Twist1 induces endothelial differentiation of tumor cells
through the Jagged1-KLF4 axis
Hsiao-Fan Chen1, Chi-Hung Huang3, Chung-Ji Liu4, Jung-Jyh Hung5, Chih-Chin Hsu6, Shu-Chun Teng7, and Kou-Juey Wu1,2,8,
1Institute of Biochemistry & Molecular Biology, and 2Genome Research Center, National Yang-Ming University, Taipei 112; 3Taiwan Advance Biopharm (TABP), Inc., Xizhi city, New Taipei City 221; and 4Department of Dentistry, Taipei Mackay Memorial Hospital, Taipei 104; 5Dept. of Surgery, Taipei Veterans General Hospital, Taipei 112; 6Dept. of Physical Medicine and Rehabilitation Medicine, Keelung Chang-Gung Memorial Hospital, Keelung 204; 7Graduate Institute of Microbiology, College of Medicine, National Taiwan University, Taipei 100; 8ResearchCenter for Tumor Medical Science, China Medical University, Taichung 40402, Taiwan
Correspondence should be addressed to:
Kou-Juey Wu, Institute of Biochemistry and Molecular Biology, National Yang-Ming University, No.155, Li-Nong St., Sec.2, Peitou, Taipei 112, Taiwan; Email: [email protected]; Tel: 886-228267328; Fax: 886-228264843
Abstract
The mechanisms controlling tumor-induced angiogenesis are presently not
clear. In principle, angiogenesis can be achieved through the activation of endothelial cells in existing vessels or by transdifferentiation of tumor cells into endothelial cells. However, whether tumor cells endothelial differentiation can go proceed through aan prior epithelial-mesenchymal transition (EMT) and further differentiate into endothelial cells remains unknown. Here we show that overexpression of Twist1, a transcriptional regulator that induces and promotes cancer metastasis, in head and neck cancer cells leads to endothelial differentiation in head and neck cancer (HNC) cells. Induction of Jagged1 expression by Twist1 is essential for Twist1-induced endothelial differentiation. The Jagged1/Notch signaling subsequently activates KLF4, inducing stem-like properties in the HNC cells and conferring them with drug resistance. Our results indicate that the Twist1-Jagged1/KLF4 axis is essential both for transdifferentiation of tumor cells into endothelial cells and also for conferring
chemoresistance acquisition.
Introduction
Tumor angiogenesis is one of the hallmark capabilities that enable tumor growth and metastasis1. The endothelial cells that constitute the tumor vasculature could can be generated from the existing endothelial cells through the “angiogenic switch” mediated by different signaling molecules2,3. Alternatively, Eendothelial stem-like cells with cancer-specific genomic alternations alterations have been described in different tumor types4,5. Recently, transdifferentiation of glioblastoma stem-like cells into endothelial cells was have been demonstrated6-8. However, it is unknown whether tumor cells can go through a prior tumor-derived endothelial differentiation can proceed in cells that have undergone an epithelial-mesenchymal transition (EMT) before further differentiation into endothelial cells.
Twist1, a master transcriptional regulator of mesoderm development, induces EMT and promotes cancer metastasis9-12. Twist1 overexpression is observed in many different types of human cancers12. The Notch signaling not only regulates vascular morphogenesis and but also communicates with existing endothelial cells to promote tumor angiogenesis13-15. KLF4 is a core component of the pluripotency transcription network that has been used to reprogram somatic cells into induced pluripotent stem (iPS) cells16. KLF4 regulates endothelial barrier and is implicated in
vasculogenesis17,18. Recent evidence showed has shown that pluripotency factors regulate endoderm specification19. The precise role of KLF4 to in the induce induction of endothelial differentiation remains to be determined.
In this report, we explored the ability of Twist1 to further induce endothelial differentiation of tumor cells after these cells went haved undergone through EMT. The signaling pathways of tumor-derived endothelial differentiation induced by Twist1 weare also investigated, which weare involved in the induction of. A signaling axis was shown to induce stem-like property and confer drug resistance. These results provide a fresh insight into the mechanisms of endothelial differentiation and chemoresistance mediated by Twist1 and have endow Twist1-overexpressing tumors with treatment a promising implications of treatment. for Twist1-overexpressing tumors.
Results
Twist1 induces endothelial differentiation of tumor cellsInduction of tumor-derived endothelial differentiation by Twist1
To test the ability of Twist1 to induce endothelial differentiation of tumor cells that go through prior to EMT, we overexpressed Twist1 in the a head and neck cancerHNC cell line, OECM-1, and examined the expressions of endothelial markers, including CD31 and CD1446-8. Western blot analysisThe results showed that CD31 and CD144 were activated in overexpressing OECM-1 cells and about 10% of Twist1-overexpressing cells expressed CD31 and CD144 as determined by using Western blot and flow cytometry analysis (Fig. 1a and Supplementary Fig. S1a). The increased expression of CD31 and CD144 are independently validated in another HNC cell line, FADU with Twist1 overexpression Another head and neck cancer cell line, FADU, also showed increased expression of CD31 and CD144 when Twist1 was overexpressed (Supplementary Fig. S1b,c). About 10% of Twist1-overexpressing cells expressed CD31 and CD144 using flow cytometry analysis. About 10% of Twist1-overexpressing OECM-1 cells co-expressed CD144Staining of Twist1 and CD144 showed that ~10% of Twist1-overexpressing OCEM-1 cells co-expressed both markers (Fig. 1b). and ~9% co-expressed vimentin and CD144Staining of vimentin and CD144 also showed that ~9% of Twist1-overexpressing OECM-1 cells
co-expressed both markers (Supplementary Fig. S2a). Further sorting by flow cytometry revealed that among Twist1-overexpressing OECM-1 cells, those (9-10%) co-expressed with CD144 or CD31 had the highest levels of Twist1 Flow cytometry sorting of the 9-10% of Twist1-overexpressing OECM-1 cells that also stained with CD144 or CD31 showed the highly expressed Twist1 levels compared to the rest of the Twist1 overexpressing OECM-1 cells (Supplementary Fig. S2b,c3). However, Iinduction of CD31 and CD144 by Twist1 overexpression was not mediated through chemokine receptor 4 (CXCR4) since CXCR4 was not activated by Twist1
(Supplementary Fig. S2ddata not shown). Twist1-overexpressing OECM-1 cells exhibited increased tube-forming ability, hemoglobin levels in xenografted tumors, and DiI-AcLDL (1,1’-dioctadecyl-3,3,3’,3’-tetramethyl-indocarbocyanide perchlorate-labeled acetylated low density lipoproteins) uptake8 (Fig. 1c,d, Supplementary Fig. S2e2b and data not shown). Other endothelial markers, including von Willebrand factor (vWF), TEK tyrosine kinase (TIEie2), CD105, and intercellular adhesion molecule 1 (ICAM1),6
,7,20 were also activated by Twist1 overexpression and Western blot analysis confirmed the activation of CD105 and ICAM1 in Twist1-overexpressing OECM-1 cells (Fig. 2a-cSupplementary Fig. S4a,b). Examination of different vascular markers by real-time PCR analysis showed the increased expression of ephrin B2 (EFNNFB2) and neuropilin -1 (NRP1) and decreased expression of EPH
receptor B4 (EPHB4) in Twist1-overexpressing OECM-1 cells (Fig. 2dSupplementary Fig. S4c). The Twist1-overexpressing and the control OECM-1 cells were labeled with green fluorescence protein (GFP) followed by subcutaneous injection into NOD-SCID nude mice to monitor tumor vessel formation in xenografted tumors. The results showed that Twist1-overexpressing OECM-1 cells contributed to the formation of tumor vessels as determinedjudged by the co-staining of CD144 and GFP (Fig. 1e). The GFP and CD144 co-expressing tumor vessels were located mostly inside the tumor mass. The hemotoxylin & eosin (H&E) staining of consecutive slides also showed the vascular structure (Supplementary Fig. S53a). Finally, immunofluorescence staining of Twist1 and CD31 showed that 91.6% (22/24) of Twist1-overexpressing head and neck cancer patient samples had tumor-derived vessel formation as determined judged by the co-expression of CD31 and Twist1 in the same cells (Fig. 1f).Immunofluorescence staining of CD144 and Twist1 also showed the co-staining of both markers in patient samples (Supplementary Fig. S53c). The H&E staining of consecutive slides showed the vascular structure (Supplementary Fig. S53b,c).Consistent with the above results, knockdown of Twist1 in H1299 (non-small cell lung cancer) and SAS (head and neck cancer) cells decreased the tube-forming ability and CD31 levels, and inhibited DiI-AcLDL uptake (Supplementary Fig. S64, S75a,b). Moreover, hemoglobin (Hb) levels were decreased
in xenografted tumors when Twist1 was knocked down in H1299 cells Knockdown of Twist1 in H1299 cells decreased the hemoglobin (Hb) levels in xenografted tumors (Supplementary Fig. S75c)., suggesting that endothelial differentiation was reduced in
vivo. In supporting of this finding, Knockdown of Twist1 in these two cell lines also
repressed the expression of expression of the endothelial markers (CD31, vWF, TIEie2, and CD105) was repressed in Twist1-knockdown H1299 and SAS cells (left panels of Supplementary Fig. S86a,b),. Noticeably, and the expression pattern of vascular markers (EFNNFB2, NRP1, EPHB4) that were regulated by Twist1 was also consistent with the Twist1 overexpression status (right panels of Supplementary Fig. S86a,b). Finally, due to the causative role of Human Papilloma virus (HPV) in head and neck cancerHNC formation21, we tested whether there is was correlation between HPV infection and Twist1 overexpression. The result showed that there was no significant correlation between the presence of HPV and Twist1 overexpression in patient samples (Supplementary Fig. S95d and Supplementary Table S1). These results indicate that Twist1 overexpression inducesd endothelial differentiation of tumor cells that favorsed arterial specification22,23 and promotesd tumor vascular formation.
differentiation
To explore the mechanism that mediates induction of endothelial differentiation by Twist1, a microarray approach was performed to search for putative Twist1 downstream targets. Among the putative Twist1 targets, we chose a Notch ligand, Jagged1, since Jagged1 is a Notch ligand and Notch signaling regulates tumor angiogenesis13,14. Further confirmation using real-time PCR and Western blot analysis showed that Twist1 overexpression induced the expressions of Jagged1 and its downstream targets, including Hes1, Hey1, and Hey2 (Supplementary Fig. S107a,b)13. Knockdown of Twist1 in H1299 cells reduced the expression of Jagged1, Hes1, Hey1, and Hey2 (Supplementary Fig. S107c,d), showing suggesting that the same signaling pathway existed in a different cell type. To test the a direct activation of Jagged1 by Twist1, we identified a Twist1-binding site in the Jagged1 promoter using transient transfection assays (Supplementary Fig. S113a,b). Chromatin immunoprecipitation (ChIP) experiments showed the direct binding of Twist1 to the Jagged1 promoter in two different cell lines (Supplementary Fig. S113c). To test the role of Jagged1 in induced endothelial differentiation, we knocked down of Jagged1 in Twist1-overexpressing OECM-1 cells decreased and this led to a decrease in CD31 and CD144 expressions and , DiI-AcLDL uptake (Fig. 24a and Supplementary Figs. S12, 138). Knockdown of Jagged1 also decreased the hemoglobin (Hb) levels in
xenografted tumors, the tube-formation ability, and the expression of CD31, CD144, vWF, CD105, and ICAM1 induced by Twist1 overexpression (Fig. 24b,c and Supplementary Fig. S149). In addition, we examined examination of various vascular markers in these cells and found that Twist1-overexpressing OECM-1 cells with Jagged1 knockdown reduced the mRNA levels of showed that the markers including
EFNB2, NRP1, and EPHB4 were regulated by Jagged1 (Fig. 24d), indicating the
essential role of Jagged1 in Twist1-mediated regulation of vascular markers. Knockdown of Jagged1 also decreased the percentage of GFP and CD144 co-staining cells or CD31 and Twist1 co-expressing cells (Fig. 24e,f). The H&E staining of consecutive sections were shown (Supplementary Fig. S1510).Finally, to test whether Jagged1 is involved in Twist1-induced metastasis, in vitro migration/invasion assays were performed. The role of Jagged1 in Twist1-induced EMT and in vitro metastatic activity was also tested. The results showed that Jagged1 knockdown in Twist1-overexpressing OECM-1 or H1299 cells reversed EMT and decreased the in vitro metastatic activity induced by Twist1 (Supplementary Fig. S116). Together, tThese results indicate the an essential role of Jagged1 in Twist1-induced endothelial differentiation, EMT, and metastasis.
Transdifferentiation into endothelial cells from tumor stem-like cells6-8 may be regulated by stemness-related transcription factors. Pluripotency factors were have been shown to regulate endoderm specification19. To further identify the transcription factors that are controlled by the Twist1-Jagged1/Notch axis to that regulate the expression of various endothelial and vascular marker genes, different stemness-related transcriptional regulators, including OCT4, SOX2, NANOG, KLF4, GFI1, WNT, and BMI1 were screened to correlate their expression with the Twist1-Jagged1/Notch axis16,24. The screening showed that oOnly KLF4 correlated with the expression of Twist1 and Jagged1 (Supplementary Fig. S127a). Real-time PCR and Western blot analysis showed that further knockdown of Jagged1 decreased the levels of KLF4 analysis of Twist1-overexpressing OECM-1 cells and Jagged1 knockdown in Twist1-overexpressing OECM-1 cells showed the correlation between Twist1, Jagged1, and KLF4 expression (Fig. 35a and Supplementary Fig. S127b). TheThis correlation was confirmed corroborated by another cell line, H1299, in which a decrease in Jagged1 also downregulated KLF4 Jagged1 knockdown in H1299 cells, which resulted in decreased KLF4 expression (Supplementary Fig. S127c). To test the a direct activation of KLF4 by Jagged1, we transiently transfected Jagged1 with a,
KLF4 promoter-driven reporter construct harboring either a wild-type or mutated
activated by Jagged1 overexpression, but mutated KLF4 reporter was unresponsive transient transfection experiments were conducted. Overexpression of Jagged1 activated the KLF4 promoter-driven reporter construct and a Notch intracellular
domain (NICD)/CSL-responsive site was identified (Fig. 35b). Moreover, Cchromatin immunoprecipitation experiments showed the a direct binding of NICD/CSL to the
KLF4 promoter in two different cell lines whereas Jagged1 depletion abolished this interaction (Fig. 35c). Together, these results indicate that KLF4 is a direct target of Jagged1 and its activation is controlled These results indicated the a direct activation of KLF4 expression by the Jagged1/Notch signaling.
KLF4 mediates Twist1-induced endothelial differentiation
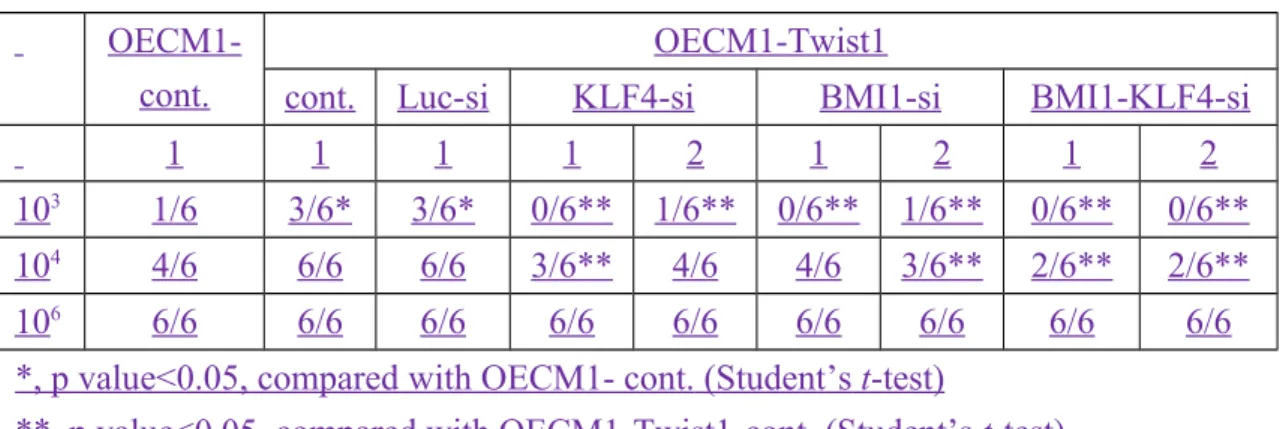
In addition to the induction of tumor-derived endothelial differentiation by Twist1, our previous results showed that Twist1 activates BMI1 expression to confer stem cell-like property24. Since both BMI1 and KLF4 are factors capable of promoting stemness16,24, we wanted to assign their contribution to Twist1-induced endothelial differentiation. To achieve this goal, KFL4-, BMI1-, or KFL4/BMI1-knockdown clones were generated in Twist1-overexpressing stable cells Twist1-overexpressing stable clones with knockdown of either KLF4 or BMI1 alone or with double knockdown of KLF4 and BMI1 were generated (Fig. 46a). KLF4 or BMI1
knockdown decreased the percentage of CD31+ and CD144+ population in Twist1-overexpressing OECM-1 cells using theby flow cytometry assay and immunofluorescence staining (Fig. 46a, b and Supplementary Figs. S18,1913). Increased in vitro tube-formation ability induced by Twist1 was also abolished by either KLF4 or BMI1 knockdown (Supplementary Fig. S2014). Other endothelial phenotypes, such as DiI-AcLDL uptake and Hb levels in xenotransplanted tumors, were also abrogated in Twist1-overexpressing cells with KLF4 or BMI1 knockdown (Fig. 46b and Supplementary Fig. S2115b). In vivo xenograft assays showed the abrogation of co-expression of Twist1 and CD31 in Twist1-overexpressing cells with KLF4 or BMI1 knockdown (Supplementary Fig. S2215a, including immunofluorescence and hematoxylin and eosin (H&E) staining of consecutive slides). However, expressions of various endothelial markers, including vWF, ICAM1, and CD105, was were abolished in KLF4 knockdown but not in BMI1 knockdown Twist1-overexpressing cells using real-time PCR analysis (Supplementary Fig. S2316a). KLF4, but not BMI1, was also responsible for the regulation of EFNB2, NRP1, and EPHB4 expressions from the KLF4 knockdown experiments (Supplementary Fig. S2316b). To further examine the role of KLF4 in the regulation of various endothelial and vascular markers, quantitative ChIP (qChIP) assays using an anti-KLF4 antibody to screen the promoter regions of various genes
with KLF4 binding were performed. The results showed that KLF4 bound to the proximal promoters of CD31, CD144, vWF, CD105, ICAM1, EFNB2, and EPHB4 genes (Fig. 46c and Supplementary Figs. S24-3017-20), which were mostly consistent with the results from KLF4 knockdown experiments (Fig. 46a and Supplementary Figs. S17-2313-16). In contrastIntriguingly, KLF4 binding to the promoters of these endothelial and vascular marker genes still existed in Twist1-overexpressing OECM1 clones with BMI1 knockdown (data not shown). Overexpression of KLF4 in OCEM-1 cells activated the expression of CD31, CD144, ICAM1, CD105, and vWF at both the mRNA and protein levels (Supplementary Fig. S3121a,b). qChIP experiments showed that BMI1 only bound to the promoters of CD31 and CD144 genes but not to the promoters of vascular markers, including vWF, CD105, ICAM1, EFNB2, and EPHB4 genes (Supplementary Figs. S3121c, 3222 and data not shown). In addition, the promoter regions of CD31 and CD144 genes that were bound by BMI1 were different from the regions bound by KLF4 (Supplementary Figs. S3121c, 3222). These results indicate that BMI1 functionsed through different mechanisms to mitigate the endothelial phenotype (i.e. expression of CD31 and CD144, increased DiI-AcLDL uptake and Hb levels) but it does not exert have no any effects on the expressions of various endothelial or vascular markers that were regulated by KLF4. The finding that Twist1-Jagged1-KLF4 can induce endothelial differentiation is consistent with our
observation that OECM-1 cells expressing CD31 and CD144 actually had the highest Twist1 expression levels (Supplementary Fig. 2b,c) The role of the Twist1-Jagged1-KLF4 axis to induce endothelial differentiation is also consistent with the very high levels of expression in the 9-10% of Twist1-overexpressing OECM-1 cells that expressed CD31 and CD144 (Supplementary Fig. S3). Next, We we also tested the correlation between among Twist1, Jagged1, and KLF4 using head and neck cancer patientin clinical samplesspecimens. Immunohistochemistry (IHC) staining of 242 cases of head and neck squamous cell carcinoma (HNSCC) patient samples showed that there was significant correlation between Twist1 and Jagged1/KLF4 expressions as well as between Jagged1 and KLF4 expressions, indicating that the Twist1-Jagged1-KLF4 axis existed may be present in real lifeclinical patient samples (Supplementary Tables S2 & S3, and Supplementary Fig. S33a,b23a). Increased expression of the Twist1-Jagged1-KLF4 signaling axis was also confirmed in 2 out of 6 primary head and neck cancer samples (Supplementary Fig. S33c23b). Finally, we tested whether other KLF family members, other KLF family members, including KLF2 and KLF5, may contribute to Twist1-Jagged1-induced endothelial differentiation25. However, quantitative real-time PCR analysis showed that knockdown of Jagged1 in Twist1-overexpressing OECM-1 cells did not decrease the
KLF5 did does not contribute to play a role in Twist1-induced endothelial differentiation.
KLF4 mediates Twist1-induced stem-like property/ and metastasis
Jagged1 was showncan to induce cancer stem cell property with an unknown mechanism26. We also wanted toTherefore, we evaluated test the involvement ofwhether KLF4 and BMI1 contribute to in Twist1-induced stem cell-like property due to the contribution of stem-like property to its tumor-derived endothelial differentiation6-8. The analysis of stem cell-like property contained CD44 staining, tumor sphere formation, and in vivo tumor-initiating ability assays24. The result showed that KLF4 knockdown did not decrease the percentage of CD44+ population induced by Twist1, whereas knockdown of BMI1 abolished the CD44+ population induced by Twist124 (Fig. 5a and Supplementary Fig. S3424a). However, knockdown of either KLF4 or BMI1 significantly decreased the size and/or numbers of tumor sphere formation as well as in vivo tumor-initiating ability (Fig. 5b,c7a,b, Table 1, and Supplementary Fig. S3524b), suggesting that both BMI1 and KLF4 may contributed to tumor-initiating ability induced by Twist1. Specifically, real-time PCR analysis showed that KLF4 was responsible for the expression of stemness genes (Wnt5A and
(Supplementary Fig. S36a,b7c,d)27-29. The qChIP assay also showed the binding of KLF4 to the promoter regions of Wnt5A gene (Supplementary Fig. S36cS25a). These findings suggest both KLF4 and BMI1 may promote stemness-like property induced by Twist1, but they may regulate cell stemness in a differential way which needs to be further characterized. These results indicate that KLF4 also regulates the stem-like property induced by Twist1.
To examine the role of KLF4 to in the regulatione of EMT marker gene expression30, Western blot analysis of various clones showed that knockdown of KLF4 did not reverse the EMT marker gene changes induced by Twist1, whereas knockdown of BMI1 abolished the EMT marker gene changes induced by Twist1but knockdown of BMI1 did (Supplementary Fig. S37a25b). However, knockdown of KLF4 still decreased the in vitro migration and invasion activity induced by Twist1 (Supplementary Fig. S37b25c), indicating that KLF4 regulates metastatic activity independent of the EMT marker gene changes. In vivo metastasis assays also showed consistent results with the in vitro migration and invasion assays (Supplementary Fig. S388).
A strategy for to guide the treatment of Twist1-overexpressing tumors
To test determine whether the information of Twist1-Jagged1-KLF4 axis can provide a guide to specific treatment strategy for patients with Twist1-overexpressing
cancers, Twist1-overexpressing OECM1 cells were used to test drug response. Two commonly used drugs for to treat head and neck cancerHNC treatment, cetuximab and cisplatin31,32, were individually tested individually together with or without a -secretase inhibitor (DAPT) that inhibits Notch signaling33. Overexpression of Twist1 increased the viability of cells treated with either cetuximab or cisplatin that could be abolished by KLF4 knockdown (Supplementary Fig. S3926a,b). Treatment with DAPT together with either cetuximab or cisplatin additively inhibited the viability of Twist1-overexpressing OECM-1 cells as well as modulated the expression of endothelial and vascular markers regulated by Twist1 (Fig. 69a and Supplementary Figs. S40a,4126c, 27). KLF4 knockdown or DAPT treatment significantly decreased the KLF4+/CD44+ population in Twist1-overexpressing OECM-1 cells (Fig. 69b). In contrast, treatment with cetuximab or cisplatin did not significantly change the percentage of cells stained with BMI1 or KLF4 (Figure 69b and Supplementary Fig. S40b26d). Treatment of xenotransplanted head and neck tumors with cetuximab and DAPT also had the an additive tumor inhibiting effects (Fig. 69c and Supplementary Fig. S4228a). These results point to the advantage of using -secretase inhibitors (e.g. DAPT) in combination with chemotherapy to treat Twist1-overexpressing human tumors.
Discussion
Although Twist1 is a master regulator of mesoderm development9, our results showed that Twist1 can also induce tumor-derived endothelial differentiation and promote tumor vascular formation. Therefore, the phenomenon of EMT induced by Twist111,12 can be extended to the epithelial-mesenchymal-endothelial transition due to the co-staining of vimentin (a mesenchymal marker) and CD144 (an endothelial marker) in the same Twist1-overexpressing head and neck tumor cells (Supplementary Fig. S2a), suggesting that ~10% of Twist1-overexpressing tumor cells can be driven into endothelial differentiation and contribute directly to the angiogenesis process. This observation is in drastic contrast to the traditional angiogenesis process contributed by surrounding endothelial progenitors that are triggered by cytokines and pro-angiogenic factors secreted from tumor-associated macrophages/neutrophils and tumor-associated fibroblasts2,3. Induction of tumor-derived endothelial differentiation by Twist1 was is different from the vasculogenic mimicry mechanism34,35 that which does not activate endothelial markers and may represent an incomplete differentiation towards the endothelial lineage. This induction of endothelial differentiation may be critical for tumor progression and metastasis since knockdown of either Jagged1 or KLF4 significantly decreased decreases the in
S11c,d, S25cS16, S37, S38). Inhibition of the angiogenesis process should be equally important to treat metastasis. Our results provide the an mechanistic explanation that why the Twist1-Jagged1-KLF4 axis reprograms head and neck cancer cells to becomeinto endothelial cells and why it promotes angiogenesis that supports facilitates tumor progression and metastasis. This result is consistent with the reports that pluripotency factors regulate endoderm specification19 and defects in the cranial vasculature are observed in Twist1 null mice36. Since knockdown of Twist1 in H1299 cells reversed reverses the vascular marker expression pattern and decreased decreases the hemoglobin levels in xenotransplanted mice (Supplementary Figs. S5c, S66-8), the same signaling pathway to mediate endothelial differentiation and vasculogenesis may be used in other tumor types.
In contrast to its role in the generation of inducible pluripotent stem cells (iPS)16, a distinct and detailed role of KLF4 in endothelial differentiation and vasculogenesis was demonstrated in this report. The regulation of KLF4 by Jagged1 also showed shows the direct connection between the Notch pathway and KLF4. It should be pointed out that the regulation of tumor-initiating ability by KLF4 is independent of the pathways mediated by BMI1, to which regulates the expression of CD44 surface marker and tumor sphere formation ability as shown previously24. The downstream targets of KLF4 (e.g. Wnt5A, CCND2) may control the stem-like
property that contributes to the tumor-initiating ability. Finally, KLF4 contributes to Twist1-induced metastatic activity through an EMT-independent mechanism, suggesting that regulation of different targets (e.g. motility genes) other than the typical EMT marker genes also contribute to the metastatic activity induced by Twist130. All these results demonstrate the versatile role of KLF4 in Twist1-induced endothelial differentiation, stem-like property, and metastasis. The knowledge gained from this report also has therapeutic implications for combining -secretase inhibitors33 with other chemotherapeutic agents to achieve additive tumor-killing effects in Twist1-overexpressing tumors, especially in head and neck cancer tumors with high a percentage (35-45%) expressingon of Twist111,24.
Jagged1 was has been shown to be a downstream target of Twist1, although the regulatory mechanism was has not been demonstrated37. This report provides the mechanism of a direct regulation of Jagged1 expression by Twist1. Negative regulation of KLF4 by Notch signaling was has been observed shown in intestinal epithelium or colorectal cancer cells38,39 and these results were areconsistent within favor of the tumor suppressor role of KLF4 in colorectal cancer cells40. In contrast to the tumor suppressor role of KLF4 in colorectal cancer cells/intestinal epithelium and pancreatic cancer38-42
0, other groups published results showed have demonstrated the oncogenic role of KLF4 in breast cancer, pancreatic cancer, and head and neck cancer
cells43-45
1-44. These results findings were are consistent with our discovery that KLF4 plays an oncogenic role, especially in head and neck squamous cell carcinoma45
4. We believe that tIn accordance with the general he overall published resultsperception, established that KLF4 is should be deemed as a context-dependent specific oncogene or tumor suppressor gene, depending on the tumor cell types.
Overall, our results propose that the Twist1-Jagged1-KLF4 axis induces tumor-derived endothelial differentiation inside the tumors to produce a favorable tumor microenvironment in addition to recruiting the existing quiescent endothelial cells. A model to summarize this report is presented (Fig. 69d), in which tumor mesenchymal cells induced by Twist1 can migrate into tumor vasculature reprogrammed by the Jagged1-KLF4 axis to initiate the metastatic process. This report also provides significant therapeutic implications to combine -scecretase inhibitors with established chemotherapeutic agents for cancer treatment.
Methods
Cell culture. The cell lines used were described including include the human tongue squamous cell carcinoma cell lines OECM1 and SAS, human hypopharyngeal squamous cell carcinoma cell line FADU, and lung cancer cell line H1299 (obtained from ATCC)11,24., and Hhuman embryonic kidney 293T cell line (all of them were obtained from American Type Culture Collection). was used in transient transfection experiments. The OECM1, SAS and FADU cell lines were used due to their low endogenous Twist1 levels and low metastatic activity (epithelial in nature). The H1299 cell line was used due to its higher Twist1 levels (mesenchymal in nature). The culture of primary head and neck tumor cells was performed by dissociating the tumor tissue in trypsin and collagenase followed by culturing in RPMI medium containing 10% fetal bovine serum24
. as described24. Informed consent was obtained from all subjects and Tthe procurement of primary HNSCC tumors was approved by the Institutional Review Board (IRB) of Taipei Mackay Memorial Hospital.
Protein extraction and, Western blot analysis,. RNA extraction, quantitative real-time PCR, in vitro migration/invasion assay, in vivo tail vein metastatic assay. For extraction of proteins from cell lines, cell lysis buffer (50 mM Tris, pH 7.5, 30 mM MgSO4, 8 mM EDTA, 2 mM DTT, 2% Triton X-100 and 0.2% SDS) containing protease inhibitors was used. Cell lysates were clarified by centrifugation
at 13000 rpm, 4°C for 10 minutes. The protein content was determined by Bradford method (Bio-Rad Laboratories, Hercules, CA). For Western blot analysis, 50-100 µg protein extracts from each clone were loaded to 10% SDS-PAGE gels and transferred to nitrocellulose filters. The filters were probed with different antibodies, and an anti-ß-actin antibody was selected as a loading control. Signals were developed using an ECL chemiluminescence kit (Millipore Corporation, Billerica, MA).
RNA extraction and quantitative real time PCR. Total RNA was isolated using Trizol reagent (Life Technologies Corporation, Carlsbad, CA) according to manufacturer's recommendations. Single-stranded cDNA was synthesized by the RevertAid™ First Strand cDNA Synthesis Kit (Fermentas International, Inc., Burlington, Canada). Real-time PCR was performed on a Bio-Rad iCycler iQ PCR detection system according to the manufacturer's instructions. The 2-ΔΔCt method of relative quantification was used to estimate the copy number of gene expression46
5, and 18Swas selected as an internal control. The sequences of the oligonucleotides for real time PCR were shown in Supplementary Table S4.
In vitro migration/invasion and in vivo metastatic assays. For in vitro migration/invasion assay, eight-µm pore size Boyden chamber was used. Cells (4 x 104) in 0.5% serum-containing RPMI wereplated in the upper chamber and 15% fetal bovine serum was added to RPMI1640 in the lower chamber as a chemo-attractant.
For invasion assay, the upper side of the filter was covered with Matrigel (BD Biosciences, San Jose, CA) (1:2 dilution with RPMI). After 12 hours for migration assay or 24 hours for invasion assay, cells on the upper side of the filter were removed, and cells that remained adherent to the undersideof membrane were fixed in 4 % formaldehyde and stainedwith Hoechst 33342 dye. The number of migrated cells wascounted using a fluorescence microscope. Ten contiguous fields of each sample were examined using a 40x objective toobtain a representative number of cells which migrated/invaded across the membrane. For in vivo tail vein metastasis assay, five six-week old female non diabetic-severe combined immunodeficiency (NOD-SCID) mice received injectionof 4 x 106 cells of different cell lines in 0.1 ml of PBS via the tail vein (6 mice for each group). Four to six weeks after injection, mice were examined grossly at necropsy for the presence of metastases in internal organs such as lung tissue and lymph node area. Microscopic examination of metastases was performed on the cross-sections of formalin-fixed, paraffin-embedded lung tissues stained with H&E. The counting of metastatic lesions in the internal organ of each mouse was evaluated by gross and microscopic examination.
Plasmids and stable transfection. The Twist1 expression vector (Flag-CMV-Twist1 or pcDNA3-Twist1) was generated by inserting a 694 bp fragment of the full length
Twist1 cDNA into the pFlag-CMV vector11
pcDNA3-Jagged1 was generated by inserting a 3657 bp fragment of the full length human Jagged1 cDNA into the HindIII/EcoRI sites of the pcDNA3 vector. The KLF4 expression vector was obtained from Dr. S.C. Teng (National Taiwan Univ.). The plasmids used and transfection methods were described using cCalcium phosphate transfection method by mixing calcium chloride and phophostate solution to generate plasmid precipitates was used for plasmid transfection11,24. The Twist1 expression vector was described11. Stable clones were generated by transfection of expression vectors or infection with lentivirus shRNA plasmids and selected by appropriate antibiotics.
Transient transfection and luciferase assays. The Jagged1 and KLF4 promoter regions were cloned and the reporter constructs were shown in Supplemental Tables S15 & S26. Both the Jagged1 promoter mutant reporter construct and KLF4 promoter mutant report construct were generated using site-directed mutagenesis. The reporter constructs were co-transfected into 293T cells using calcium phosphate transfection method with different expression vectors and an internal control plasmid. Luciferase assays were performed using the same amount of cell extracts and corrected for transfection efficiency using an internal control (GFP)11,24.
Standard and quantitative chromatin immunoprecipitation47
(ChIP and qChPip). ChIP assay was performed as described46. Briefly, cCells were cross-linked
with 2% formaldehyde for 10 min and stopped by adding glycine to a final concentration of 0.125 M. Fixed cells were washed twice with TBS (20 mM Tris, pH 7.5, 150 mM NaCl) and harvested in 5 ml of SDS buffer (50 mM Tris, pH 8.0, 0.5% SDS, 100 mM NaCL, 5 mM EDTA, and protease inhibitors). Cells were pelleted by centrifugation and suspended in 2 ml of IP buffer (100 mM Tris, pH 8.6, 0.3% SDS, 1.7% Triton X-100, 5 mM EDTA). Cells were sonicated with a 0.25-inch diameter probe for 15s twice using an MSE-soniprep 1500 sonicator (setting 18). For each immunoprecipitation, 1 ml of lysate was precleared by adding 50 ml of blocked protein A beads (50% protein A-Sepharose, Amersham Biosciences; 0.5 mg /ml-1
bovine serum albumin, 0.2 mg/ml-1
salmon sperm DNA) at 4 0C for 1h. Samples were spun, and the supernatants were incubated at 4 0C for overnight with no antibody, IgG, anti-twist, anti-NOTCH-ICD, anti-KLF4 or anti-BMI1 antibody. Immune complexes were recovered by adding 50 ml of blocked protein A beads and incubated overnight at 4 0C. Beads were successively washed with 1) mixed micelle buffer (20 mM Tris, pH 8.1, 150 mM NaCl, 5 mM EDTA, 5% w/v sucrose, 1% Triton X-100, 0.2% SDS), 2) buffer 500 (50 mM Hepes, pH 7.5, 0.1% w/v deoxycholic acid, 1% Triton X-100, 500 mM NaCl, 1 mM EDTA), 3) LiCl detergent wash buffer (10 mM Tris, pH 8,0.5% deoxycholic acid, 0.5% Nonidet P-40, 250 mM LiCl, 1mM EDTA), and 4) TE buffer (10 mM Tris, 1 mM EDTA) and then eluted with 1% SDS and 0.1 M
NaHCO3. 20 ml of 5 M NaCl was added to the elutes, and the mixture was incubated at 65 0C for 5 h to reverse the cross-linking. After digestion with proteinase K, the solution was phenol/chloroform-extracted and ethanol-precipitated. For standard ChIP, DNA fragments were resuspended in 50 l of water and 5l was used in a PCR reaction for 30 cycles. For qChIP analysis, DNA fragments were resuspended in 400 l of water and 5 l was used by real-time PCR. Each sample was calculated as the percentage of input sample. The sequences of PCR regions and primers used in ChIP assay are listed (Supplementary Tables S1-35-7). The source and concentration of the antibodies used are listed (Table S58).
Immunofluorescence staining and flow cytometry. For immunofluorescence staining, cells on glass coverslips or chamber slides were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100 . After washing 3 times for 10 min with PBS, fixed cells or slides were blocked with blocking buffer (PBS, 0.1% Tween-20, with 3% goat serum) for 1 hour and incubated with primary antibody diluted in blocking buffer overnight at 4℃. After washing 3 times with PBS for 10 min, the fixed cells were treated with the appropriate secondary antibody (fluorescein isothiocyanate (FITC)-conjugated mouse IgG or rhodamine-conjugated anti-rabbit IgG) (Sigma) that was diluted in blocking buffer for 1 h at room temperature. Finally, the fixed cells were washed 3 times for 10 min with TBS, and their nuclei
were counterstained, mounted and observed by using fluorescence microscope or confocal microscope. The paraffin-embedded tissue specimens from patient samples and mouse samples were de-paraffinized in xylene, rehydrated through graded ethanol, and rinsed in PBS buffer. Antigen retrieval processes were then performed by pressure cooker at 1.2 atm for 15 min with 1 mM sodium citrate buffer (pH 6.0) and then incubated with 20 g/ml-1
Proteinase K in TE buffer at 37 ºC for 20 min. These slides would be used for immunofluorescence staining by using hCD31, CD144, Twist1 and GFP antibody, the detailed antibody information were shown in the supplementary table S58. The characterizations of the specificity of endothelial marker antibodies such as CD31 and CD144 were shown (Supplementary Fig. S4328b). The CD31 antibody is human specific since staining of mouse tissue was negative. The CD144 antibody could stain both human and mouse CD144. Informed consent was obtained from all subjects and Tthe usage of patient samples was approved by the IRB of Taipei Mackay Memorial Hospital. To analyze the expression of specific cell surface markers, cells were resuspended and incubated with FITC-conjugated anti-CD44 antibody, FITC-conjugated anti-CD31 antibody and PE-conjugated anti-CD144 antibody, respectively. The stained cells were evaluated with a FACSCalibur flow cytometer (BD Biosciences). At least ten thousand cells were analyzed per sample and each staining experiment was repeated four times.
CELLQuest analysis software was used for fluorescence determination and data analysis. Flowjo was used to generate the dot plots.
Lentiviral infection. Lentivirus containing short hairpin RNAs (shRNAs) expressed in a lentiviral vector (pLKO.1-puro) were generated in 293T cells as previously described48
7. Plasmids pLKO-Luciferase-i, pLKO-KLF4-i-3, pLKO-KLF4-i-934, pLKO-GFP, and packaging plasmid pCMVΔR8.91 were obtained from S.C. Teng (National Taiwan University, Taiwan). Plasmids 2 and pLKO-BMI1-i-3 were provided by National RNAi Core Facility of Academia Sinica, Taipei, Taiwan. For lentivirus production, 293T cells were transfected with 15 μg pLKO.1-puro lentiviral vectors expressing different shRNAs along with 1.5 μg of envelope plasmid pMD.G and 15 μg of packaging plasmid pCMVΔR8.91. Virus was collected 48 h after transfection. To prepare KLF4 or Bmi1 knockdown cells, OECM-1-Twist1 cells were infected with lentivirus for 24 h, and stable clones were generated by selection with appropriate antibiotics. The sequences for the lentivirus were shown in Supplementary Table S4.
2.5 Dimensional (2.5D) tube formation assay49
8. For experiments in which cells
were cultured on top of the Matrigel, 50 l growth factor reduced Matrigel were plated in 8-well chamber slides. The chambers were then incubated at 37°C for 30 min to allow Matrigel to polymerize. 1 × 104 cells were added to the top of the
Matrigel in each well. After incubation for 5 days, the slides were examined for endothelial tubes using light microscope. Five random images per well were quantified by the MetaMorph Imaging System, an image analysis program a digital imaging software to analyze biological images. The Angiogenesis Tube Formation Module was used to analyze tube formation activity. The image was run through the software to identify tubes or nodes. A pixel is a single point in a graphic image as a unit of measurement.
Three dimensional (3D) tube formation assay. For 3D culture, tumor cells were resuspended in 5 mg/ml-1
growth factor reduced Matrigel (BD Biosciences) on ice. 1 × 104 cells each well were seeded in 8-well chamber slides and allowed to polymerize at 37°C for 30 min followed by adding 10% serum-containing MCDB131 culture medium to the top of the wells. The chambers were then incubated at 37°C. After incubation for 5 days, the slides were used for DiI-AcLDL uptake analysis and immunofluorescence assay.
DiI-AcLDL uptake analysis. The functional assay for endothelial cells was performed by incubation of cells with 10 g/ml of DiI-labeled acetylated low density lipoproteins (DiI-AcLDL)(Molecular Probes, Invitrogen) for 4 h followed by observation by fluorescence microscope. A picture with 3D tumor formation assay was usually accompanied with the LDL uptake picture to indicate the presence of cell
clones tested.
In vivo sSubcutaneous implantation and, hemoglobin quantification., and drug treatment of xenografted mice. For in vivo monitoring of tumor-associated angiogenesis, 1 x 106 cells of different cell lines in 0.1 ml Matrigel were injected under the abdominal skin of nude 6 week old female Balb/c nude mice. After tumor formation (4 to 6 weeks after injection), animals were sacrificed and matrigel implants were collected, weighed, and fixed in 4% paraformaldehyde for IHC analysis or in PBS for hemoglobin quantification. Hemoglobin contents of matrigel implants were measured using a Drabkin’s reagent kit (Sigma) as described previously49
8. The tissue was transferred into the Eppendorf containing 0.5 ml PBS buffer under overnight incubation at 37℃ to lyse RBC and release hemoglobin. The tissue samples were homogenized and spun down to collect the clear supernatant. Samples (50 l) of tissue supernatant were added to 1 ml Drabkin’s reagent and then incubated for 15 minutes at room temperature. Absorbance of each sample was measured at 540 nm. The results are divided by tissue weight and expressed as hemoglobin concentration (mgmg-1
).
Drug treatment of xenografted mice. In the xenotransplanted tumor model, the OECM1-Twist1 cells (2x106) in 0.2 mL PBS were injected into the right lateral flank of 6 week old female Balb/c Nude mice. Treatment was started when the tumors grew
to certain size. Mice were treated once a week by intraperitoneal injections (i.p.) of 10 mg/kg-1
cetuximab (Merck KGaA, Darmstadt, Germany) or 100 mg/kg-1
DAPT31 (Selleck Chemicals., Houston, USA) or both together. The tumor size of each group were monitored and measured every week. Tumor volume (mm3) was estimated according to the formula of length x (width) 2 x 0.5 in mm3.
Tumor sphere formation24
. The procedure was performed as described24. Cells (1 ×103) were seeded and incubated with serum-free medium composed of DMEM/F-12 (Thermo Fisher Scientific Inc. United States), N2 supplement (Thermo Fisher Scientific Inc. United States), 10 ng/ml-1
human recombinant bFGF (R&D Systems, Inc. United States) and 10 ng/ml ml-1
EGF (R&D Systems, Inc. United States) at 24 well plate. The spheroids were resuspended to form secondary and tertiary spheroids. Only the tumor spheres with a size greater than 100m were counted. The number of spheroids was counted after 14 days.
Tissue microarray construction50
. A high-density tissue microarray (TMA) was constructed using formalin-fixed, paraffin-embedded specimens of HNSCC patient samples as previously described11. H&E-stained sections were made from each paraffin block to define representative tumor regions. Tissue cylinders with a diameter of 0.6 mm were punctured from these regions in the paraffin block and arrayed into a recipient block using the tissue chip microarrayer (Beecher Instruments, Silver
Spring, MD). The recipient block was serially cut into 5-μm sections on silanized slides (Dako, Glostrup, Denmark). Informed consent was obtained from all subjects and Tthe protocol was approved by the IRB of the Taipei Veterans General Hospital. In situ hybridization. The paraffin-embedded tissue specimens were de-paraffinized in xylene, rehydrated through 100%, 90%, 70%, 50% ethanol, and rinsed in PBS buffer. Antigen retrieval processes were then performed twice by pressure cooker at 1.2 atm for 15 min with target retrieval solution (#S1700, Dako Corporation, Denmark) and then blocked with 0.3% hydrogen peroxide for 30 minutes. 20 uL of the biotinylated probes for general screening (HPV 6, 11, 16, 18, 30, 31, 33, 45, 51, 52) (#Y1404, DAKO) were applied on separate specimen, and were covered with coverslips. Denaturation of DNA was performed in 92° C for 5 minutes in an oven. Hybridization was performed in a humid chamber at 37° C overnight. After washing, the GenPoint Kit (#K0620; DAKO) was used according to the manufacturer's instruction.
Immunohistochemstry (IHC), scoring, and validation of antibodies11,24,47. The sample processing and IHC procedure for determining the immunoreactivity of Twist1, Jagged1, and KLF4 were described as the following.11,24,46. Briefly, The paraffin-embedded tissue specimens were de-paraffinized in xylene, rehydrated through 100%, 90%, 70%, 50% ethanol, and rinsed in PBS buffer. Antigen retrieval
processes were then performed by pressure cooker at 1.2 atm for 15 min with 1 mM sodium citrate buffer (pH 6.0) and then blocked with 3% hydrogen peroxide for 30 minutes. The specimens were incubated with primary antibodies in 5% BSA buffer in a humidity chamber overnight at 40C. Detection of the staining was used by the Super Sensitive Non-Biotin Polymer HRP Detection System according to the manufacturer’s instructions (BioGenex, San Ramon, CA). The immunoreactivity of Twist1, Jagged1 and KLF4 was graded from 0 to 3+ (0, no staining; 1+, 1 25%; 2+, 26 50%; 3+, >50% nuclear staining) according to nuclear expression, and only 3+ (>50% nuclear staining) was considered as a positive immunohistochemistry result. All IHC staining was independently scored by two experienced pathologists. If there was discordance with IHC scoring, a pathologic peer review will be performed to consolidate the result into a final score. The pathologists scoring the IHC were blinded to the clinical data. The interpretation was performed in five high power views for each slide, and 100 cells per for each view were counted for analysis. Validation of Twist1 antibody was described11. Validation of antibodies against Jagged1 and KLF4 was performed from various overexpression and knockdown experiments (e.g. Jagged1 from Supplementary Figs. 1611a,b, 1712b,c; KLF4 from Fig. 46a, Supplementary Fig. S3121b) as well as from the manufacturer’s protocols.
continuous variables between two groups, and the 2 test was applied for comparison of dichotomous variables in tables. The control groups of all the statistical analyses were specified in the figure legends. The level of statistical significance was set at 0.05 for all tests unless specified.
Deposition of microarray data. The raw data of microarray (H1299 control vs. H1299-Twist1-si) were deposited to GenBank in MIAME format and the accession number is GSE57756.
References
1. Hanahan, D. & Weinberg, R.A. Hallmarks of cancer: the next generation. Cell 144, 646-674 (2011).
2. Le Bras, A. et al. Molecular mechanism of endothelial differentiation. Vasc. Med. 15, 321 (2010).
3. Weis, S.M. & Cheresh, D.A. Tumor angiogenesis: molecular pathways and therapeutic targets. Nature Med. 17, 1359-1370 (2011).
4. Pezzolo, A. et al. Tumor origin of endothelial cells in human neuroblastoma. J.
Clin. Oncol. 25, 376-383 (2007).
5. Streubel, B. et al. Lymphoma-specific genetic aberrations in microvascular endothelial cells in B-cell lymphoma. N. Engl. J. Med. 351, 250-259 (2004). 6. Ricci-Vitiani, L. et al. Tumor vascularization via endothelial differentiation of
glioblastoma stem-like cells. Nature 468, 824-828 (2010).
7. Soda, Y. et al. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc. Natl. Acad. Sci. USA 108, 4274-4280 (2011).
8. Wang, R. et al. Glioblastoma stem-like cells give rise to tumor endothelium.
Nature 468, 829-833 (2010).
9. Furlong, E.E. et al. Patterns of gene expression during Drosophila mesoderm development. Science 293, 1629-1633 (2001).
Cell 139, 871-890 (2009).
11. Yang, M.H. et al. Direct regulation of TWIST by HIF-1promotes metastasis.
Nat. Cell Biol. 10, 295-305 (2008).
12. Yang, M.H. & Wu, K.J. TWIST activation by hypoxia inducible factor-1 (HIF-1): implications in metastasis and development. Cell Cycle 7, 2090-2096 (2008). 13. Dufraine, J. et al. Notch signaling regulates tumor angiogenesis by diverse
mechanisms. Oncogene 27, 5132-5137 (2008).
14. Bridges, E. et al. Notch regulation of tumor angiogenesis. Future Oncol. 7, 569-588 (2011).
15. Zeng, Q. et al. Crosstalk between tumor and endothelial cells promotes tumor angiogenesis by MAPK activation of Notch signaling. Cancer Cell 8, 13-23 (2005).
16. Yamanaka, S. & Blau, H.M. Nuclear reprogramming to a pluripotent state by three approaches. Nature 465, 704-712 (2010).
17. Suzuki, T. et al. Vascular implications of the Kruppel-like family of transcription factors. Arterioscler. Thromb. Vasc. Biol. 25, 1135-1141 (2005).
18. Yet, S.F. et al. Human EZF, a Kruppel-like zinc finger protein, is expressed in vascular endothelial cells and contains transcriptional activation and repression domains. J. Biol. Chem. 273, 1026-1031 (1998).
through eomesodermin. Genes Dev. 25, 238-250 (2011).
20. Lawson, C. & Wolf, S. ICAM-1 signaling in endothelial cells. Pharmacol. Rep. 61, 22-32 (2009).
21. Panwar, A. et al. Human papilloma virus positive oropharyngeal squamous cell carcinomas: a growing epidemic. Cancer Treat. Rev. 40, 215-219 (2014).
22. Adams, R.H. Molecular control of arterial-venous blood vessel identity. J. Anat. 202, 105-121 (2003).
23. You, L.R. et al. Suppression of Notch signaling by the COUP-TFII transcription factor regulates vein identity. Nature 435, 98-104 (2005).
24. Yang, M.H. et al. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat. Cell Biol. 12, 982-992 (2010).
25. Suzuki, T. et al. Vascular implications of the Kruppel-like family of transcription factors. Arterioscler. Thromb. Vasc. Biol. 25, 1135-1141 (2005).
26. Lu, J. et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell. 23,171-185 (2013).
27. Lee, J. et al. Genetic reconstruction of mouse spermatogonial stem cell self-renewal in vitro by Ras-cyclin D2 activation. Cell Stem Cell 5, 76-86 (2009). 28. Yang, D.H. et al. Wnt5a is required for endothelial differentiation of embryonic
stem cells and vascularization via pathways involving both Wnt/-catenin and protein kinase C. Circ. Res. 104, 372-379 (2009).
29. Yeh, J.R. et al. Wnt5a is a cell-extrinsic factor that supports self-renewal of mouse spermatogonial stem cells. J Cell Sci. 124, 2357-2366 (2011).
30. Wu, C.Y. et al. Epigenetic reprogramming and post-transcriptional regulation of the epithelial-mesenchymal transition. Trends Genet 28, 454-463 (2012).
31. Gerber, D.E. & Choy, H. Cetuximab in combination therapy: from bench to clinic.
Cancer Metastasis Rev. 29, 171-180 (2010).
32. Vokes, E.E. Induction chemotherapy for head and neck cancer: recent data.
Oncologist 15 (Suppl 3), 3-7 (2010).
33. Sjolund, J. et al. Suppression of renal cell carcinoma growth by inhibition of Notch signaling in vitro and in vivo. J. Clin. Invest. 118, 217-228 (2008).
34. Kirschmann, D.A. et al. Molecular pathways: vasculogenic mimicry in tumor cells: diagnostic and therapeutic implications. Clin. Cancer Res. 18, 2726-2732 (2012).
35. Sun, T. et al. Expression and functional significance of Twist1 in hepatocellular carcinoma: its role in vasculogenic mimicry. Hepatology 51, 545-556 (2010). 36. Chen, Z.F. & Behringer, R.R. twist is required in head mesenchyme for cranial
neural tube morphogenesis. Genes Dev. 9, 686-699 (1995).
37. Yen, H.Y. et al. Jagged1 functions downstream of Twist1 in the specification of the coronal suture and the formation of a boundary between osteogenic and non-osteogenic cells. Dev. Biol. 347, 258-270 (2010).
38. Ghaleb, A.M. et al. Notch inhibits expression of the Kruppel-like factor 4 tumor suppressor in the intestinal epithelium. Mol. Cancer Res. 6, 1920-1927 (2008). 39. Miyamoto, S. et al. Suppression of colon carcinogenesis by targeting Notch
signaling. Carcinogenesis 34, 2415-2423 (2013).
40. Zhao, W. et al. Identification of Kruppel-like factor 4 as a potential tumor suppressor gene in colorectal cancer. Oncogene 23, 395-402 (2004).
41.Wei, D. et al. Kruppel-like factor 4 induced p27Kip1 expression in and suppresses the growth and metastasis of human pancreatic cancer cells. Cancer Res. 68, 4631-4639 (2008)
42.Zammarchi, F. et al. KLF4 is a novel candidate tumor suppressor gene in pancreatic ductal carcinoma. Am. J. Pathol. 178, 361-372 (2011).
43. Rowland, B.D. et al. The KLF4 tumor suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nat. Cell Biol. 7, 1074-1082 (2005).
44. Yu, F. et al. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene 30, 2161-2172 (2011).
Wei, D. et al. KLF4 up-regulation promotes cell cycle progression and reduces survival time of patients with pancreatic cancer. Gastroenterology 139, 2135-2145 (2010).
45. Tai, S.K. et al. Persistent Kruppel-like factor 4 expression predicts progression and poor prognosis of head and neck squamous cell carcinoma. Cancer Sci. 102, 895-902 (2011).
46.Livak, K.J. & Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 25, 402-408 (2001).
47.Wu, M.Z. et al. Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Molecular Cell 43, 811-822 (2011). 48.Moffat, J. et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124, 1283-1298 (2006).
49.Passaniti, A. et al. A simple, quantitative method for assessing angiogenesis and antiangiogenic agents using reconstituted basement membrane, heparin, and fibroblast growth factor. Lab. Invest. 67, 519-528 (1992).
50.Freier, K. et al. Tissue microarray analysis reveals site-specific prevalence of oncogene amplifications in head and neck squamous cell carcinoma. Cancer Res. 63, 1179-1182 (2003).
Acknowledgments
We appreciate Dr. L.R. You for critical comments on the manuscript. We thank Dr. H.W. Wang, Dr. T.Y. Chou, C.Y. Hsieh and Yang-Ming Genome Research Center for technical assistance. This work was supported in part to K.J.W. by National Science Council Frontier grant (NSC101-2321-B-010-002, NSC102-2321-B-010-001), Keelung Chang-Gang Memorial Hospital (CMRPG2D0031), National Research Program for Biopharmaceuticals (NSC102-2325-B-010-004), a grant from Ministry of Education, Aim for the Top University Plan (102AC-TC13, 103AC-T301), center of excellence for cancer research at Taipei Veterans General Hospital (DOH102-TD-C-111-007, MOHW103-TD-B-111-02), Taichung Veterans General Hospital (TCVGH-YM1000301, TCVGH-YM1010301), and National Health Research Institutes (NHRI-EX102-10230SI, NHRI-EX103-10230SI).
Author contributions
K.J.W. conceived the project and designed the experiments, H.F.C., C.H.H., J.J.H, and C.C.H. carried out experiments and statistical analysis, C.J.L. provided the tumor samples, K.J.W., H.F.C. and S.C.T. analyzed the data, K.J.W. wrote the manuscript with the help of S.C.T.
Author information
materials should be addressed to K.J.W. ([email protected]).
Competing financial interest
Figure legends
Figure 1. Induction of tumor-derived endothelial differentiation by Twist1. (a) InductionExpression of CD31 and CD144 expression by Twist1 in OECM-1 Twist1-overexpressing OECM-1 cells using Western blot analysis. The middle panel showed the dot plots from flow cytometry analysis and the rightmost panel showed the percentage of cells expressing bar graph analysis of CD144 expression in overexpressing OECM-1 cells. (b) Co-expression of Twist1 and CD144 in Twist1-overexpressing OECM-1 cells. Red: Twist1;, Ggreen: CD144. (left panels: images; right panel: the percentage of cells co-expressing Twist1 and CD144). Scale bar: 50m. (c) Increased t2.5D Tube-forming ability assays in Twist1-overexpressing OECM-1 cells by 2.5D tube-formation assays (left panels: images; right panel: pixels representing tube formation as described in the method section (bar graph)). bar graph analysis). Scale bar: 50m. (d) Increased DiI-AcLDL uptake assays in Twist1-overexpressing OECM-1 cells. Rred: DiI-AcLDL staining. The AcLDL staining photos were accompanied by 3D tube-formation ability photos (on top of Dil-AcLDL staining) for each clone. Red: DiI-AcLDL. Scale bar: 50m. (e) Participation of GFP-labeled Twist1-overexpressing cells in tumor vessel formation. Green: GFP; Red: CD144; Bblue: DAPI. Scale bar: 50m. (f) Co-expression of Twist1 and CD31 in
head and neck cancer patient samples. Examples from two different patient samples were shown. Green: hCD31; Rred: Twist1; Bblue: DAPI. Scale bar: 50m. Data shown in panels a-c are mean±s.d. of three independent experiments. The asterisk (*) indicates statistical significance (P<0.05 by Student's t-test) between experimental and control clones. The asterisk (*) indicates statistical significance (P<0.05) between experimental and control clones. The OECM-1 control clone was selected as the control group in panels a-c.
Figure 2. Regulation of various endothelial and vascular markers by Twist1. (a,b) Real-time PCR analysis of various endothelial markers including vWF, TIE2, CD105, and ICAM1 in Twist1-overexpressing OECM-1 clones. The fold change of relative RNA expression was shown (bar graph). (c) Western blot analysis of CD105 and ICAM1 in Twist1-overexpressing OECM-1 cell clones vs. the control clones. (d) Real-time PCR analysis of various vascular markers including EFNB2, NRP1, and EPHB4 in Twist1-overexpressing OECM-1 clones. The OECM1 control clone was selected as the control group, respectively, in panels a-d. Data shown in panels a,b and d are mean±s.d. of three independent experiments. The asterisk (*) indicates statistical significance (P<0.05 by Student's t-test) between experimental and control clones.
region of the Jagged1 gene containing a putative Twist1-responsive site. (b) Transient transfection experiments were performed using a Twist1 expression vector with a
Jagged1 promoter-driven reporter construct harboring either a wild-type or mutated
Twist1-responsive site. The pcDNA3 control transfected lane was selected as the control group vs. the pcDNA3-Twist1 transfected lane for each reporter construct. Data shown in panel b are mean±s.d. of three independent experiments. The asterisk (*) indicates statistical significance (P<0.05 by Student's t-test) between experimental and control clones. (c) Chromatin immunoprecipitation experiments were performed using the IgG or anti-Twist1 antibody to detect their binding to two regions in the
Jagged1 gene promoter. Two different cell lines (OECM-1, H1299) with different
Twist1 expression status were tested. The binding of Twist1 to the N-cadherin promoter was used as a positive control.
Figure 24. Jagged1 is essential for Twist1-induced endothelial differentiation. (a) Expression of CD144 in Twist1-overexpressing OECM-1 cells with Kknockdown of Jagged1 decreased the CD144 levels induced by Twist1 overexpression. Dot plots from flow cytometry and the percentage of cells expressing bar graph analysis of the CD144 (bar graph) levels were shown. (b) Knockdown of Jagged1 decreased tube-formation ability induced by Twist1 using 2.5D tube-tube-formation ability assays in
Twist1-overexpressing OECM-1 cells with knockdown of Jagged1. Images of tube formation and pixels representing tube formation ability (bar graph) were shown. Scale bar: 50m. (c) Knockdown of Jagged1 decreased the eExpression of various endothelial markers including vWF, TIEie2, ICAM1, and CD105 in Twist1-overexpressing OECM-1 cells. The fold change of the levels of various endothelial markers were shown. (d) Knockdown of Jagged1 decreased the eExpression of
ENFFNB2, and NRP1, and increased the expression of EPHB4 in
Twist1-overexpressing OECM1 cells with knockdown of Jagged1. (e) Inhibition of tumor vessel formation by Jagged1 knockdown Expression of CD144 in GFP-labeled Twist1-overexpressing OECM-1 cells with knockdown of Jagged1. Green: GFP; Red: CD144; Bblue: DAPI. Scale bar: 50m. (f) Abrogation of cCo-expression of Twist1 and CD31 by Jagged1 knockdown in Twist1-overexpressing cells with knockdown of Jagged1. The anti-human-CD31 antibody was used due to its ability to discriminate between human and mouse CD31. Green: hCD31; Rred: Twist1; Bblue: DAPI. Data shown in panels a-d are mean±s.d. of three independent experiments. The asterisk (*) indicates statistical significance (P<0.05 by Student's t-test) between experimental and control clones. The asterisk (*) indicates statistical significance (P<0.05) between experimental and control clones. The OECM1-Twist1 control clone was selected as the control group in panels a-df.
Figure 35. Direct activation of KLF4 expression by the Jagged1/Notch signaling. (a) Western blot to examine the expression levels of Jagged1, BMI1, KLF4, and Twist1 in various stable clones. Actin was used as an internal control. Correlation between Jagged1 and KLF4 expression in Twist1-overexpressing OECM-1 cells. (b) Transient transfection assays were performed using a Jagged1 expression vector with a KLF4 promoter-driven reporter construct harboring either a wild-type or mutated NICD/CSL-responsive site.showed the activation of KLF4 promoter-driven luciferase constructs by the Jagged1 expression vector and identification of a NICD/CSL binding site. The pcDNA3 control transfected lane was selected as the control group vs. the pcDNA3-Jagged1 transfected lane for each reporter construct. The asterisk (*) indicates statistical significance (P<0.05) between experimental and control transfections. (c) Chromatin immunoprecipitation (ChIP) experiments were performed using an IgG or anti-NICD antibody to detect their binding to two regions in the
KLF4 gene promoter. The cell clones of OECM-1 control, OECM1-Twist1, and
OECM1-Twist1 with Jagged1 knockdown were used. qChIP results of NICD binding to the KLF4 promoter regions were also shown. The OECM1 control clone was selected as a control. showed the direct binding of NICD to the NICD/CSL-responsive site located in the KLF4 promoter region. The Hes1 promoter was used as