Theoretical Electronic Structure and Properties of Alternating
Fluorene
YAcceptor Conjugated Copolymers and Their
Model Compounds
Wen-Chung Wu
1and Wen-Chang Chen
1,2,*
1Department of Chemical Engineering, National Taiwan University, Taipei 106, Taiwan
2
Institute of Polymer Science and Engineering, National Taiwan University, Taipei 106, Taiwan
(*Author for correspondence; Tel.: +886-2-23628398; Fax: +886-2-23623040; E-mail: [email protected])
Received 16 February 2006; accepted in revised form 12 May 2006; published online 21 June 2006
Key words: conjugated polymers, fluoreneYacceptor, electronic properties, geometry, intramolecular charge transfer
Abstract
The theoretical geometries and electronic properties of fluorene (F) based alternating donorYacceptor conjugated copolymers and their model compounds were studied by the density function theory (DFT) at the B3LYP level with 6Y31G or 6Y31G** basis set. The acceptors investigated in this study include thiazole (TZ), thiadiazole (TD), thienopyrazine (TP), thienothiadiazole (TT), thiadiazolothienopyrazine (TTP), quinoxaline (Q), benzothiadiazole (BT), pyrazinoquinoxaline (PQ), thiadiazoloquinoxaline (TQ), and benzobisthiathiadiazole (BB). The torsional angle, bridge bond length, bond length alternation, and intramolecular charge transfer were simulated and correlated with the electronic properties, i.e., HOMO, LUMO level, and band gap. The geometries of fluorene-based donorYacceptor alternating copolymers and their model compounds are significantly affected by the structure of acceptors, particularly the ring size on the backbone. The electronic properties of the polymers and their model compounds are well correlated with the acceptor strength, coplanarity of the backbone, and intramolecular charge transfer. The theoretical study suggests that the electronic properties of alternating fluoreneYacceptor conjugated copolymers could be tuned by the geometries or acceptor strength. Hence, these proposed copolymers could have potential applications as light-emitting diodes (LEDs), transparent conductor, or photovoltaic devices.
Introduction
Conjugated polymers have been extensively studied for their potential applications in electroluminescence displays [1], photovoltaic devices [2], and thin film transistors [3]. They provide the advantages of tunable physical proper-ties, processibility, flexibility, and cost over the traditional inorganic semiconductors. We are particularly interested in the alternating donorYacceptor conjugated copolymers since their electronic and optoelectronic properties can be tuned efficiently by intramolecular charge transfer (ICT) for various applications [4,5].
Polyfluorenes (PFs) have been considered as potential materials for polymer light-emitting diodes (PLEDs) among many conjugated polymers because of their pro-cessibility, high quantum yield, good charge transport properties, and good thermal and oxidative stabilities [6,
7]. Recently, donorYacceptor conjugated copolymers based on fluorene with various acceptors were reported in the literature, and the light-emitting color or efficiency of fluoreneYacceptor copolymers were easily tuned to blue, green or red by the acceptor strength, as demonstrated by us or others [8Y10]. Besides, photovoltaic devices with good efficiencies was demonstrated by
poly(9,9-dioctyl-fluorene-alt-benzothiadiazole) (F8BT) [11]. High electron mobility from the F8BT-based field effect transistor was also reported recently [12]. The important optoelectronic properties of polyfluorenes also stimulate theoretical analysis on the geometry and electronic properties [13]. However, a systematic study on the effects of different acceptor structures on the electronic structures of poly-fluorenes has not been fully explored yet. A comprehensive understanding of the electronic structures and the electron-ic properties of fluorene-based alternating donorYacceptor copolymers would be helpful for their future applications.
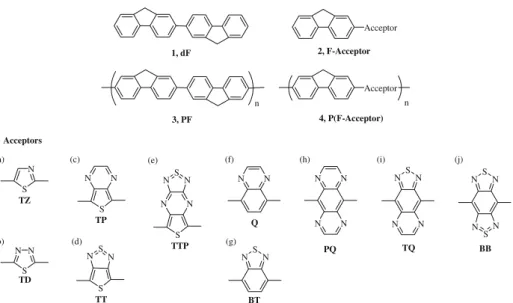
In this paper, a theoretical study of the optimized geometries and electronic structures of fluorene-based alternating donorYacceptor copolymers and their model compounds is reported, whose molecular structure are shown in Figure 1. The 10 electron acceptors investigated can be grouped into two categories based on the ring structure on the backbone: (1) Five-member ring: Thiazole (TZ), thiadiazole (TD), thienopyrazine (TP), thienothiadia-zole (TT), and thiadiazolothienopyrazine (TTP); and (2) six-member ring: Quinoxaline (Q), benzothiadiazole (BT), pyrazinoquinoxaline (PQ), thiadiazoloquinoxaline (TQ), and benzobisthiadiazole (BB). The model compounds and their corresponding polymers are named as dF (1),
F-acceptor (2), PF (3), and P(F-F-acceptor) (4), respectively. The series of 10 acceptors provide a basis for a compre-hensive understanding of the effects of the backbone ring, heteroatom, and fused rings on the geometry and electronic properties of the model compounds and copolymers. The electronic structure and properties were investigated by the density functional theory (DFT) at the B3LYP level and 6Y31G or 6Y31G** basis set. The bond length of the bridge, bond length alternation, and intramolecular charge transfer of the materials were analyzed and correlated with their chemical structures. The effects of the acceptor strength on the electronic properties, including the HOMO level, LUMO level, and band gap, were also studied.
Theoretical analysis
Methodology
The ground-state geometries and electronic structures of the studied fluorene-based donorYacceptor alternating copoly-mers and their model compounds were optimized by means of the hybrid density functional theory (DFT) method at the B3LYP level of theory (Becke-style 3-Parameter Density Functional Theory [14] using the LeeYYangYParr correla-tion funccorrela-tional [15]) with the 6Y31G and 6Y31G** basis set performed on Gaussion03 program package [16]. This method B3LYP//B3LYP (started with B3LYP geometry optimization followed by B3LYP electronic structure calculation) is more reliable than other methodology when applied to the system where the equilibrium geometries deviate substantially from planar structures [17]. In the case of the model compounds, both basis set (6Y31G and 6Y31G**) were used. Similar theoretical results were observed from these two basis set. We note that the 6Y31G** basis set considers the orbital polarization whereas the 6Y31G does not. Hence, only the DFT/B3LYP/6Y31G was used to analyze the copolymers since the DFT/B3LYP/ 6Y31G** would be time-consuming. Adjacent repeating
units of the alternating fluoreneYacceptor copolymers were arranged in a fashion so that each point in opposite direction without further considering single-bond torsional potentials by ab initio method [18].
In this study, the starting unit cell geometries were taken from the central portion (two repeating units) of the cor-responding polymers, and then fully optimized to the equilibrium geometries inside a given lattice length on the constraints of periodic boundary conditions by assuming that the unit cell is repeated identically an infinite number of times along the translation vector. Both the lattice parameter and molecular structures were varied to locate the lowest energy position in the unit cell. Band structures were calculated along the k vector of these one-dimensional polymers with 30 k points after the optimization using the same level of theory. The lowest 4 unoccupied and highest 4 occupied bands in the first Brillouin zone were plotted. All the computed polymer orbital energies were directly used without further shifting or scaling.
Geometrical parameters and electronic properties analyzed
The definitions of torsional angle (8), bond length alternation of donor (dD), bridge length (LB), and
intramo-lecular charge transfer (DCT) of fluorene based alternating
donorYacceptor conjugated copolymers and their model compounds are shown in Figure 2 by using compound FYPQ as an example. Torsional angle, 8, is the deviation from coplanarity between donor and acceptor, as shown in Figure 2a. Bond length alternation (BLA) is the bond length difference between single and double bonds, and represents a major contribution to the existence of a finite band gap in conjugated polymers. Since there is no obvious definition for the complicated structures of the model compounds, it might be feasible to just investigate the BLA of the invariable fluorene donor, which is defined as dD. As
shown in Figure2a, the dD is calculated as the difference Acceptor Acceptor n n 1, dF 2, F-Acceptor 3, PF 4, P(F-Acceptor) Acceptors N S N N S N S N N N S N N S S N N N N N N N S N N N N S N N S N TZ TQ BB TD TP Q TT BT PQ N N S TTP N S N (a) (b) (c) (e) (f) (d) (g) (h) (i) (j)
between the average bond length of C2YC3, C4YC11,
C5YC12, and C6YC7 and the average bond length of
C3YC4, C11YC12, and C5YC6. Bridge length, LB, is the bond
length between donor and acceptor, as shown in the C7YC14
of Figure2a. Intramolecular charge transfer, DCT, was
cal-culated as the summation of Mulliken charge distribution of donor or acceptor. For example, the DCT of FYPQ is a
summation of all charges for the fluorene ring (j0.163 j 0.123 j 0.141 j 0.121 j 0.144 j 0.101 + 0.079 j 0.116 j 0.372 + 0.045 + 0.029 + 0.043 + 0.021 + 0.123 + 0.121 + 0.122 + 0.129 + 0.128 + 0.130 + 0.131 + 0.168 + 0.169 = 0.157), as shown in Figure2b.
Band structures were calculated along the k vector of these one-dimensional polymers with 30 k points in the first Brillouin zone. The lowest 4 unoccupied and highest 4 occupied bands in the first Brillouin zone were plotted. The energy levels of the HOMO and LUMO were determined
from the maximum point of the highest occupied molecular orbital and the minimum point of the lowest unoccupied molecular orbital, respectively. The band gap was then obtained from the minimum difference between the HOMO and LUMO energy levels at a constant k. The bandwidth was calculated from the spread of energy levels available to carriers within each band. The effective mass of carriers at the band edge representing mobility was obtained as the square of multiplied by the reciprocal of the curvature from E(k) with k.
Results and discussions
Justification of the theoretical analysis
Our theoretical calculation shows that the optimized geometry of dF is almost identical with that reported in
the literature [13]. For example, the bond length, LC11YC12,
of dF is 1.470 and 1.468 A˚ from the 6Y31G and 6Y31G** basis, respectively, which is almost identical with that calculated by time-dependent density functional theory (TD-DFT) and ZINDO[13]. The HOMO level of the dF with the methyl groups at C9 position estimated by cyclic voltammetry was at j5.2 eV [19], which was very close to the theoretical values of j5.38 and j5.35 eV from 6Y31G and 6Y31G** basis in the present study, respectively. The comparisons suggest that the accuracy of the calculated geometry and electronic structure in this theoretical study is fairly good.
In the reported polyfluorene copolymers, alkyl groups, such as hexyl or octyl groups, were attached to the C9 position of fluorene to improve the solubility. However, such alkyl substitution results in time-consuming for the theoretical calculation. Thus, we compared the optimized geometries and electronic structures of the PF without the alkyl substitution and poly(9,90-dihexyl-2,7-fluorene) at the DFT/B3LYP/6Y31G level to rationalize the effect of alkyl groups on the electronic structure. The calculated electronic structures, (HOMO, LUMO, Eg) in eV, of the PF and
poly(9,90-dihexyl-2,7-fluorene) are (j5.05, j1.58, 3.47) and (j4.99, j1.55, 3.44), respectively. The almost identical electronic properties of the PF and poly(9,90 -dihexyl-2,7-fluorene) suggest that the alkyl groups at the C9 position makes no significant difference in theoretical analysis.
We note that the DFT/B3LYP method was utilized to calculate the optimized geometries and electronic struc-tures of fluorene-based donorYacceptor alternating copoly-mers in ground state in this study. Although the optical band gap estimated from the UVYVisible spectra is not exactly the same as the energy gap in the ground state, they are well-correlated in conjugated polymer systems. Hence, the optical band gap could provide the examination on the accuracy of the obtained theoretical electronic properties.
Optimized geometries and electronic properties of model compounds
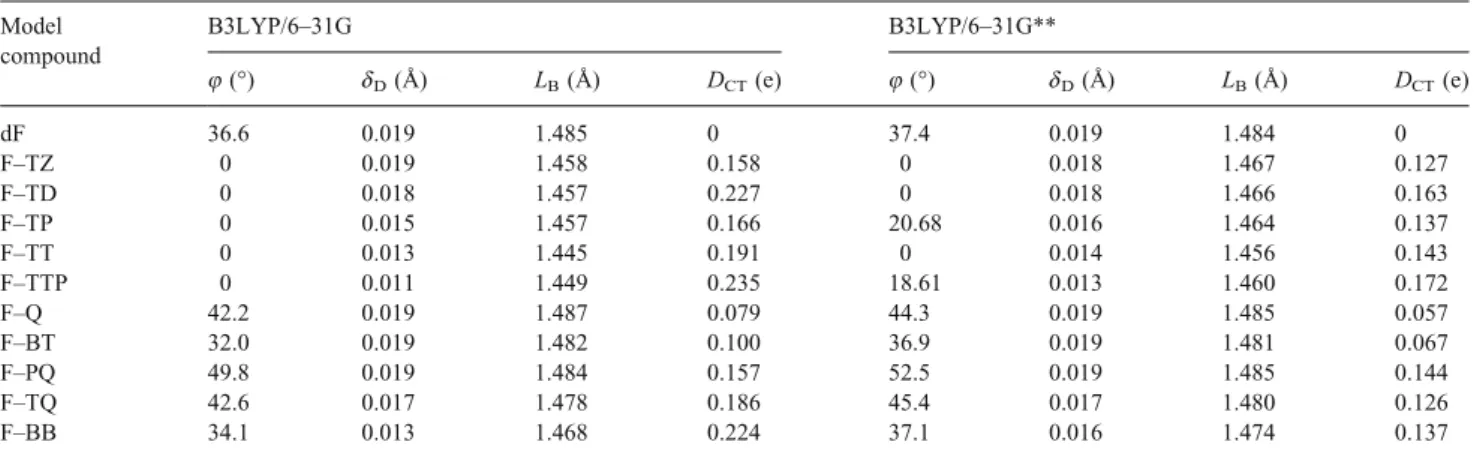
The torsional angle (8), bond length alternation of donor (dD), bridge length (LB), and intramolecular charge transfer
(DCT) of the model compounds obtained with the 6Y31G
and 6Y31G** basis sets are listed in Table1. The trend of geometries obtained from the 6Y31G and 6Y31G** is similar, as shown in the table.
The optimized geometries of the model compounds with five-member ring acceptors (2aY2e) obtained from both 6Y31G and 6Y31G** basis sets suggest that they have a coplanar conformation, as shown in torsional angles of Table 1. Note that the LUMO levels of the acceptors, TZ, TD, TP, TT, and TTP, obtained from the 6Y31G basis set are j1.05, j1.67, j2.52, j3.42, and j4.14 eV, respec-tively, which suggests that the acceptor strength is in the order of TZG TD G TP G TT G TTP. Both the trend of dD
and that of LBobtained from the 6Y31G and 6Y31G** basis
sets are in the reverse as that of the acceptor strength, except for the LBof FYTT. This exception is probably due
to the possible mesomeric structure, DYA @Y D+=Aj, induced by intramolecular charge transfer. The order on the intramolecular charge transfer, DCT, obtained from the
Mulliken charge distribution with 6Y31G and 6Y31G** basis sets is FYTTP 9 FYTD 9 FYTT 9 FYTP 9 FYTZ, which is similar to that of the acceptor strength except for the FYTD. The unusually large DCT of the FYTD is
probably resulted from the two adjacent imine nitrogen atoms in the thiadiazole ring on the backbone. The intramolecular charge transfer significantly enhances the
p-electron delocalization and thus decreases the bond length alternation of the model compounds. The optimized geometries suggest that the dD, LB, and DCT can be
controlled by the acceptor strength in the coplanar fluoreneYacceptor model compounds. The trends of dD,
LB, and DCT obtained from the 6Y31G and 6Y31G** basis
sets are the same for the model compounds with five-member ring acceptor.
The twisted conformation of the model compounds with six-member ring acceptors (2fY2j) suggests that strong steric hindrance exists between the fluorene and acceptor moieties, as shown in the large torsional angles of Table1. The order of the torsional angels, 8, obtained from 6Y31G and 6Y31G** basis sets is FYPQ 9 FYTQ 9 FYQ 9 FYBB 9 FYBT. The large value of 8 and its variation among these acceptors result from the difference in acceptor structure, including the different size and number of fused rings. The above order suggests that the torsional angles of the model
Table 1. The optimized geometries of model compounds. Model
compound
B3LYP/6Y31G B3LYP/6Y31G**
8 (-) dD(A˚ ) LB(A˚ ) DCT(e) 8 (-) dD(A˚ ) LB(A˚ ) DCT(e) dF 36.6 0.019 1.485 0 37.4 0.019 1.484 0 FYTZ 0 0.019 1.458 0.158 0 0.018 1.467 0.127 FYTD 0 0.018 1.457 0.227 0 0.018 1.466 0.163 FYTP 0 0.015 1.457 0.166 20.68 0.016 1.464 0.137 FYTT 0 0.013 1.445 0.191 0 0.014 1.456 0.143 FYTTP 0 0.011 1.449 0.235 18.61 0.013 1.460 0.172 FYQ 42.2 0.019 1.487 0.079 44.3 0.019 1.485 0.057 FYBT 32.0 0.019 1.482 0.100 36.9 0.019 1.481 0.067 FYPQ 49.8 0.019 1.484 0.157 52.5 0.019 1.485 0.144 FYTQ 42.6 0.017 1.478 0.186 45.4 0.017 1.480 0.126 FYBB 34.1 0.013 1.468 0.224 37.1 0.016 1.474 0.137
compounds with acceptors fused with the pyrazine ring are larger than those fused with the thiadiazole ring, and that those with a tricyclic ring are larger than those with a bicyclic ring. The model compounds with five-member ring acceptors are more planar than those with six-member ring acceptors due to the small size of five-member ring and the lack of possible HYH repulsion. Since the LUMO level of the Q, BT, PQ, TQ, and BB obtained from 6Y31G basis set are j2.03, j2.80, j2.98, j3.65, and j4.32 eV, respectively, the order of the acceptor strength is suggested to be BB 9 TQ 9 PQ 9 BT 9 Q. Both the dD and LB
obtained from the 6Y31G and 6Y31G** basis sets decrease as the acceptor strength increases except for the LB of the
FYBT. This is probably due to the smallest torsional angle of the FYBT among these model compounds. The longer LB of 2fY2j than that of 2aY2e indicates that single bond
character is required for the model compound with a larger torsional angle to rotate. The trend of intramolecular charge transfer is exactly the same as that of the acceptor strength. The above results also suggest that dD, LB, and DCT are
controlled by the acceptor strength in fluoreneYacceptor model compounds with six-member ring acceptors.
Figure 3 shows the electronic structures of the model compounds calculated from the 6Y31G basis set. The HOMO, LUMO, and energy band gap (Eg) from both
6Y31G and 6Y31G** are summarized in Table 2. The HOMO, LUMO, and Eg calculated from the 6Y31G and
6Y31G** basis sets show a similar trend among these 10 model compounds, and those based on the former basis set are smaller.
As shown in Figure3a, the HOMO energy level of mod-el compounds (2aY2e) is in the order of FYTT 9 FYTP 9 FYTTP 9 FYTZ 9 FYTD, which is in consistence with the order of HOMO energy level of acceptors from 6Y31G: TT (j6.03 eV) 9 TP (j6.41 eV) 9 TTP (j6.72 eV) 9 TZ (j7.14 eV) 9 TD (j7.67 eV). The LUMO energy level decreases as the acceptor strength increases. The above results suggest that the electronic structures of coplanar fluoreneYacceptor model compounds are controlled by the electronic structures of acceptors. The much lower Egof the
-9 -8 -7 -6 -5 -4 -3 -2 -1 0 -5.44 -4.15 -5.24 -5.40 -6.04 -3.35 -2.60 -2.01 -5.71 -1.69 -5.38 En er g y (eV ) -1.17 LU MO HOM O dF N S F-TZ N N S F-TD N N S F-TP NSN S F-TT N N S N S N F-TTP -9 -8 -7 -6 -5 -4 -3 -2 -1 0 -5.63 -5.70 -4.02 -3.49 -5.52 -2.90 -5.66 -2.75 -2.06 -5.61 -5.38 -1.17 HOM O LUMO En er g y ( eV ) dF N N F-Q N S N F-BT N N N N F-PQ N S N N N F-TQ N S N N SN F-BB
a
b
Figure 3. HOMO and LUMO energy levels (unit: eV)of the model compounds by DFT/B3LYP/6Y31G basis set (a) acceptors with five-member ring, and (b) six-member ring, respectively.
FYTP, FYTT, and FYTTP compared to that of dF implies significant effects of the low-lying LUMO energy level of acceptor and intramolecular charge transfer between fluorene and acceptor on the electronic structure. However, the Eg values of the FYTZ and FYTD are only a little
smaller than that of the dF. The lowering of the LUMO level by the incorporation of these two acceptors is compensated by the lowering of the HOMO level. This result is probably resulted from the nitrogen atoms on the backbone which break the symmetry of the structures [20], thereby increasing the bond length alternation with consequent widening of the band gap of these two model compounds.
The HOMO energy level of model compounds (2fY2j) shown in Figure 3b is almost the same in the range of j5.52 to j5.70 eV. The LUMO energy level decreases in the order of FYQ 9 FYBT 9 FYPQ 9 FYTQ 9 FYBB, which is in consistence with the acceptor strength. The Eg of
model compounds (2fY2j) is in the range of 1.50 to 3.57 eV, which is significantly lower than that of the dF. This decrement of band gap also suggests that the low-lying LUMO level of the acceptor and intramolecular charge transfer are dominant effects in the electronic structures. The theoretical electronic properties of the F-benzene (HOMO, LUMO, and Eg) calculated from the 6Y31G basis
set are j5.59, j1.01, and 4.58 eV, respectively. By comparing the electronic structures of the F-benzene,
FYQ, and FYBT, we find that the LUMO is lowered by 1.05 and 1.74 eV as the benzene ring is fused with the pyrazine and thiadiazole rings, respectively. Furthermore, the correlation between the Egand the chemical structures
of these model compounds also suggests that the thiadia-zole ring is more effective in lowing Egthan the pyrazine
ring. In addition to the consideration of the acceptor strength, the lower LUMO energy level of the model compounds containing thiadiazole ring than those contain-ing pyrazine rcontain-ing is probably due to larger torsional angle in the later.
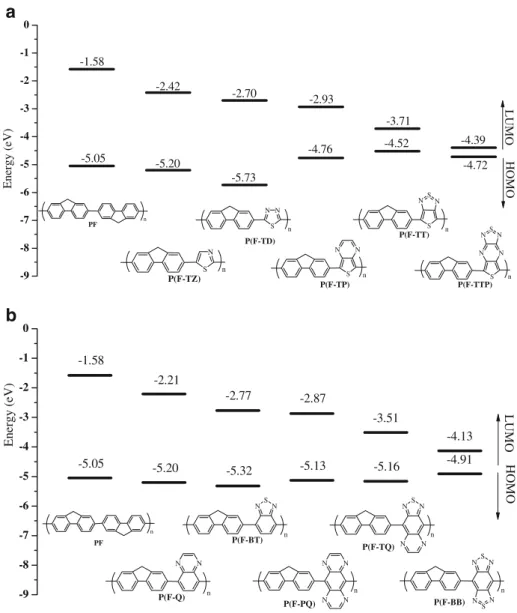
Optimized geometries and electronic properties of fluoreneYacceptor copolymers
Table 3 lists the optimized geometrical and intramolec-ular charge transfer parameters of the alternating fluo-reneYacceptor copolymers at the DFT/B3LYP/6Y31G level. The zero torsional angles of the copolymers with a five-member ring acceptor (4aY4e) suggest that these copolymers are coplanar, except for the P(FYTP). This deviation from coplanar conformation is probably due to the HYH repulsion between the fluorene and TP seg-ments. The order on dDand LB of these polymers shown
in Table 3 is similar with their corresponding model compounds. Note that the smaller LB of these polymers
Table 3. The optimized geometries and the theoretical electronic properties of fluoreneYacceptor copolymers.
Polymer 8 (-) dD(A˚ ) LB(A˚ ) DCT(e) HOMO (eV) LUMO (eV) Eg(eV)
PF 35.2 0.015 1.485 0 j5.05 j1.58 3.47 P(FYTZ) 0 0.011 1.457 0.306 j5.20 j2.42 2.78 P(FYTD) 0 0.010 1.456 0.412 j5.73 j2.70 3.03 P(FYTP) 14.7 0.010 1.455 0.268 j4.76 j2.93 1.83 P(FYTT) 0 0.001 1.438 0.307 j4.52 j3.71 0.81 P(FYTTP) 0 0.002 1.443 0.360 j4.72 j4.39 0.33 P(FYQ) 41.7 0.010 1.487 0.153 j5.20 j2.21 2.99 P(FYBT) 47.64 0.008 1.484 0.192 j5.32 j2.77 2.56 P(FYPQ) 53.0 0.010 1.485 0.306 j5.13 j2.87 2.26 P(FYTQ) 46.6 0.007 1.478 0.341 j5.16 j3.51 1.64 P(FYBB) 36.6 0.001 1.468 0.385 j4.91 j4.13 0.78
Table 2. The theoretical electronic properties of model compounds.
Model compound B3LYP/6Y31G B3LYP/6Y31G**
HOMO (eV) LUMO (eV) Eg(eV) HOMO (eV) LUMO (eV) Eg(eV)
dF j5.38 j1.17 4.21 j5.35 j1.19 4.16 FYTZ j5.71 j1.69 4.02 j5.60 j1.56 4.04 FYTD j6.04 j2.01 4.03 j5.91 j1.81 4.10 FYTP j5.40 j2.60 2.80 j5.35 j2.36 2.99 FYTT j5.24 j3.35 1.89 j5.22 j2.82 2.40 FYTTP j5.44 j4.15 1.29 j5.38 j3.66 1.72 FYQ j5.63 j2.06 3.57 j5.59 j1.97 3.62 FYBT j5.70 j2.75 2.95 j5.63 j2.35 3.28 FYPQ j5.61 j2.90 2.71 j5.57 j2.77 2.80 FYTQ j5.66 j3.49 2.17 j5.58 j3.10 2.48 FYBB j5.52 j4.02 1.50 j5.50 j3.41 2.09
than that of the corresponding model compounds suggests the formation of mesomeric structures due to enhanced intramolecular charge transfer, as shown in Figure 4 with the P(FYTT) and P(FYTTP) as examples. The order of intramolecular charge transfer (DCT) is P(FYTD) 9
P(FYTTP) 9 P(FYTT) 9 P(FYTZ) 9 P(FYTP). This trend is not in consistence with the acceptor strength, particularly in the case of P(FYTZ) and P(FYTD). The deviation is likely due to the presence of the highly electronegative nitrogen atoms localized on the backbone of P(FYTZ) and P(FYTD), as discussed previously.
The copolymers with six-member ring acceptors (4fY4j) show a twisted conformation with a large torsional angle of 36.6Y53.0-, like their corresponding model compounds (2fY2j), as shown in Table3. The order of the dD and LB,
similar to their corresponding model compounds, decreases as the acceptor strength increases. The order of intramolec-ular charge transfer: P(FYBB) 9 P(FYTQ) 9 P(FYPQ) 9 P(FYBT) 9 P(FYQ) is exactly the same as the acceptor strength, like that of the corresponding model compounds. The above results suggest that the geometry undergoes a very small change from model compound to copolymer in the case of these polymers with six-member acceptors and that the dD, LB, and DCTare also controlled by the acceptor
strength.
The electronic structure of the copolymers, including HOMO, LUMO, and Eg are also listed in the Table 3.
Figure5represents the HOMO and LUMO energy level of copolymers with five-member ring acceptors (4aY4e). As shown in Figure 5, the HOMO energy level of these copolymers does not show a clear trend with that of the acceptor strength. However, the LUMO energy level decreases as the acceptor strength increases. For these copolymers, the HOMO level is elevated while the LUMO is lowered compared to the corresponding model com-pounds (2aY2e). This is an indication of extension of conjugation length. The order of the calculated Eg(eV) is
P(FYTD) (3.03) 9 P(FYTZ) (2.78) 9 P(FYTP) (1.83) 9 P(FYTT) (0.81) 9 P(FYTTP) (0.33). The Egalso decreases
as the acceptor strength increases except for the P(FYTD). This deviation is probably due to the exceptional low-lying HOMO level of TD, which compensates the lowering of
the LUMO level. For the P(FYTP), P(FYTT), and P(FYTTP), the calculated Eg decreases with increasing
DCT. The exceptional small band gap of the P(FYTT) and
P(FYTTP) is probably resulted from the coplanar confor-mation, the low-lying LUMO level of acceptor (j3.42 and j4.14 eV, respectively), and large intramolecular charge transfer (0.307 and 0.360 e, respectively). It has been shown that ICT effects would result in reduced band gaps of conjugated donorYacceptor copolymers [21], which is in good agreement with our theoretical results.
As shown in Figure5b, the HOMO energy level of the copolymers with six-member acceptors (4fY4j) is almost the same in the range of j4.91 to j5.32 eV. The LUMO energy level decreases with increasing acceptor strength. The order of the calculated Eg (eV) is P(FYQ) (2.99) 9
P(FYBT) (2.56) 9 P(FYPQ) (2.26) 9 P(FYTQ) (1.64) 9 P(FYBB) (0.78). The Eg also decreases with increasing
acceptor strength and intramolecular charge transfer. The properties of the PF and P(FYBT) have been theoretically analyzed and reported by Cornil et al. [22]. The band gaps estimated by them were 3.08 and 2.24 eV for the PF and P(FYBT), respectively. The same trend of band gap decrement is predicted after the incorporation of the BT unit. The small theoretical underestimations compared to our results could be attributed to the assumption of planar chain conformation they hypothesized [22]. The excep-tional small Eg of the P(FYBB) is probably due to the
smallest torsional angle (36.6-), smallest bond length alternation of donor (0.001 A˚ ), low-lying LUMO of acceptor (j4.32 eV), and largest intramolecular charge transfer (0.385 e) among these copolymers.
Figure 6 shows the one-dimensional band structure in the first Brillouin zone of the P(FYTT) and P(FYTTP). The calculated upper valence bandwidths of P(FYTT) and P(FYTTP) are 1.03 and 1.04 eV, respectively, which are larger than that of the PF of 0.588 eV. Thus, the hole mobility of these copolymers is expected to be higher than the PF homopolymer and could have potential applications as thin film transistors for organic electronics.
The electronic structures of the P(FYTD) and P(FYTP) with alkyl side-chain at C9 position have been character-ized by cyclic voltammetry. The (HOMO, LUMO, Eg)
N S N S P(F-TT) N N S P(F-TTP) N S N n n N S N S n δ + δ -N N S N S N n δ + δ
-8 -7 -6 -5 -4 -3 -2 -1 0 LUMO HOMO
Energy level (eV)
(a) Wave vector (k) 0 π/a -7 -6 -5 -4 -3 -2 -1 LUMO π/a 0 Wave vector (k)
Energy level (eV)
(b)
HOMO
Figure 6. Band structures of the relatively small band gap polymers, (a) P(FYTT) and (b) P(FYTTP).
-9 -8 -7 -6 -5 -4 -3 -2 -1 0 HOM O LU MO -4.72 -4.39 -4.52 -4.76 -5.73 -3.71 -2.93 -2.70 -5.20 -2.42 -5.05 Ene rgy (eV) -1.58 n PF N S n P(F-TZ) N N S n P(F-TD) N N S n P(F-TP) NSN S n P(F-TT) N N S N S N n P(F-TTP) -9 -8 -7 -6 -5 -4 -3 -2 -1 0 -5.20 -5.32 -4.13 -3.51 -4.91 -2.87 -5.16 -2.77 -2.21 -5.13 -5.05 -1.58 HOM O LUM O E n er gy (e V) N N n P(F-Q) n PF N S N n P(F-BT) N N N N n P(F-PQ) N S N N N n P(F-TQ) N S N N SN n P(F-BB)
a
b
Figure 5. HOMO and LUMO energy levels (unit: eV)of the fluoreneYacceptor alternating copolymers by DFT/B3LYP/6Y31G basis set (a) acceptors with five-member ring, and (b) six-member ring, respectively.
(eV) of these two copolymer are (j6.4, j2.6, 3.8) and (j5.13, j3.33, 1.80), respectively [9, 23]. By comparing with the calculated electronic structures of the P(FYTD) and P(FYTP), it suggests that the experimental and theoretical electronic structures are very similar and have the same trend of variation. The (HOMO, LUMO, Eg) (eV)
of the P(FYQ) and P(FYBT) with alkyl side-chain at C9 position obtained by cyclic voltammetry are (j5.51, j2.65, 2.86) and (j5.49, j3.14, 2.35), respectively [9]. The theoretical electronic properties of the two polymers shown in Table3 show a higher LUMO energy level and band gap than those experimental values. This deviation could be attributed to the intermolecular interaction which was not considered for an isolated single chain polymer in our calculations. This deviation is especially apparent for more twisted polymers, namely copolymer 4fY4j, because the degree of twisting could usually be reduced by the intermolecular interaction in the solid state. However, the trend on the theoretical electronic properties shown in Table3could provide the synthetic chemists for designing new conjugated polymers.
The studied fluoreneYacceptor copolymers could be used for polymer light diodes by tuning the emitting color through the acceptor type. Another potential application is transparent conductors or photovoltaic devices since extremely small band gaps of 0.81 and 0.33 eV are obtained for the P(FYTT) and P(FYTTP), respectively.
Conclusions
We have theoretically analyzed the optimized geometries and electronic properties of fluorene based alternating donorYacceptor conjugated copolymers and their model compounds by density function theory. The geometries of these compounds are significantly by the structure of acceptors, particularly the ring size on the backbone. The electronic properties of these compounds are well corre-lated to the acceptor strength. The coplanar conformation, low-lying LUMO level of acceptor, or efficient intramo-lecular charge transfer of the P(FYTTP) and P(FYBB) result in an extremely small band gap of 0.33 and 1.0 eV, respectively. The theoretical study suggests that the electronic properties of alternating fluoreneYacceptor con-jugated copolymers are determined by both the geometries and acceptor strength.
Acknowledgements
The financial support by the National Science Council, Industrial Technology Research Institute, and the Ministry of Economic Affairs of Taiwan, R.O.C. are highly appreciated.
References
1. J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 347, 539 (1990).
2. G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 270, 1789 (1995).
3. K. M. Coakley and M. D. McGehee, Chem. Mater., 16, 4533 (2004). 4. H. A. M. van Mullekom, J. A. J. M. Vekemans, E. E. Havinga and E.
W. Meijer, Mater. Sci. Eng., 32, 1 (2001).
5. T. Yasuda, T. Imase, Y. Nakamura and T. Yamatomo, Macro-molecules, 38, 1500 (2005).
6. W.-J. Lin, W.-C. Chen, W.-C. Wu, Y. H. Niu and A. K.-Y. Jen, Macromolecules, 37, 2335 (2004).
7. M. C. Hung, J. L. Liao, S. A. Chen, S. H. Chen and A. C. Su, J. Am. Chem. Soc., 127, 14576 (2005).
8. J. Yang, C. Jiang, Y. Zhang, R. Yang, W. Yang, Q. Hou and Y. Cao, Macromolecules, 37, 1211 (2004).
9. X. Chen, H.-E. Tseng, J.-L. Liao and S.-A. Chen, J. Phys. Chem. B., 109, 17496 (2005).
10. W.-C. Wu, C.-L. Liu and W.-C. Chen, Polymer, 47, 527 (2006). 11. M. Svensson, F. Zhang, S. C. Veenstra, W. J. H. Verhees, J. C.
Hummelen, J. M. Kroon, O. Inganas and M. R. Andersson, Adv. Mater., 15, 988 (2003).
12. L.-L. Chua, J. Zaumseil, J.-F. Chang, E. C.-W. Ou, P. K.-H. Ho, H. Sirringhaus and R. H. Friend, Nature, 434, 194 (2005).
13. J.-F. Wang, J.-K. Feng, A.-M. Ren, X. D. Liu, Y.-G. Ma, P. Lu and H.-X. Zhang, Macromolecules, 37, 3451 (2004).
14. A. D. Becke, J. Chem. Phys., 98, 5648 (1993).
15. B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 157, 200 (1989).
16. Gaussian 03, Revision B.4, Gaussion Inc.: Pittsburgh, Pennsylvania, 2003.
17. G. R. Hutchison, Y. J. Zhao, B. Delley, A. J. Freeman, M. A. Ratner and T. J. Marks, Phys. Rev. B, 68, 035204 (2003).
18. P. M. Viruela, R. Viruela, E. Ortı´ and J. L. Bre´das, J. Am. Chem. Soc., 119, 1360 (1997).
19. P. Hapiot, C. Lagrost, F. L. Floch, E. Raoult and J. Rault-Berthelot, Chem. Mater., 17, 2003 (2005).
20. G. Brocks and E. E. Havinga, Synth. Met., 119, 93 (2001). 21. S. A. Jenekhe, L. Lu and M. M. Alam, Macromolecules, 34, 7315
(2001).
22. L. Cornil, I. Gueli, A. Dkhissi, J. C. Sancho-Garcia, E. Hennebicq, J. P. Calbert, V. Lemaur, D. Beljonne and J. L. Bre´das, J. Chem. Phys., 118, 6615 (2003).
23. S. Janietz, J. Barche, A. Wedel and D. Sainova, Macromol. Chem. Phys., 205, 187 (2004).