Discovery of Recurrent Structural Motifs for Approximating
Three-Dimensional Protein Structures
Ta-Tsen Soonga,b( ), Ming-Jing Hwanga* ( ) and Chung-Ming Chenb( ) a
Institute of Biomedical Sciences, Academia Sinica, Taipei, 115, Taiwan, R.O.C. bInstitute of Biomedical Engineering, National Taiwan University, Taipei, 106, Taiwan, R.O.C.
The scope of conformation space that protein molecules can adopt is a problem of significant interest. Previous studies by other groups have shown that there are stereochemical constraints that confine local pro-tein structures to a limited range of conformations. Furthermore, the results of many groups have demon-strated that the sequence-to-structure relationship remains detectable to some extent on a local level. By studying the conformational space of local protein structures, we may obtain more information concerning the constraints on local structural space and the sequence-to-structure mapping, hence facilitate ab initio struc-ture prediction. In this study, we propose a novel algorithm that automatically discovers recurrent pentamer structures in proteins.
The algorithm starts by applying Expectation-Maximization (EM) clustering to the distances between non-adjacent backbone Caatoms in a large set of pentamer fragments. A rough partition of the conformation space can thus be derived. In the second stage, by applying a split-and-merge algorithm, we can obtain a finite number of clusters and guarantee the homogeneity and distinctiveness of each one. Each cluster of protein structures is represented by a centroid structure. The results show that, with 40 major representative tures, we can approximate most of the protein fragments with an error of 0.378 Å. With only 20 types of struc-tures, the fragment structures can still be modeled at 0.44 Å, which is comparable to or better than the perfor-mance of previous methods. We term the representatives “building blocks.” On the global level, we demon-strate that by concatenating different combinations of building blocks, we can model whole protein structures at high resolution: a resolution of 2.54 Å can be achieved simply by using 10 types of building blocks. This finding suggests that the study of molecular structures can be hugely simplified using this reduced representa-tion.
Keywords: Structural approximation; Clustering; Expectation-Maximization (EM); Local structural al-phabets; Protein structure prediction.
INTRODUCTION
Structural genomics is a systematic and large-scale ef-fort towards structural characterization of all proteins. To fa-cilitate this research, it is important to predict the three-dimensional structure from the amino acid sequence. The ex-periments conducted by Anfinsen in the 1950s concluded that a protein’s three-dimensional structure can be determined by its amino acid sequence alone.1Ever since then scientists, in-cluding the authors of this journal,6,12have shown great inter-est in the relationship between sequence and structure, which is the protein folding, or structure prediction problem.
The most difficult case in the protein structure predic-tion problem occurs when the sequence identity between the
target and the template falls below 30%. In this case, protein structures are predicted using ab initio methods. Although these methods still are far from being successful, they have shown promising advances in recent years. For example, a method called Rosetta has demonstrated great potential in CASP contests (Critical Assessment of techniques for pro-tein Structure Prediction).16In some cases, it can predict the basic topology of a protein or domain reasonably well.
Rosetta first characterizes the major types of structural motifs that occur in proteins, calculates different combina-tions of structural motifs for a given sequence using scoring functions and Monte Carlo simulation, and finally pieces to-gether the best set of motifs and generates a whole protein structure.2,5Its success demonstrated the existence of the
se-Dedicated to Professor Sunney I. Chan on the occasion of his 67thbirthday and his retirement from professional life.
quence-structure relationship on the local level and high-lighted the importance of using short structural motifs for protein structure prediction.
The first systematic characterization of short structural motifs was done by Unger et al.22Following the assumption that topological, steric, and chemical features can effectively reduce the space of viable conformation,18they questioned whether the structures of short protein fragments vary contin-uously. Their findings suggest that short protein fragments seem to form specific clusters in the conformation space. They further proposed the idea of using a limited set of stan-dard fragments as structural building blocks to construct the whole protein structure.
Among other followers of this approach were Rooman et al,19Oliva et al,17and Bystroff et al.5The most recent re-search before our work21was done by Micheletti et al.14Later on Kolodny and coworkers applied similar approaches and acquired comparable results.11
In this paper, we present a clustering algorithm for dis-covering recurrent structural motifs for accurate representa-tion of protein structures. As a proof of principle, we here demonstrate the procedure and results for discovering pen-tamer motifs. The extension of this algorithm to discover 7-mer motif libraries with different compactness and sizes will be presented in detail in Lee et al.13In general, our algorithm is capable of finding a small set of motifs that cover most of the conformation space of local protein structures. By using this set of motifs, we can represent three-dimensional whole structures by concatenating consecutive motifs. This serial representation of protein structures, which we call “structural alphabet representation,” is a simplified one-dimensional string of structural motifs and can hugely reduce computa-tional cost while maintaining structural information. Our pre-liminary results also suggest that we may treat this reduced representation as a variant of the lattice model for generating decoys when predicting protein structures (Soong and Hwang, unpublished data).
METHODS AND MATERIALS Rationale
Clustering algorithms are a widely used approach to finding recurrent local structures of proteins. Some of the typical clustering approaches include mode-seeking tech-niques,14,17K-nearest-neighbor,22K-means,11hyper-cosine,10 and hierarchical agglomerative algorithms.19A sequence-based clustering algorithm complemented with iterative
structural refinement also came into play.5
In addition to the previous approaches, one effective and popular clustering method is the Expectation-Maximiz-ation (EM) algorithm,7which has sound statistical founda-tion and is more robust than other methods.4,15However, the clustering features used by previous approaches such as discretized torsion (Ramachandran) angles, embracing sec-ondary structure distances, and curvature are generally diffi-cult to be utilized by the EM algorithm. One way around this obstacle while maintaining the benefits of the EM algorithm is to employ the distance matrix as the clustering feature.
As proposed by the popular structure comparison pro-gram DALI,9the distance matrix, which is composed of all pairwise distances between backbone Caatoms, is a good fea-ture for evaluating similarity between proteins. Similar struc-tures have similar Ca-Cadistances and can thus be clustered using the EM algorithm. We hereby develop an algorithm that discovers the major partitions of the conformation space based on the EM algorithm.
Protein database
The set of proteins for deriving the structural alphabets was chosen randomly from the non-redundant pdb_select_25 list which was generated by the algorithm of Hobohm et al.8 as of March 2001. Among the set of proteins, 1,059 chains were chosen for training and the remaining 405 chains were used for testing. The training set proteins were cut into over-lapping pentamers which add up to 136,800 fragments. The spatial coordinates were downloaded from the Protein Data Bank (PDB).3
Structural motif discovery algorithms
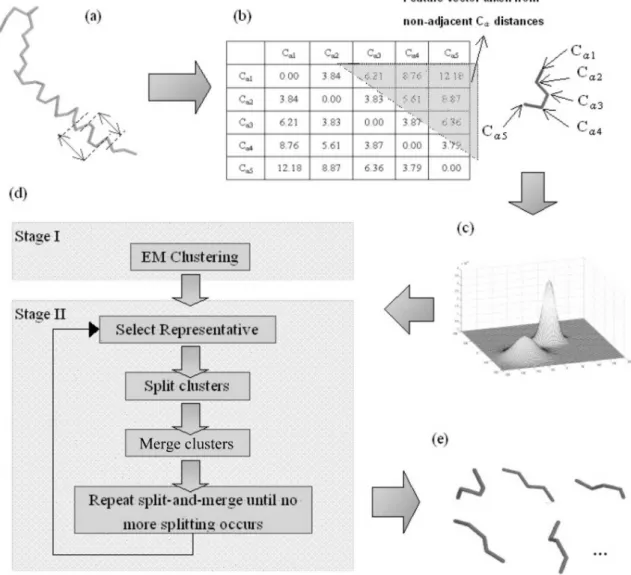
The first crucial step is to generate a set of structural motifs for sufficient coverage of the conformation space of local protein structures. Here we adopt a two-stage algorithm that first partitions the conformation space by an Expecta-tion-Maximization (EM) algorithm, and then refines the re-sults by a split-and-merge procedure (see Fig. 1).
For EM clustering in the first stage, we transform the fragment coordinates into feature vectors. In each fragment there are six intra-molecular distances between non-adjacent backbone Caatoms. By stacking the six distances into a vec-tor, we can then treat the fragments as six-dimensional data which we cluster using the EM algorithm.
The second stage of the algorithm is split-and-merge, which refines the clusters obtained by the EM algorithm. The split-and-merge procedure is as follows:
distance between fragments fAand fB, cutoff as the threshold
below which two fragments are considered similar. Denote ri
as the representative of cluster ci, C = {c1, c2, c3,…, cN} as the
set of all clusters, and |C| as the total number of clusters for the iteration. The cutoff value is set to 0.65 Å according to Micheletti et al.14
(2) Select representatives: Within every cluster ciin C,
select fragment fj as the representative of ci such that
u dist fj fk cutoff fj fl ci
( ( , )
-¹ Î
å
) is maximized, where u is the unit step function.
(3) Split: For every fragment fjof cluster ci, if dist(ri, fj)
> cutoff, then put fjin cmsuch that dist(rm, fj) is minimized and
dist(rm, fj) < cutoff, otherwise put fjin a new cluster ck, k = |C| + 1.
(4) Merge: For every pair of clusters ciand cjin C, if
dist(ri, rj) < cutoff, merge ciand cjinto cluster ck, k = |C| + 1. Then
remove clusters ciand cj.
(5) Repeat steps (2) through (4) until no more clusters can be split and merged.
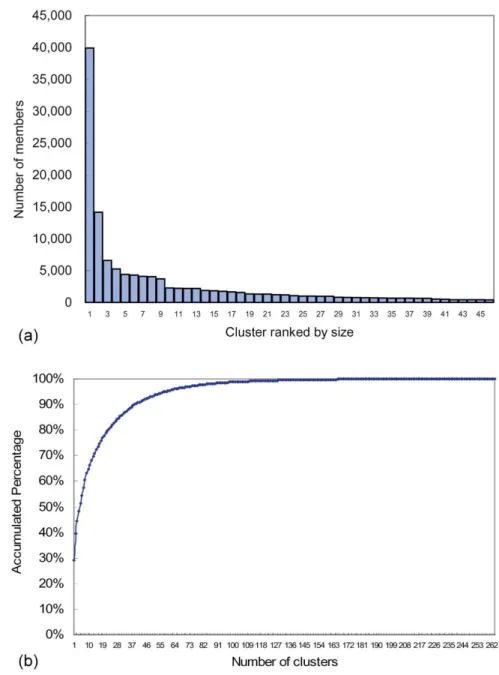
The algorithms return a set of structural motifs, most of which are trivial and contain a very small proportion of the database (see Fig. 2). The set shows that a small number of representative motifs can cover most of the conformation space, as approximated by our cluster representatives. As
Fig. 1. The procedure of structural alphabet discovery. (a) Protein structures are cut into overlapping 5-mer fragments. (b) For each protein fragment, the distances between non-adjacent backbone Caatoms are used as a feature vector for clustering. (c) The different protein fragments are now treated as points in a high-dimensional space. (d) The protein structures are clustered using the Expectation-Maximization algorithm and refined by the split-and-merge algorithm. (e) The algorithms converge and generate a representative set of protein structures. The top 5 representatives are shown here and correspond with secondary structure elements.
each such motif can be labeled with an alphabet, we also call these motifs “structural alphabets.”
RESULTS AND DISCUSSION Structural alphabets
For each number of clusters obtained by the EM
algo-rithm, we calculated the quality of the clustering according to the Bayesian Information Criteria (BIC).20As the BIC scores began to saturate from 40 clusters, we chose to use these 40 clusters for refinement using the split-and-merge algorithm. In total, 262 clusters of various sizes were found.
Fig. 2(a) shows the number of fragments each cluster contains. The largest cluster, which corresponds to ana he-lix, contains more than 40,000 fragments, equal to about 30
Fig. 2. (a) Size distribution of representative clusters. The clusters are ranked according to size. Most of them contain very few fragments, so only those with more than 400 members are shown for clarity. The largest cluster, which corre-sponds to thea helix, contains over 40,000 fragments. The following three clusters, which are detailed representa-tions of secondary structures, also contain large numbers of fragments. (b) The percentage of database that different numbers of clusters can cover. The top 5 clusters cover more than half of the database; top 20 clusters for 80 percent; and top 40 for 90 percent of the database.
percent of the database. The following three clusters, each containing more than 5,000 fragments, are detailed descrip-tions ofb strands after closer inspection. Fig. 2(b) illustrates the proportion of the database that different numbers of clus-ters can cover. The most populated 40 clusclus-ters cover up to 90 percent of the data set and 20 clusters for about 80 percent, while with only the top five clusters, more than 50 percent of the database can be represented. The percent coverage of the top five clusters, which consist of detaileda-helices and b strands, conforms with the common knowledge that
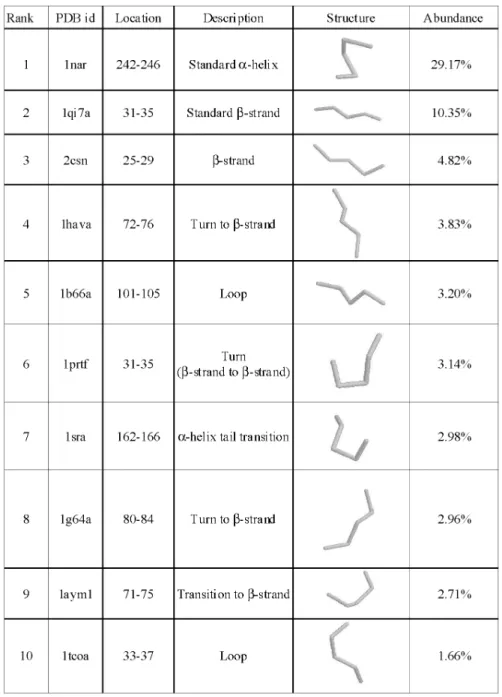
second-ary structures occur in about half of the protein structures. We present the fragment structures of the top 10 clus-ters in Fig. 3 along with their description and abundance level in the database for brevity. The coordinates of the structures in our fragment library can be obtained at http://gln.ibms. sinica.edu.tw/jccs. Beside the regular secondary structures described above, specific types of turns and loops also occur frequently in proteins. And some of the fragment structures mark the transition between secondary structures and loops (see Fig. 3).
Local-fit approximations
To demonstrate the ability of using a small set of struc-tural alphabets to represent the conformation space of local protein structures, we used different numbers of structural motifs to approximate protein fragments in the test set data-base. The results are compared with those of Micheletti and Kolodny in Table 1. By using the top 40 representatives, we can fit the test set at 0.38 Å and the top 20 at 0.43 Å. The data in Table 1 show that our set of structural motifs can represent the conformation space with high accuracy and is comparable with or better than other methods.
Global-fit approximations
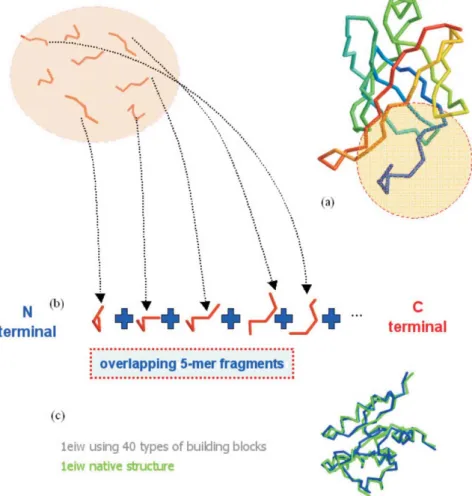
We also fit the whole native protein structures with the local structural motifs to assess the feasibility of using the structural representatives as a simplified representation. Here we used a brute-force algorithm similar to that used by
previ-ous studies.11,14,22The procedure is shown in Figs. 4(a) and 4(b). An illustration of the global-fit approximation is shown
Table 1. Performance of clustering algorithms in rmsd
Algorithm # Representatives rmsd (Å) (Local) rmsd (Å) (Global) Micheletti et al* 40 0.44 1.64 40 0.40 1.41 30 0.43 1.59 20 0.47 1.85 Kolodny et al** 10 0.57 2.57 40 0.38 1.17 30 0.40 1.25 20 0.43 1.53 EM + split-and-merge 10 0.54 2.81
* Calculated with the libraries published by Micheletti et al.14
** Data are provided as in the paper of Kolodny et al.11
Fig. 4. Global-fit protein structure approximation. (a) The native protein structure to be approximated. (b) Starting with the first 5-mer fragment at the N-terminal, select a structural motif from the structural library that best resembles the na-tive fragment. Then extend the fragment by adding the structural motifs that make the concatenated structure best ap-proximate the corresponding parts of the native structure. (c) An example of fitting pdb1eiw with 40 types of building blocks.
in Fig. 4(c), with the rmsds in approximating the global pro-tein structures given in Table 1. A resolution of 2.81 Å can be obtained by using only 10 types of structural motifs and 1.53 Å using 20 types. The results suggest that the structural al-phabets can also be used as an alternative lattice model for generating decoys when predicting protein structures. Comparison with other fragment libraries
As detailed elsewhere,13we have also conducted thor-ough research on the performance of this algorithm in cluster-ing 5 and 7-mer motif. In general, this algorithm generated sets of structural representatives that performed slightly better than others’ methods in terms of protein 3D structure approximation. Furthermore, our studies showed that our fragment library can better reduce approximation errors inb structures than do other libraries such as that of Hunter and Subramaniam.10
A brief description of the clustering procedures of vari-ous groups is provided in Table 2. In comparison with other groups, our method could handle a much larger training set for deriving the structural motifs and thus could obtain a wider coverage of local conformation space. Our algorithm also employs the distance matrix-based measure to take ad-vantage of the statistical robustness of the EM algorithm. In combination with the root-mean-square deviation (rmsd) measure in the split-and-merge procedure, we can derive a good fragment library.
CONCLUSION
In this study we developed a method for finding recur-rent local protein structures. The algorithm starts with EM
clustering to provide a rough partition of the conformation space. Then the clusters are subjected to the split-and-merge algorithm for refinement. The results demonstrate that with a small set of representative structures, proteins can be approx-imated with high resolution.
In comparison with the work of Micheletti et al.14and of Kolodny et al.,11our structural motifs provide a more par-simonious representation in covering the conformation space. Each of the representative structural motifs discovered by our method demonstrates high homogeneity within its cluster and high discriminability between other motifs. The results suggest that we may approximate structures at atomic resolution using less than 20 types of motifs.
From each of the structural motifs, we have compiled a position-specific profile composed of the frequencies of amino acids making up the motif structures. Preliminary ex-amination of the profiles indicates that many unique amino acid patterns exist and they may help investigate the se-quence-structure relationship. We have also been developing algorithms to analyze the information embedded in the struc-tural motifs. By unveiling the relationship, we could further map local sequences onto corresponding motifs and predict protein structures ab initio.
ACKNOWLEDGEMENT
This work was supported by Academia Sinica’s the-matic program project. We are indebted to Professor Sunney Chan’s unceasing support and encouragement.
Received March 23, 2004.
Table 2. Training set size, clustering method, and similarity measure of various methods
Work TS Description Similarity measure
Unger et al.22 4 Variant K-nearest neighbor clustering
with a subcluster refinement procedure
rmsd
Rooman et al.19 75 Hierarchical ascending rmsd
Micheletti et al.14 75 Greedy density-seeking rmsd
Kolodny et al.11 200 Simulated annealing K-means with
split-and-merge
rmsd
Hunter et al.10 790 Hypercosine clustering Hypercosine value (inner
product of coordinate vectors of two fragment vectors)
This research21 1,059 EM + split-and-merge Distance matrix + rmsd
REFERENCES
1. Anfinsen, C. Science 1973, 181, 223. 2. Baker, D.; Sali, A. Science 2001, 294, 93.
3. Berman, H. M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T. N.; Weissig, H.; Shindyalov, I. N.; Bourne, P. E. Nucleic
Acids Research 2000, 28, 235.
4. Bradley, P.; Fayyad, U.; Reina, C. Research Technical
Report, MSR-TR-98-35 1999.
5. Bystroff, C.; Baker, D. Journal of Molecular Biology 1998,
281, 565.
6. Chan, C. H.; Lyu, P. C.; Hwang, J. K. Journal of the Chinese
Chemical Society 2003, 50, 677.
7. Dempster, A. P.; Laird, N. M.; Rubin, D. B. Journal of the
Royal Statistical Society: Series B 1977, 39, 1.
8. Hobohm, U.; Scharf, M.; Schneider, R.; Sander, C. Protein
Science 1993, 1, 409.
9. Holm, L.; Sander, C. Trends in Biochemical Sciences 1995,
20, 478.
10. Hunter, C.; Subramaniam, S. Proteins: Structure, Function,
and Genetics 2003, 50, 580.
11. Kolodny, R.; Koehl, P.; Guibas, L.; Levitt, M. Journal of
Molecular Biology 2002, 323, 297.
12. Kumar, T. K. S.; Sivaraman, T.; Samuel, D.; Sriailam, S.;
Ganesh, G.; Hsieh, H.-C.; Hung, K. W.; Peng, H. J.; Ho, M. C.; Arunkumar, A. I.; Yu, C. Journal of the Chinese
Chemi-cal Society 2000, 47, 1009.
13. Lee, I. Y.; Soong, T. T.; Ho, J. M.; Hwang, M. J. IEEE Fourth
Symposium on Bioinformatics and Bioengineering (BIBE2004)
2004, pp 516-521.
14. Micheletti, C.; Seno, F.; Martin, A. Proteins: Structure,
Function, and Genetics 2000, 40, 662.
15. Moore, A. Advances in Neural Information Processing
Sys-tems 1999, April, 543.
16. Moult, J.; Hubbard, T.; Fidelis, K.; Pedersen, J. Proteins
1999, 37 (Supplement 3), 2.
17. Oliva, B.; Bates, P. A.; Querol, E.; Aviles, F. X.; Sternberg, M. J. E. Journal of Molecular Biology 1997, 266, 814. 18. Ramachandran, G. N.; Sasisekharan, V. Advances in Protein
Chemistry 1968, 23, 283.
19. Rooman, M.; Rodriguez, J.; Wodak, S. Journal of Molecular
Biology 1990, 213, 327.
20. Schwarz, G. The Annals of Statistics 1978, 6, 461.
21. Soong, T.-T. M. S. Thesis, National Taiwan University, June 2002.
22. Unger, R.; Harel, D.; Wherland, S.; Sussman, J. L. Proteins