Synthesis and characterization of spiro-linked poly(terfluorene): a
blue-emitting polymer with controlled conjugated length
Fang-Iy Wu, Rajasekhar Dodda, D. Sahadeva Reddy and Ching-Fong Shu*
Department of Applied Chemistry, National Chiao Tung University, Hsin-Chu, Taiwan, 30035, R. O. C.Received 31st May 2002, Accepted 5th August 2002
First published as an Advance Article on the web 5th September 2002
The synthesis and characterization of a fluorene–spirobifluorene alternating copolymer, P(OF-SBF), are described. In the case of the spiro-segment, the two fluorene rings are orthogonally arranged and connected through a tetrahedral bonding carbon atom (the spiro center). As a consequence, the polymer chain periodically zigzags, with an angle of 90u at each spiro center. This structural feature not only preserves the rigidity of the polymer chain but also prevents the p-stacking of the polymer backbone, resulting in an improvement in both thermal and spectroscopic stabilities. Comparing the absorption and emission spectra of P(OF-SBF) and of a terfluorene model compound, it was revealed that P(OF-SBF) possesses a well-defined conjugation length. The polymer can serve as a host matrix to effectively transfer its excitation energy to a derivatized perylene dopant, yielding an efficient blue-light-emitting layer.
Introduction
Since the discovery of poly(phenylene vinylene)-based LEDs in 1990,1considerable efforts have been made in the
develop-ment of new conjugated polymers and in the performance of the related LEDs.2–4 The ability to fine-tune the luminescent properties of polymers by manipulation of the chemical structure, along with the feasibility of utilizing spin-coating and printing processes for large area display devices, makes organic luminescent polymers very attractive. Polymers with large energy band gaps that emit blue light efficiently are of special interest, because these materials are desired for full color display applications and can also serve as energy-transfer donors in the presence of lower energy fluorophores.5–8 Polyfluorenes are among the most promising candidates for blue-emitting polymers due to their high photoluminescence and electroluminescence efficiencies.9–12In addition, the facile functionalization at the C-9 position of the fluorene unit not only provides the possibility to improve the solubility and processability of polymers, but also offers the ability to control interchain interactions,13,14 cross-linking,15 and optical and electrochemical properties.16,17 Although bright LEDs based on polyfluorenes have been fabricated, a major problem with polyfluorenes concerns their tendency to form long wave-length aggregates/excimers in the solid state upon exposure to heat.18–20This leads to the issue of color stability of the light emitted from LEDs fabricated with polyfluorenes.
Previous studies have shown that the introduction of a spirobifluorene linkage into the structure of small mole-cules leads to a reduction in crystallization tendency, an enhancement in solubility, and an increase in glass transition temperature.21–25Such spiro-structures have also been applied
to polymeric materials, leading to enhancements in both glass transition temperature (Tg) and luminescent stability in
alternating polyfluorene copolymers26 and amorphous poly-2,7-fluorene networks.27Recently, we have synthesized
amor-phous aromatic polyquinolines and polyimides, containing spirobifluorene units, which also demonstrate improved solubility and thermal stability.28–30
In light of these observations, the goal of this research was to synthesize a novel fluorene–spirobifluorene alternating copolymer, poly(9,9-dioctylfluorene)-alt-co-(9,9’-spirobifluorene) (P(OF-SBF)), which may possesses a low tendency toward
crystallization and a high Tg. Thereby, the stability of the
amorphous state in solid films is enhanced and the formation of aggregates and interchain excimers can be suppressed. In the spiro-segment, the rings of the bifluorene entities are ortho-gonally arranged and are connected through an sp3 carbon atom (the spiro center).31 The resulting polymer would be expected to have a polymer backbone that is periodically twisted with an angle of 90u at each spiro center. This structural feature restricts the close packing of the polymer chains and reduces the probability of interchain interactions, resulting in a more highly soluble amorphous polymer,28–30 in which the
tendency to form aggregate/excimers in the solid state would be suppressed. In addition, the rigidity of the main chain of the polymer would be preserved due to the spiro-structure, leading to a significant increase in both Tgand thermal stability.
28–30
Moreover, the tetrahedral bonding carbon at the center connects the conjugated moieties via a s-bonded network, which serves as a spacer and a conjugation interrupt, thus effectively controlling the conjugation length of this polyfluor-ene derivative.32 While this paper was in preparation, Kim, Miller, and Chen, and their co-workers, reported the synthesis of spirobifluorene-linked orthogonal polymers and oligofluor-enes and showed that these spiro-polymers (oligomers) have enhanced thermal and luminescent stabilities over the corres-ponding poly(dioctylfluorene).33–36
Experimental section
Materials
Compounds 1,372,383,38and 5,39were synthesized according to literature procedures. Diethyl ether and toluene were refluxed with sodium and distilled. Acetonitrile was distilled from CaH2. All other reagents and solvents were used as
received from commercial sources unless otherwise stated. Characterization
1
H and13C NMR spectra were recorded on a Varian Unity 300 MHz or a Bruker-DRX 300 MHz spectrometer using CDCl3as solvent. Mass spectra were obtained on a JEOL
JMS-SX/SX 102A mass spectrometer. Size exclusion chromatogra-phy (SEC) was carried out on a Waters chromatograchromatogra-phy unit
DOI: 10.1039/b205334a J. Mater. Chem., 2002, 12, 2893–2897 2893
interfaced to a Waters 410 differential refractometer. Three 5 mm Waters styragel columns (300 6 7.8 mm) connected in series in decreasing order of pore size (104, 103and 102A˚ ) were
used with THF as the eluent, and standard polystyrene samples were used for calibration. Differential scanning calorimetry (DSC) was performed on a SEIKO EXSTAR 6000 DSC unit using a heating rate of 20 uC min21
and a cooling rate of 40 uC min21. Samples were scanned from 30 to 400 uC and
then cooled to 30uC, and scanned for a second time from 30 to 400uC. Glass transition temperatures (Tg) were determined
from the second heating scan. Thermogravimetric analysis (TGA) was made on a PerkinElmer Pyris 1 TGA instrument. The thermal stability of the samples was determined in nitrogen, by measuring weight loss while heating at a rate of 10 uC min21
. UV-visible spectra were measured with an HP 8453 diode array spectrophotometer. Photoluminescence spectra were obtained on a Hitachi F-4500 luminescence spectrometer. IR spectra were obtained on a Nicolet 360 FT-IR spectrometer.
2,2’-Dibromo-9,9’-spirobifluorene (4)
To a stirred mixture of dry CuBr2(1.60 g, 7.16 mmol) in dry
acetonitrile (20 mL) at 0uC, tert-butylnitrite (0.80 g, 7.76 mmol) was added dropwise over 10 min. After stirring for a further 10 min, solid 2,2’-diamino-9,9’-spirobifluorene (3) (0.90 g, 2.60 mmol) was added in three portions. The reaction mixture was then heated at 60 uC for 30 min. After this period, the reaction mixture was cooled and poured into a cold, 20% HCl aqueous solution (30 mL), and extracted with diethyl ether (2 6 30 mL). The organic extracts were dried over MgSO4,
evaporated and purified with column chromatography (hexane– EtOAc, 98 : 2) followed by recrystallization (hexane–CH2Cl2)
to afford 4 (0.71 g, 58%) as colorless crystals. 1H NMR (300 MHz, CDCl3): d 6.71 (d, 2H, J ~ 7.6 Hz), 6.84 (d, 2H, J ~
1.7 Hz), 7.14 (ddd, 2H, J ~ 7.5, 7.5, 0.7 Hz), 7.38 (ddd, 2H, J ~ 7.5, 7.5, 0.7 Hz), 7.50 (dd, 2H, J ~ 8.1, 1.7 Hz), 7.70 (d, 2H, J ~ 8.1 Hz), 7.80 (d, 2H, J ~ 7.6 Hz).13C NMR (75 MHz, CDCl3): d 65.6, 120.2, 121.5, 124.1, 127.3, 128.2, 128.4, 131.2,
140.6, 140.7, 147.6, 149.9. HRMS (EI): calc. for C25H14 79
Br2:
471.9463, found 471.9475; calc. for C25H1479Br81Br: 473.9442,
found 473.9449; calc. for C25H1481Br2: 475.9421, found
475.9330.
Preparation of P(OF-SBF)
To a solution of 4 (200 mg, 0.422 mmol) and 5 (271 mg, 0.422 mmol) in toluene (3.0 mL), aqueous potassium carbonate (2.0 M, 0.7 mL) and aliquate 336 (40 mg) was added. The above mixture was degassed and tetrakis(triphenylphosphine)palla-dium (5 mg, 1.0 mol%) was added in one portion under a nitrogen atmosphere. The solution was then refluxed under nitrogen for 3 days. The end groups were capped by refluxing for 6 h each with phenylboronic acid (108 mg, 0.89 mmol) and bromobenzene (140 mg, 0.89 mmol). After cooling, the resulting polymer was precipitated into a mixture of methanol and water (80 mL, 50% v/v). The crude polymer was collected, washed with methanol, and dissolved in THF (2.0 mL). The resulting solution was filtered and precipitated into methanol. The polymer was collected and washed with acetone using a Soxhlet apparatus for 24 h. The polymer was then dried under vacuum to give P(OF-SBF) (230 mg, 78%).1H NMR (300 MHz, CDCl3): d 0.55 (br, 4H), 0.73 (t, 6H, J ~ 6.8Hz), 0.93–1.11 (m, 20H), 1.90 (br, 4H), 6.75 (d, 2H, J ~ 7.6 Hz), 7.08 (br, 2H), 7.11 (dd, 2H, J ~ 7.4, 7.4 Hz), 7.31–7.39 (m, 6H), 7.52 (d, 2H, J ~ 7.8 Hz), 7.67 (d, 2H, J ~ 7.8Hz), 7.87 (d, 2H, J ~ 7.2 Hz), 7.92 (d, 2H, J ~ 7.8 Hz).13C NMR (75 MHz, CDCl3): d 14.1, 22.6, 23.7, 29.2, 29.9, 31.7, 31.8, 40.3, 55.3, 66.3, 119.8, 120.1, 120.3, 121.2, 122.7, 124.2, 126.2, 127.2, 127.9, 139.7, 140.0, 141.1, 141.5, 141.6, 149.4, 151.6.
Results and discussion
Synthesis and characterization
The dibromo monomer, 2,2’-dibromo-9,9’-spirobifluorene (4) was synthesized in three steps starting from 9,9’-spirobifluorene (1), as shown in Scheme 1. The precursor, 9,9’-spirobifluorene (1) was prepared by the Clarkson and Gomberg method, which involved reacting 9-fluorenone with a Grignard reagent prepared from 2-iodobiphenyl, followed by dehydrative ring closure of the resulting carbinol in acetic acid.37The nitration
of 1 with nitric acid in an acetic acid medium gave the dinitro
Scheme 1 J. Mater. Chem., 2002, 12, 2893–2897
derivative 2, which subsequently, on reduction, afforded the spirodiamine 3.38 Substitutive deamination of the diamine 3 with tert-butylnitrite and copper(II) bromide in acetonitrile40 produced the desired dibromo monomer, 2,2’-dibromo-9,9’-spirobifluorene (4), with one bromine substituent located in each orthogonal ring. The preparation of 4 by direct bromination of 9,9’-spirobifluorene (1) in the presence of a catalytic amount of ferric chloride has been reported in the literature.41 However, our attempts to prepare 4 by direct
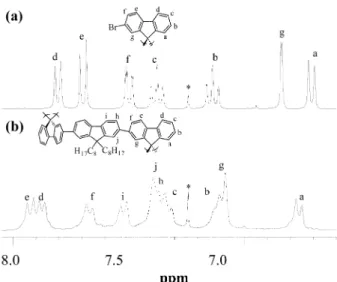
bromination methods led to an inseparable mixture of mono-, di- and polybromides. The structure of compound 4 was confirmed by 1H and13C NMR spectroscopy. As shown in
Fig. 1, the 1H NMR spectrum of 4 exhibited seven equally integrated aromatic proton signals. The chemical shift assign-ments of aromatic protons in 4 were established from the detailed analysis of1H and13C NMR data of 4, aided by 2D NMR experiments including 1H-1H correlation spectroscopy (1H-1H COSY) and long-range1H-13C heteronuclear correla-tion spectroscopy (HMBC). In the13C NMR spectrum, the central spiro carbon signal at d 65.6 (C-9) was indicative of the presence of a spiro skeleton in 4. In addition, the high resolution mass spectral data also verified the structure of 4.
As illustrated in Scheme 1, the fluorene–spirobifluorene alternating copolymer, poly(9,9-dioctylfluorene)-alt-co-(9,9’-spirobifluorene) (P(OF-SBF)) was prepared from a Suzuki coupling reaction between the dibromide 4 and the diboronate 5,42 2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9,9-dioctylfluorene.39 The copolymerization was carried out using Pd(PPh3)4 as catalyst in a mixture of toluene and
aqueous potassium carbonate (2.0 M) in the presence of aliquate 336 as a phase-transfer reagent. At the end of polymerization, the end groups of the polymer chain were capped by refluxing the reaction mixture for a period of 6 h each with phenylboronic acid and bromobenzene. The result-ing polymer solution was precipitated repeatedly into metha-nol–water and methanol several times, followed by Soxhlet extraction with acetone to give P(OF-SBF). The 1H and13C NMR spectra of P(OF-SBF) are consistent with the expected fluorene–spirobifluorene structure of the polymer. The signals between d 0.55 and 1.90 ppm correspond to protons of the n-octyl chain. The resonances of the aromatic protons of the fluorene–spirobifluorene repeating unit appear between d 6.75 and 7.92 ppm. As expected, the protons He, Hfand Hgshow a
down-field shift compared with those of the monomer 4. Based on the observed HMBC connectivities of C-9 (d 66.3) with Ha
and Hgin the 9,9’-spirobifluorene unit and of C-9 (d 55.3) and
Hjin the 9,9-dioctylfluorene unit, and with auxiliary 2D1H-1H
COSY data, the positions of the chemical shift of aromatic protons in P(OF-SBF) were assigned, as shown in Fig. 1. P(OF-SBF) was readily soluble in common organic solvents, such as THF, chloroform, toluene, xylene. The highly soluble nature of this copolymer can be attributed to the presence of the spiro-fused bifluorene segment in the polymer backbone along with the fluorene unit containing two flexible n-octyl chains at C-9. In the spiro-segment, the two fluorene rings were orthogonally arranged and connected via a common tetra-coordinated carbon. This structural feature provides assistance in reducing interchain interactions and enhances the solubility of the polymer chains. The molecular weights of the polymer were determined by gel permeation chromatography (GPC) analysis in a THF solution calibrated against polystyrene standards. P(OF-SBF) has a number-average molecular weight (Mn) of approximately 1.4 6 10
4
g mol21, with a polydispersity index of 1.5.
Thermal properties
The thermal properties of P(OF-SBF) were investigated by DSC and TGA. DSC was performed in the temperature range from 30 to 400uC. A distinct Tgwas observed at 185uC, which
is much higher than that of poly(9,9-dioctylfluorene) (POF) (Tgy 51 uC),43and no crystallization and melting transition
was found upon heating beyond the Tg. It is evident that the
incorporation of rigid 9,9’-spirobifluorene units into the polymer backbone increases the chain rigidity and results in a higher Tg.28–30The relatively high Tgis essential for polymers
as emissive materials in light-emitting applications.44In contrast
to our spiro-linked poly(terfluorene), the spiro-linked terfluor-ene oligomer reported recently shows, in succession, a glass transition at 60uC, a crystallization exothermal peak at 84 uC, and a melting endothermal peak at 194uC.36
This comparison illustrates the fact that polymeric materials usually have superior morphological stability of the amorphous state in solid films over the corresponding oligomers or small molecules. As revealed by TGA, polymers containing spirobifluorene units have excellent thermal stability. A 5% weight loss was observed at 429uC, with a 10% weight loss occurring at 451 uC. Optical properties
The absorption and photoluminescence (PL) spectra of P(OF-SBF) in diluted solution are shown in Fig. 2. The spiro-polymer in chloroform solution exhibits an absorption due to a p–p* transition with a lmaxat 352 nm, while the emission spectrum
displays a vibronic fine-structure with two sharp bands at 398 and 419 nm, with a shoulder at 443 nm. In general, the presence Fig. 1 1H NMR spectra in the aromatic region of: (a) compound 4 and
(b) P(OF-SBF) in CDCl3. * indicates a signal arising from CHCl3.
Fig. 2 UV-vis absorption and PL spectra in CHCl3of P(OF-SBF) and
terfluorene 6.
J. Mater. Chem., 2002, 12, 2893–2897
of a well-defined vibronic structure in the emission spectrum indicates that the polymer has a rigid and well-defined backbone structure.
Because the p-conjugation of the polymer chain is inter-rupted by the spiro-carbon atom, P(OF-SBF) shows a substantial blue-shift in absorption (42 nm) and PL (20 nm) as compared to the fully conjugated counterpart, POF. To demonstrate that the tetrahedral bonding carbon at the center of the spiro moiety could serve as a conjugation interrupt to control the conjugation length of the spiro-polymer, a model compound, 9,9,9’,9’,9@,9@-hexaoctyl-7,2’,7’,2@-terfluorene (6),45 was pre-pared as the repetitive conjugated unit of polymer P(OF-SBF). As illustrated in Fig. 2, the solution UV-vis absorption and PL spectra of compound 6 in CHCl3and those obtained
from P(OF-SBF) are almost superimposable. This result implies that the effective conjugation length of P(OF-SBF) is the same as that of terfluorene and that the tetrahedral bonding carbon at the center of the spiro moiety efficiently confines the conjugation length of the polymer.
Fig. 3 shows the absorption and PL spectra of P(OF-SBF) film spin-coated from toluene solution onto a quartz plate. In comparison to dilute solution, the absorption spectrum of the thin film is broadened and has a 3 nm red-shift while the emission spectrum shows a 6 nm red-shift. The small red-shift of the emission observed in the solid state is probably due to the different relative permittivity of the environment. The solid state PL quantum yield was estimated to be 0.42 by comparing the fluorescence intensity of the polymer film with that of a sample of POF excited at 365 nm (Wf~ 0.55). To examine the
thermal stability of the spiro polymer, the polymer film was heated on a hot-plate at 150uC in a nitrogen atmosphere for 20 h. After thermal treatment, both absorption and PL spectra of the P(OF-SBF) film remain almost unchanged with the appearance of a long wavelength tail in the emission spectrum, as shown in Fig. 3. For comparison, the same experiments were conducted for the film of POF. Annealing of the POF film led to not only a spectral shift, but also the appearance of an additional emission band between 500 and 600 nm. It is apparent that the thermal stability of P(OF-SBF) is improved over that of POF. The formation of interchain excimers was suggested as the cause of the undesirable emissive color instability of the POF film.18–20Attributed to the presence of spiro-fused orthogonal bifluorene segments, which restrict the close packing of the polymer chains and reduce the probability of interchain interactions, the tendency to form aggregates and excimers in P(OF-SBF) film upon thermal treatment is
suppressed. Recently, List et al.46 studied the photo- and electrooxidative degradation of mono- and dialkylated poly-fluorenes. They reported that the keto-defects may be the source of low-energy emission bands. In this work, the IR transmission spectra of polymer films on silicon wafer for P(OF-SBF) and POF were also investigated. However, we did not observe the appearance of the fluorenone site (wCLO stretching) at 1721 cm21after thermal treatment.
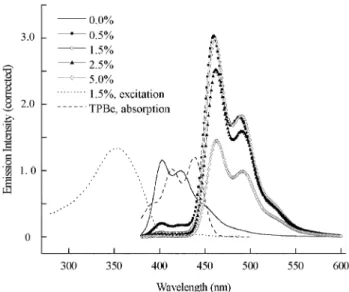
P(OF-SBF) exhibits a purplish-blue emission with the color coordinates in CIE 1931 chromaticity measured to be (0.165, 0.128), which is less sensitive to human eyes. Therefore, this polymer may not be suitable for use as a pure blue emitter but may be useful as a host material doped with dyes having high PL efficiency. The color of PL can be then tuned by the proper choice of a dopant dispersed in the polymer, which is excited by energy transfer from the host matrix and emits strongly at the desired wavelength.5–8,47In this study, a nonplanar derivative of perylene, 2,5,8,11-tetra-tert-butylperylene (TBPe), which is a prototypical dopant for blue emitting electroluminescent devices,48,49was chosen as the dopant. In order to ensure an efficient energy transfer from P(OF-SBF) to TBPe by dipole coupling (Fo¨rster transfer), the emission spectrum of the polymer and the absorption spectrum of the dopant must overlap.50,51 As shown in Fig. 4, the spectral overlap of the
emission spectrum of P(OF-SBF) and the absorption spectrum of TBPe is substantial. Fig. 4 also shows the PL spectra of P(OF-SBF) films doped with various amounts of TBPe. The intensity of emission was corrected with the absorbance at the excitation wavelength, 355 nm. The variation in absorbance at 355 nm of P(OF-SBF) films doped with various amounts of TBPe is within 3% (not shown in the figure), indicating that the absorption at 355 nm is mainly contributed from the host polymer and the thickness of the films prepared is similar. At 0.5% dopant concentration, there is a small shoulder at around 406 nm, which is attributed to the host emission of P(OF-SBF) due to an incomplete energy transfer. Raising the doping level to 1.5% results in complete elimination of this shoulder. In a comparison of the emission intensity of an undoped P(OF-SBF) film, the emission intensity of the P(OF-P(OF-SBF) film doped with 1.5% TBPe is enhanced 2.7-fold. The emission of doped films (doping level w 0.5%) is blue in color, with the color coordinates in CIE 1931 chromaticity measured to be (0.147, 0.186). The PL spectra of TBPe-doped P(OF-SBF) films are nearly identical to the EL spectrum of a OLED fabricated with
Fig. 3 UV-vis absorption and PL spectra of the P(OF-SBF) film before and after annealing at 150uC for 20 h in a nitrogen atmosphere. Inset: UV-vis absorption and PL spectra of the POF film before and after annealing at 150uC for 20 h in a nitrogen atmosphere.
Fig. 4 PL spectra of P(OF-SBF) films doped with various amounts of TBPe excited at 355 nm (the intensity was corrected with the variation in absorbance at the excitation wavelength); also included are the absorption spectrum of TBPe in THF and the excitation spectra of the P(OF-SBF) film doped with 1.5% TBPe, monitored emission at 500 nm. J. Mater. Chem., 2002, 12, 2893–2897
a TBPe-doped emitting layer,52implying the origin of the blue emission of doped P(OF-SBF) films to be the dopant. In addition, the excitation spectra of TBPe-doped P(OF-SBF) films are perfectly superimposable to the absorption spectrum of the undoped P(OF-SBF) film. These results demonstrate that the excitation energy of P(OF-SBF) is efficiently trans-ferred to the TBPe dopant to furnish a blue emission layer with improved PL yield. It is noted that the emission intensity drops in going from 1.5% to 5% dopant concentration. This may be due to self-quenching of the dopant emission at higher concentration.53,54
Summary
A spiro-linked poly(terfluorene) was synthesized by copoly-merization of 2,2’-dibromo-9,9’-spirobifluorene (4) and the diboronate 5, via a palladium-catalyzed Suzuki coupling reaction. In the spiro-fused bifluorene segment, the two mutually perpendicular fluorene rings were connected via a common tetracoordinated carbon atom, which served as a conjugation interrupt to effectively control the conjugation length of the polymer. Attributed to the spiro structure, the polymer exhibits a higher glass transition temperature and a more stable emission spectrum in comparison with the conventional dialkyl polyfluorene. In TBPe-doped P(OF-SBF) films, the excitation energy of P(OF-SBF) is efficiently trans-ferred to the TBPe dopant to furnish a blue emission layer with the color coordinates in CIE 1931 chromaticity measured to be (0.147, 0.186). The utilization of P(OF-SBF) as a host matrix for an emitting layer in electroluminescence applications is in progress.
Acknowledgements
We thank the National Science Council of the Republic of China for financial support. We also thank Dr. Banumathy Balaganesan for providing the TBPe samples.
References
1 J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539.
2 A. Kraft, A. C. Grimsdale and A. B. Holmes, Angew. Chem. Int. Ed. Engl., 1998, 37, 402.
3 M. T. Bernius, M. Inbasekaran, J. O’Brien and W. Wu, Adv. Mater., 2000, 12, 1737.
4 U. Mitschke and P. Ba¨urele, J. Mater. Chem., 2000, 10, 1471. 5 J. Kido, K. Hongawa, K. Okuyama and K. Nagai, Appl. Phys.
Lett., 1994, 64, 815.
6 J. Kido, H. Shionoya and K. Nagai, Appl. Phys. Lett., 1995, 67, 2281.
7 M. D. McGehee, T. Bergstedt, C. Zhang, A. P. Saab, M. B. O’Regan, G. C. Bazan, V. I. Srdanov and A. J. Heeger, Adv. Mater., 1999, 11, 1349.
8 F.-C. Chen, Y. Yang, M. E. Thompson and J. Kido, Appl. Phys. Lett., 2002, 80, 2308.
9 Q. Pei and Y. Yang, J. Am. Chem. Soc., 1996, 118, 7416. 10 M. Leclerc, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2867. 11 D. Neher, Macromol. Rapid Commun., 2001, 22, 1365.
12 S. Becker, C. Ego, A. C. Grimsdale, E. J. W. List, D. Marsitzky, A. Pogantsch, S. Setayesh, G. Leising and K. Mu¨llen, Synth. Met., 2002, 125, 73.
13 S. Setayesh, A. C. Grimsdale, T. Weil, V. Enkelmann, K. Mu¨llen, F. Meghdadi, E. J. W. List and G. Leising, J. Am. Chem. Soc., 2001, 123, 946.
14 D. Marsitzky, R. Vestberg, P. Blainey, B. T. Tang, C. J. Hawker and K. R. Carter, J. Am. Chem. Soc., 2001, 123, 6965.
15 G. Kla¨rner, J.-I. Lee, V. Y. Lee, E. Chan, J.-P. Chen, A. Nelson,
D. Markiewicz, R. Siemens, J. C. Scott and R. D. Miller, Chem. Mater., 1999, 11, 1800.
16 F. Uckert, S. Setayesh and K. Mu¨llen, Macromolecules, 1999, 32, 4519.
17 F.-I. Wu and C.-F. Shu, unpublished work.
18 J. I. Lee, G. Klaerner and R. D. Miller, Synth. Met., 1999, 101, 126.
19 J. Teetsov and M. A. Fox, J. Mater. Chem., 1999, 9, 2117. 20 K.-H. Weinfurtner, H. Fujikawa, S. Tokito and Y. Taga, Appl.
Phys. Lett., 2000, 76, 2502.
21 J. Salbeck, N. Yu, J. Bauer, F. Weisso¨rtel and H. Bestgen, Synth. Met., 1997, 91, 209.
22 J. Salbeck, J. Bauer and F. Weisso¨rtel, Macromol. Symp., 1997, 125, 121.
23 N. Johansson, D. A. dos Santos, S. Guo, J. Cornil, M. Fahlman, J. Salbeck, H. Schenk, H. Arwin, J. L. Bre´das and W. R. Salenek, J. Chem. Phys., 1997, 107, 2542.
24 N. Johansson, J. Salbeck, J. Bauer, F. Weisso¨rtel, P. Bro¨ms, A. Andersson and W. R. Salaneck, Adv. Mater., 1998, 10, 1136.
25 Y.-H. Kim, D.-C. Shin, S.-H. Kim, C.-H. Ko, H.-S. Yu,
Y.-S. Chae and S.-K. Kwon, Adv. Mater., 2001, 13, 1690. 26 W.-L. Yu, J. Pei, W. Huang and A. J. Heeger, Adv. Mater., 2000,
12, 828.
27 D. Marsitzky, J. Murray, J. C. Scott and K. R. Carter, Chem. Mater., 2001, 13, 4285.
28 C.-L. Chiang and C.-F. Shu, Chem. Mater., 2002, 14, 682. 29 D. S. Reddy, C.-F. Shu and F.-I. Wu, J. Polym. Sci., Part A:
Polym. Chem., 2002, 40, 262.
30 C.-H. Chou, D. S. Reddy and C.-F. Shu, J. Polym. Sci., Part A: Polym. Chem, in press.
31 R. Wu, J. S. Schumm, D. L. Pearson and J. M. Tour, J. Org. Chem., 1996, 61, 6906.
32 G. Klaerner and R. D. Miller, Macromolecules, 1998, 31, 2007. 33 D. J. Park, Y.-Y. Noh, J.-J. Kim and D.-Y. Kim, Polym. Prepr.
(Am. Chem. Soc., Div. Polym. Chem.), 2002, 43, 71.
34 R. D. Miller, F. Do¨tz, A. R. Murphy, V. Y. Lee, J. C. Scott, L. Bozano, R. Smith and C. Bacilieri, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2002, 43, 116.
35 D. Katsis, Y. H. Geng, J. J. Ou, S. W. Culligan, A. Trajkovska, S. H. Chen and L. J. Rothberg, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2002, 43, 118.
36 D. Katsis, Y. H. Geng, J. J. Ou, S. W. Culligan, A. Trajkovska, S. H. Chen and L. J. Rothberg, Chem. Mater., 2002, 14, 1332. 37 R. G. Clarkson and M. Gomberg, J. Am. Chem. Soc., 1930, 52,
2881.
38 J. H. Weisburger, E. K. Weisburger and F. E. Ray, J. Am. Chem. Soc., 1950, 72, 4253.
39 M. Ranger, D. Rondeau and M. Leclerc, Macromolecules, 1997, 30, 7686.
40 M. P. Doyle, B. Siegfried and J. F. Dellaria, Jr., J. Org. Chem., 1977, 42, 2426.
41 F. K. Sutcliffe, H. M. Shahidi and D. Patterson, J. Soc. Dyes Colors, 1978, 94, 306.
42 N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457. 43 J. Ding, M. Day, G. Robertson and J. Roovers, Macromolecules,
2002, 35, 3474.
44 S. Tokito, H. Tanaka, K. Noda, A. Okada and Y. Taga, Appl. Phys. Lett., 1997, 70, 1929.
45 S. Beaupre´, M. Ranger and M. Leclerc, Macromol. Rapid
Commun., 2000, 21, 1013.
46 E. J. W. List, R. Guentner, P. S. de Freitas and U. Scherf, Adv. Mater., 2002, 14, 374.
47 C. W. Tang, S. A. VanSlyke and C. H. Chen, J. Appl. Phys., 1989, 65, 3610.
48 B. X. Mi, Z. Q. Gao, C. S. Lee, S. T. Lee, H. L. Kwong and N. B. Wang, Appl. Phys. Lett., 1999, 75, 4055.
49 J. Shi and C. W. Tang, Appl. Phys. Lett., 2002, 80, 3201. 50 T. Fo¨rster, Discuss. Faraday Soc., 1959, 27, 7.
51 A. Dogariu, R. Gupta, A. J. Heeger and H. Wang, Synth. Met., 1999, 100, 95.
52 J. Shi, C. W. Tang and C. H. Chen, US Pat., 5 935 721, 1999. 53 G. Yu, Y. Liu, X. Wu, D. Zhu, H. Li, L. Jin and M. Wang, Chem.
Mater., 2000, 12, 2537.
54 Y. T. Tao, E. Balasubramaniam, A. Danel, B. Jarosz and
P. Tomasik, Chem. Mater., 2001, 13, 1207.
J. Mater. Chem., 2002, 12, 2893–2897