Unexpected regioselectivity in the coupling of p-coordinated tritylallene with
an amido ligand in molybdenum complex
Bor-Chen Huang, Ying-Chih Lin,* Yi-Hong Liu and Yu Wang
Department of Chemistry, National Taiwan University Taipei, Taiwan 106, Republic of China. E-mail: [email protected] Coupling of p-coordinated tritylallene with an amido ligand
was unexpectedly found to take place at the terminal carbon in the reaction of [Cp(CO)3Mo(h2-CH2NCNCHCPh3)][BF4]
1, with three secondary amines (dimethylamine, piperidine,
morpholine).
The synthesis and reactivity of organometallic complexes containing h3-allyl,1h1-allenyl2and h1-propargyl3ligands have
attracted a great deal of attention owing to their wide applications in organic synthesis. We recently reported distinc-tive regiospecificity of C–C bond formation in the reactions of tungsten allenyl and propargyl complexes. In the allenyl system,4reactions with amines and with alcohols afforded high
yields of azametallacycles and oxametallacycles, respectively. The C–C bond formation takes place solely at Caof the allenyl
ligand in both cases. By contrast, the corresponding propargyl complex afforded exclusively the b-coupled allylic complex,
the latter regiospecificity was assumed to proceed via a
h2-allene intermediate.5 In a particular system, the h2-tritylallene complex [Cp(CO)
3M(h2-CH2NCNCHCPh3
)]-[BF4], (M = Mo 1, M = W 1A, Cp = h5-C5H5) could be isolated
and displays coupling reactivity with the expected regiospeci-ficity in reactions with alcohols and some amines.6However,
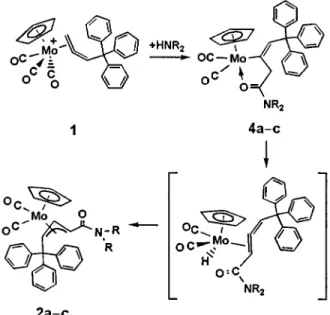
when we studied more reactions of 1 with amines, three amines were found to display different regioselectivity. Herein we report the unexpected regioselectivity in the reaction of 1 with these three amines, yielding the a-amido substituted allylic
complex as the major product and the b-amido allylic complex
as the minor product.
Reaction of 1 with neat piperidine at room temperature for 1 h afforded two amido-substituted allylic products. The major product Cp(CO)2Mo[h3-CH(CONC5H10)CHCHCPh3] 2a,† has
a surprising a-amido-substituted geometry, and the minor
product Cp(CO)2Mo[h3-CH2C(CONC5H10)CHCPh3] 3a, a
normal b-amido-substituted geometry (Scheme 1). The two
isomers can be separated by chromatography over silica gel. Complexes 2a and 3a were collected as orange–yellow and light-yellow microcrystalline powders upon re-crystallization from hexane–CH2Cl2 in ca. 65 and 17% yields, respectively.
Similar results were found with morpholine and dimethylamine to yield the a-amido-allylic complexes 2b, c† respectively as
the major product and the b-amido-allylic complexes 3b, c as
the minor product and an X-ray analysis was carried out on a crystal of 2b.‡ An ORTEP drawing of 2b is shown in Fig. 1. The most salient feature of the molecule is the presence of an amido-substituted tritylallyl ligand. The amido substituent is attached to the a-carbon C(5) of the allyl ligand with a geometry syn to
the central hydrogen and the trityl moiety is in an anti configuration.
Two possible mechanisms are proposed to account for the formation of 2a. In both cases, it is necessary to consider nucleophilic attack of amine to the terminal carbonyl giving the amido ligand. Deprotonation7 of the tritylallene ligand in the
presence of amine may result in formation of an allenyl ligand and coupling of the amido ligand with the a-carbon of the s-allenyl followed by protonation would give the major
product.8Alternatively, coupling of the amido group with allene
leading to C–C bond formation may precede hydrogen
migration and the selectivity would be controlled by the presence of the trityl group. To better understand the detail and with the hope to see an intermediate the reaction was monitored
Scheme 1 HNR2 = piperidine a, morpholine b or dimethylamine c
Fig. 1 ORTEP drawing of 2b with thermal ellipsoids shown at the 50%
probability level. Selected bond distances (Å) and angles (°): Mo–C(3) 2.379(2), Mo–C(4) 2.212(2), Mo–C(5) 2.359(2), C(3)–C(4) 1.417(3), C(4)–C(5) 1.409(3), C(5)–C(6) 1.500(3), C(6)–N(1) 1.356(3), C(6)–O(3) 1.228(3); C(4)–C(3)–C(11) 125.1(2), C(4)–C(5)–C(6) 115.4(2), C(1)–Mo– C(2) 77.13(11).
spectroscopically. When the reaction was carried out at 260 °C,
an intermediate was indeed observed. Upon addition of piperidine at 260 °C, the light yellow complex 1 dissolved and
the solution turned deep red, to give a mixture of an unstable intermediate 4a as well as 3a. In the IR spectrum of the mixture the intermediate displays two peaks at 1927 and 1828 cm21as
well as one amido CO stretching absorption at 1577 cm21. The
latter suggested the presence of O-coordinated amido carbonyl.9
Complex 4a† transforms to 2a in 1 h at room temperature but at lower temperature this process is slowed and the structure of 4a can be assigned on the basis of the spectroscopic data of the mixture obtained at 260 °C. In the 1H NMR spectrum, two
doublet resonances at d 2.28 and 2.74 with JHH22.4 Hz indicate the presence of a CH2group while a singlet resonance at d 6.55
is assigned to the NCH– group for 4a. Two-dimensional HSQC10data confirms the CH
213C resonance at d 47.2 and 13CH group at d 147.5. In the HMBC11spectrum, the cross-peak
between the CH2(dH2.28, 2.74) and the CON (dC180) groups12
indicate C–C bond formation at the terminal CH2group. These
observations imply that the intermediate could be a vinyl13
complex, (Scheme 1) and the first mechanism is thus ruled out. Hydrogen migration of 4a may proceed through b-elimination
to give the metal hydride allene followed by coupling of the hydride at Cbof the allene to give the final product 2a.
Reactions of 1 with other amines such as methylamine, ethylamine, propylamine, phenylamine, benzylamine, diethyl-amine, diisopropyl diethyl-amine, di-sec-butyldiethyl-amine, diisobutyl amine and hydrazine gave only the b-coupled product. The pKa values
of the three unique amines (8.30 for morpholine, 10.90 for Me2NH and 11.20 for piperidine) giving the a-coupled product
are in the range of regular amines (4.69 for aniline to 11.1 for diisopropyl amine) while no striking steric effect is seen for these three amines. While we cannot explain their different reactivity, this is the first case where coupling at the a-position
of a h2-allene has been found. A detailed mechanism for this
unusual coupling, the reactivity of compound 1 with other nucleophiles and the corresponding reaction for the tungsten system is currently under investigation.
We are grateful for support of this work by the National Science Council, Taiwan, the Republic of China.
Notes and References
† Selected spectroscopic data: 1H and 13C{1H} NMR were recorded in
CDCl3relative to SiMe4and IR in CH2Cl2. 2a: IR, 1954s, 1873s, 1605m
cm21. 1H NMR, d 7.28–7.11 (m, 15H, aromatic H), 5.39 (t, J
HH10.0 Hz,
1H, Hcentre), 5.28 (5H, s, Cp), 5.06 (d, JHH10.0 Hz, 1H, CHsyn), 3.60, 3.24,
2.83 (m, 4H, H2CNCH2), 1.52 (m, 6H, CH2CNCH2C3H6), 0.99 [1H, d, JHH
10.0 Hz, HCC(O)N]. 13C{1H} NMR, d 241.4, 238.4 (CO), 169.9 (CNO),
130.3, 127.2, 126.1 (Ph), 94.1 (Cp), 70.1 (CHcentre), 68.6 (CHsyn), 61.2 (CPh3), 50.2 (CHanti), 46.3, 43.3 (CH2NC2H), 26.8, 25.7, 24.7 (NC2H4C3H6). FAB MS: m/z 614 (M++ 1), 585 (M+2 CO), 557 (M+2 2CO). 2b: IR (KBr), 1937s, 1858s, 1623m cm21. 1H NMR, d 7.29–7.15 (m, Ph), 5.40 (t, JHH10.2 Hz, 1H, Hcentre), 5.29 (s, 5H, C5H5), 5.07 (d, JHH10.2 Hz, 1H, Hsyn), 3.57–2.73 (m, 8H, NC4H8O), 0.88 (d, JHH10.2 Hz, 1H,
Hanti); 13C{1H} NMR, d 241.6, 237.8 (CO), 170.5 (CNO), 130.3–126.1 (Ph),
94.2 (Cp), 69.9 (CHcentre), 68.8 (CHsyn), 66.9 (CH2OCH2), 61.2 (CPh3),
49.0 (CHanti), 45.8, 42.5 (CH2NC2H). FAB MS: m/z 616 (M++ 1), 587 (M+
2 CO), 559 (M+2 2CO). 2c: IR (KBr), 1939s, 1855s, 1611m cm21. 1H
NMR, d 7.28–7.15 (m, Ph), 5.36 (t, JHH10.2 Hz, 1H, Hcentre), 5.29 (s, 5H,
C5H5), 5.05 (d, JHH10.2 Hz, 1H, Hsyn), 2.81, 2.46 (s, 2H, NCH3), 1.12 (d, JHH10.2 Hz, 1H, Hanti). 13C{1H} NMR, d 241.8, 238.3 (CO), 171.8 (CNO),
130.3–126.2 (Ph), 94.2 (Cp), 70.3 (CHcentre), 61.1 (CPh3), 50.8 (CHsyn),
37.1, 36.1 (NCH3). FAB MS: m/z 574 (M++ 1), 545 (M+2 CO), 515 (M+
2 2CO). 4a: 1H NMR (CDCl3): d 7.27–7.08 (m, Ph), 6.55 (s, 1H, NCH),
5.33 (s, 5H, C5H5), 2.74 (d, JHH22.4 Hz, 1H, CHH), 2.28 (d, JHH22.4 Hz,
1H, CHH), 3.26–2.66 (m, 4H, CH2NCH2), 1.96 (m, 6H,
CH2NCH2CH2CH2CH2). 13C NMR [(CD3)2CO], d 180 (CON), 162.5 (Mo–
C), 147.5 (NCH–), 47.2 (CH2).
‡ Crystal data for 2b: C34H31O4NMo, M = 613.54, monoclinic, space
group P21/c, a = 13.6809(4), b = 9.8539(3), c = 21.6322(7) Å, b =
104.061(1), V = 2828.9(2) Å3, Z = 4, DC= 1.441 g cm23, m = 5.03 cm21, F(000) = 1264, 20 869 reflections collected on Smart CCD [T = 295(2) K],
6481 independent reflections (Rint = 0.0436) observed with I > 2s(I), 362
parameters, no restraints. The final discrepancy indices R1and wR2were
0.0357 and 0.0734 respectively. CCDC 182/980.
1 C.-C. Su, J.-T. Chen, G.-H. Lee and Y. Wang, J. Am. Chem. Soc., 1994,
116, 4999; J.-C. Choi and T. Yamamoto, J. Am. Chem. Soc., 1997, 119,
12 390; R.-H. Hsu, J.-T. Chen, G.-H. Lee and Y. Wang,
Organome-tallics, 1997, 16, 1159; K. Okuro and H. Alper, J. Org. Chem., 1997, 62,
1566.
2 K.-W. Liang, G.-H. Lee, S.-M. Peng and R.-S. Liu, Organometallics, 1995, 14, 2353; P. Blenkiron, J. F. Corrigan, N. J. Taylor and A. J. Carty,
Organometallics, 1997, 16, 297; S. Doherty, M. R. J. Elsegood, W.
Clegg, N. H. Rees, T. H. Scanlan and M. Waugh, Organometallics, 1997, 16, 3221; S. Doherty, M. R. J. Elsegood, W. Clegg, M. F. Ward and M. Waugh, Organometallics, 1997, 16, 4251; M. A. Esteruelas, F. J. Lahoz, M. Martin, E. Onate and L. A. Oro, Organometallics, 1997,
16, 4572.
3 M.-C. Chen, R.-S. Keng, Y.-C. Lin, Y. Wang, M.-C. Cheng and G. H. Lee, J. Chem. Soc., Chem. Commun., 1990, 1138.
4 T.-W. Tseng, I.-Y. Wu, J.-H. Tsai, Y.-C. Lin, D.-J. Chen, G.-H. Lee, M.-C. Cheng and Y. Wang, Organometallics, 1994, 13, 3963. 5 T.-W. Tseng, I.-W. Wu, Y.-C. Lin, C.-T. Chen, M.-C. Chen, Y.-J. Tsai,
M.-C. Chen and Y. Wang, Organometallics, 1991, 10, 43; I.-Y. Wu, T.-W. Tseng, Y.-C. Lin, M.-C. Cheng and Y. Wang, Organometallics, 1993, 12, 478.
6 L. Lee, I.-Y. Wu, Y.-C. Lin, G.-H. Lee and Y. Wang, Organometallics, 1994, 13, 2521.
7 H. A. Brune, W. Eberius and H. P. Wolff, J. Organomet. Chem., 1968,
12, 485.
8 K. Hiraki, N. Ochi, Y. Sasada, H. Hayashida, Y. Fuchita and S. Yamanaka, J. Chem. Soc., Dalton Trans., 1985, 873; E. Hernandez and H. Hoberg, J. Organomet. Chem., 1986, 315, 245; R. Vac, J. H. Nelson, E. B. Milosavljevic, L. Solujic and J. Fischer, Inorg. Chem., 1989, 28, 4132.
9 D. Hedden, D. M. Roundhill, W. C. Fultz and A. L. Rheingold,
Organometallics, 1986, 5, 336; R. D. Adams and S. Wang, Organome-tallics, 1987, 6, 45; M. Shakij, S. P. Varkey and P. S. Hameed, Polyhedron, 1994, 13, 1355.
10 G. Bodenhausen and D. J. Ruben, Chem. Phys. Lett., 1980, 69, 185. 11 W. Adam, J. Rutterlik, R.-M. Schuhmann and J. Sundermeyer,
Organometallics, 1996, 15, 4586; G. Jia, W.-F. Wu, R. C. Y. Yeung and
H.-P. Xia, J. Organomet. Chem., 1997, 539, 53.
12 A. Bax and M. F. Summers, J. Am. Chem. Soc., 1986, 108, 2093; A. Bax and D. Marion, J. Magn. Reson., 1988, 78, 186.
13 F. Muller, G. van Koten, K. Vrieze and D. Heijdenrijk,
Organome-tallics, 1989, 8, 33.
Received in Cambridge, UK, 21st July 1998; 8/05689G