence is uncertain. Comparing the results of phase transition tempera-tures among all H-bonded polymer networks (inFigs. 5–8), i.e. blend(PD1/pA1)s, blend(PD2/PA2)s, poly(D1-co-A1)s, and poly(D2-co-A2)s, it is clear that poly(D1-co-A1)s have the broadest composition of F, i.e. the broadest crosslinking density, to possess the mesophase and have the broadest ranges of the mesophase at most of the same F values, i.e. crosslinking density, in all these H-bonded networks. Overall, the highest crosslinking density of stilbazole and benzoic acid, i.e. 100% crosslinking, in most H-bonded physical blending polymer and (chemically blending) copolymer networks possesses the longest layer spacing values and the widest ranges of mesophases.
4. Conclusions
† Various kinds of H-bonded side-chain and crosslinking LC polymers are developed and investigated thoroughly, which have seldom been discussed in covalently crosslinking LC polymers. The LC polymer networks are easily to form by H-bonds, and the supramolecular crosslinks in liquid crystals have rarely been surveyed.
† With various combinations of H-bonded side-chain and crosslinking LC polymers, the structural effects of the H-bonded copolymers and physical blending homopoly-mers on mesomorphism are easy to be tuned in order to adjust their supramolecular properties.
† The isotropization temperatures of the H-bonded blends and copolymers increase as the molar ratios of benzoic acids increase, while the higher H-bonded crosslinking density stabilizes the liquid crystalline phase.
† It is evidenced that more homogenous phases occur in the H-bonded networks for chemically mixing of donor and acceptor moieties in H-bonded copolymers than those for H-bonded physical blending homopolymers.
Acknowledgements
Department of Materials Science and Engineering at National Chiao Tung University, Institute of Chemistry at Academia Sinica, and the National Science Council of Taiwan
(ROC) through NSC 89-2113-M-001-045 and NSC 92-2113-M-009-016 are acknowledged for the financial support of this project.
References
[1] Lehn JM. Angew Chem Int Ed Engl 1988;27:89. [2] Lehn JM. Angew Chem Int Ed Engl 1990;29:1304. [3] Stupp SI, Son S, Lin HC, Li LS. Science 1993;259:59.
[4] Lin HC, Lin YS, Lin YS, Chen YT, Chao I, Li TW. Macromolecules 1998;31:7298.
[5] Ikkala O, Ruokolainen J, ten Brinke G, Torkkeli M, Serimaa R. Macromolecules 1995;28:7088.
[6] Ruokolainen J, Tanner J, ten Brinke G, Ikkala O, Torkkeli M, Serimaa R. Macromolecules 1995;28:7779.
[7] Kotera M, Lehn JM, Vigneron JPJ. Chem Soc Chem Commun 1994;197. [8] Kumar U, Kato T, Freche´t JMJ. J Am Chem Soc 1992;114:6630. Sato A, Kato T, Uryu TJ. Polym Sci, Polym Chem 1996;34:503. Kato T, Ihata O, Ujie S, Tokita M, Watanabe J. Macromolecules 1998;31:3551. [9] Vuillaume PY, Bazuin CG, Galon JC. Macromolecules 1995;28:8877.
Stewart D, Imrie CT. Macromolecules 1997;30:877. Kaneko T, Yamaoka K, Gong JP, Osada Y. Macromolecules 2000;33:412. [10] Kawakami T, Kato T. Macromolecules 1998;31:4475.
[11] Lin HC, Lin YS. Liq Cryst 1998;24:315. Lin HC, Shiaw JM, Liu RC, Tsai C, Tso HH. Liq Cryst 1998;25:277. Lin HC, Ko CW, Guo K, Cheng TW. Liq Cryst 1999;26:613. Lin HC, Shiaw JM, Wu CY, Tsai C. Liq Cryst 2000;27:1103.
[12] Lin HC, Sheu HY, Chang CL, Tsai C. J Mater Chem 2001;11:2958. [13] Stewart D, Imrie CT. Liq Cryst 1996;20:619.
[14] Barmatov EB, Bobrovsky AY, Barmatova MV. Liq Cryst 1999;26:581. [15] Ilhan F, Gray M, Rotello VM. Macromolecules 2001;34:2597. [16] Shandryuk GA, Kuptsov SA, Shatalova AM, Plate’ NA, Talroze RV.
Macromolecules 2003;36:3417.
[17] Medvedev AV, Barmatov EB, Medvedev AS, Shibaev VP, Ivanov SA, Kozlovsky M, et al. Macromolecules 2005;38:2223.
[18] Aoki K, Nakagawa M, Ichimura KJ. Am Chem Soc 2000;122:10997. [19] Wiegel KN, Griffin AC, Black MS, Schiraldi DA. J Appl Polym Sci 2004;
92:3097.
[20] Feldstein MM, Shandryuk GA, Plate´ NA. Polymer 2001;42:971. Feldstein MM, Kuptsov SA, Shandryuk GA, Plate´ NA. Polymer 2001; 42:981. Feldstein MM, Roos A, Chevallier C, Creton C, Dormidontova EE. Polymer 2003;44:1819.
[21] Yount WC, Loveless DM, Craig SL. Angew Chem Int Ed Engl 2005;44: 2746.
[22] Yamaoka K, Kaneko T, Gong JP, Osada Y. Macromolecules 2001;34: 1470.
[23] Kato T, Kihara H, Kumar U, Uryu T, Freche´t JMJ. Angew Chem Int Ed Engl 1994;33:1644.
[24] Kihara H, Kato T, Uryu T, Freche´t JMJ. Chem Mater 1996;8:961. [25] Kihara H, Kato T, Uryu T, Freche´t JMJ. Liq Cryst 1998;24:413. [26] Kurihara S, Mori T, Nonaka T. Macromolecules 1998;31:5940. [27] Kelly SM. J Mater Chem 1995;5:2047. Kelly SM. Liq Cryst 1998;24:71. [28] Portugall M, Ringsdorf H, Zentel R. Makromol Chem 1982;183:2311. [29] Yanqian T, Akiyama E, Nagase Y, Kanazawa A, Tsutsumi O, Ikeda T.
Macromol Chem Phys 2000;201:1640.
[30] Review articles of H-bonded LC complexes Kato T. Struct Bond 2000;96: 95. Paleos CM, Tsiourvas D. Liq Cryst 2001;28:1127.
[31] Akashi R, Inoue A. Mol Cryst Liq Cryst 1994;250:269. [32] Poths H, Zentel R. Liq Cryst 1994;16:749.