Trace Analysis of Amikacin in Human Plasma
by High-Performance Liquid Chromatography
2001,53, Suppl.,S-213
S-217C. H. Feng / S. J. Lin / H. L. Wu / S. H. Chen*
School of Pharmacy, Kaohsiung Medical Universif,/, Kaohsiung, 807,Taiwan; E-Maih [email protected]
Key Words
Column liquid chromatography
Amikacin in plasma
Naphthyl isothiocyanate derivatization
Summary
A simple and sensitive liquid chromatographic method is described for the determination of
amikacin in human plasma. The amikacin is derivatized with 1-naphthyl isothiocyanate (NITC)
at 70 ~
derivatization, a methylamine acetonitrile solution is added to the reaction mix-
ture to eliminate excess derivatizing agent. The resulting derivative was analysed by HPLC on
a LiChroCART RP-Cls column with water-acetonitrile
(57:43, v/v)
mobile phase and detection
at 230 nm. Parameters affecting derivatization of amikacin, including reaction temperature,
reaction time and amount of derivatizing agent, were investigated. The linear range for the
determination of amikacin in spiked plasma was over 10- 52 nmol mL 1; the detection limit
(signal-to-noise ratio = 3; injection volume, IOI~L) was ca. 1 nmol mL 1. The relative standard
deviation was < 2.37% for intra-day assay (n = 6),
5.80%
for inter-day assay (n = 6) and rela-
tive recoverywas > 91%.
Introduction
Amikacin is an aminoglycoside antibiotic and commonly administered parenterally to treat infections caused by aerobic gram-negative bacteria. Amikacin is semi- synthesized from kanamycin B and devel- oped to resolve drug resistance to genta- micin, kanamycin and tobramycin. It binds irreversibly to the 30S and 50S ribo- somal subunits resulting in a defective,
Presented at: 23 rd International Symposium on
Chromatography, London, UK, October 1 5, 2000
bacterial-cell membrane-synthesis of pro- tein. Like other aminoglycosides, amika- cin has a comparatively narrow safety margin. It may cause both ototoxicity and nephrotoxicity in patients, especially dur- ing long-term therapy. Its therapeutic plasma concentration is in the range: 8 161xgmL 1. Monitoring of amikacin le- vels in plasma is required for therapeutic and toxic control, hence, a simple, sensi- tive and specific method for trace analysis in plasma is essential [1 4].
Several methods, including microbio- logical assay [5 6], immunoassay [7 18] and chromatographic [19 35] methods,
have been applied to the analysis of ami- kacin in various matrices. Microbiological assay is inexpensive and simple but time- consuming. Several parameters influence the accuracy of this method, such as test strain and presence of other antibiotics. Radioimmunoassay (RIA) methods [10 13] and fluorescence polarization immu- noassay (FPIA) [14 18] are common as- says for amikacin in human plasma. These methods are more specific and sensitive than microbiological assay, but they de- pend on suitable specific antibiotics. Cross-reactions are sometimes noted with the immunoassay kits. In addition, hand- ling of radioactive materials and radioac- tive waste and high cost are inhibitory fac- tors. HPLC is widely used and is the most accurate technique for the analysis of ami- noglycoside antibiotics in various ma- trices.
The structure of amikacin, shown in Figure 1, indicates that amikacin has four primary amino groups, one primary OH group, one secondary amino group, and seven secondary OH groups. Due to lack of any chromophore in the molecule, di- rect HPLC of amikacin is not straightfor- ward. Chemical derivatization can modify drugs as detector-oriented derivatives by reacting with a reagent to enhance detec-
CH2NH2 CH2OH
H O ~ / HO v - ~ O
?.
O 1 1 0 ~ / ~ - ~
N.cocc.2C.2N.2
NH2 H
Figure 1. Structure of amikacin.
Original Chromatographia Supplement Vol. 53,2001 S-213
RNH 2 H N C S N C S
~
ItR + lm 1- naphthyl isothiocyanateFigure 2. Putative reaction scheme between amikacin and NITC.
tion sensitivity. To ensure amikacin has high UV absorption and enhanced chro- matographic characteristics, derivatiza- tion is often applicable. Commonly used reagents are 2,4,6-trinitrobenzene sulfonic acid [22 24], 1-fluoro-2, 4-dinitrobenzene [25 27] and o-phthaldehyde [28 34]. However, the derivatives obtained with these agents were unstable.
In this paper, a simple plasma pretreat- ment and sensitive HPLC method is de- scribed for trace analysis of amikacin in human plasma. The method is based on chemical derivatization of amikacin with NITC in pyridine and the use of methyla- mine to eliminate excess NITC after deri- vatization. The proposed method also can be applied to other aminoglycoside anti- biotics in clinical drug monitoring.
Experimental
Chemicals and Reagents
Amikacin was from Sigma (St. Louis MO, USA). 1-Naphthyl isothiocyanate (NITC) was from TCI (Tokyo, Japan). Naphtha- lene (internal standard, I.S.), methyla- mine and pyridine were from E. Merck (Darmstadt, Germany). Acetonitrile and other reagents were of analytical-reagent grade. Blank plasma from healthy donors was obtained from the Department of Transfusion Medicine, University of Kaohsiung Medicine. Solutions of amika- cin at various concentrations were pre- pared by dissolving a suitable amount of amikacin in deionized water. The deriva- tizing agent, NITC, was fresh prepared in pyridine. Methylamine was prepared in acetonitrile.
HPLC Conditions
A Waters 717 plus autosampler, Model 486 UV-Vis detector, Beckman program- mable solvent module 126 pump and sys- tem Gold software were used. A LiChro- CART Rpls column (55 • 4mm; 3 ixm)
and water-acetonitrile (57:43,
v/v)
at 0.8 mL min 1 were used. The column elu- ate was monitored at 230 nm. The solvent was filtered through a Millipore, HVLP, 0.45 ixm filter under vacuum for degassing before use.Sample Preparation
and Derivatization Procedures
Amikacin spiked plasma at various con- centrations was prepared as follow: 0.4mL human plasma was pipetted into a 10mL glass-stoppered test tube, and 0.1 mL of an aqueous solution containing various amounts of amikacin were added to each tube to prepare final amikacin concentrations in plasma sample over the range 10 5 2 n m o l m L 1. Tubes were mixed for 10 s. A 0.5 mL sample of aceto- nitrile was added and mixed by vortexing for 1 min. Tubes were then centrifuged (1000g) for 10min. A 0.8mL sample of supernate was transferred to a 10mL glass-stoppered test tube and 0.3mL NITC pyridine solution (80mM) and 0.1 mL of naphthalene pyridine solution (1.25mM) added. The reaction mixture was shaken for 1 h at 70 ~ in a thermo- statted water bath. After reaction, 0.1 mL methylamine acetonitrile solution (0.5 M) was added and the reaction mixture sha- ken for 5 min at 70 ~ to eliminate excess derivatizing agent, then centrifuged at 1000g for 5min. A 101xL aliquot of the supernate was injected into the HPLC for analysis.
Precision and Accuracy Test
The reproducibility and reliability of the proposed method were determined by ex- tracting the amikacin from plasma, spiked at four different levels (10, 20, 30 and
1
52 n m o l m L ), then treated according to the procedure described under Sample Preparation and Derivatization Proce- dure.
Results and Discussion
The structure of amikacin is shown in Fig- ure 1 (see Introduction also). Since it lacks a chromophore 1-naphthyl isothiocyanate (NITC) was chosen as derivatization re- agent for its high UV sensitivity. NITC can react with primary and secondary amines and alcohols. The reactivity of this
agent towards primary amino groups seems greater than towards other groups. Primary amino groups can be added to isothiocyanate to give isothiourea deriva- tives hence the primary amino groups on amikacin react with NITC by addition to form naphthyl isothioureas. The putative reaction scheme for amikacin with NITC is illustrated in Figure 2. After derivatiza- tion, methylamine was added to eliminate excess NITC and stop further addition re- actions. The effect of the tested para- meters on the extraction-derivatization of amikacin was evaluated from the peak area ratios of the resulting derivative to the naphthalene (I. S.).
Effect of Organic Solvents
Because of the high polarity of aminogly- coside antibiotics, we considered using water-miscible organic solvents, e.g. N, N-dimethylformamide, dimethylsulfox- ide, acetonitrile and pyridine for derivati- zation of extracted amikacin from spiked plasma. The solvent effect on the yield of amikacin derivative was studied accord- ing to the Derivatization Procedures. To prevent solvents from boiling, reaction temperatures were set lower than the re- spective boiling point of the solvents tested. Derivatization of amikacin with NITC in N, N-dimethylformamide or di- methylsulfoxide as reaction solvent leads to a more complicated chromatogram with interfering peaks. The solubility of NITC in acetonitrile is inadequate and a fronting peak of the amikacin derivative was obtained. Peak fronting is sometimes indicative of incomplete derivatization and hence failure of the assay method. The solubility of NITC in pyridine is very good. When pyridine is used as reaction solvent, complete derivatization of amika- cin is assured. In this system, pyridine serves a dual role as a medium to maintain a basic reaction environment for the addi- tion reaction and as a co-solvent for the non-polar amikacin-naphthyl adduct. It can improve the yield of amikacin- naphthyl adduct and ensure symmetrical chromatographic peaks. The results indi- cated that pyridine is the best solvent for derivatization of amikacin in human plas- ma.
Effect of Amount
of Derivatizing Agent
To optimize the amount of derivatizing agent for amikacin in spiked plasma ( 5 2 n m o l m L 1), different amounts of N I T C were examined. Over the range: 6 301xmol N I T C (0.3mL, 20 100mM) for 1 h at 70 ~ required for derivatization to a maximum formation of the amikacin de- rivative is shown in Figure 3. The forma- tion of the amikacin derivative increased with increasing derivatizing agent. Maxi- mum formation of the derivative is attain- able using N I T C in amounts > 18 ixmol. An excess amount of 241xmol of N I T C was used to compensate for possible con- sumption of derivatizing agent by water or other components of the human plas- ma. The result indicates that a suitable molar ratio of N I T C to amikacin ~ 350 is needed.
Effects of Reaction
Temperature and Time
Amikacin has several reactive sites. It will give rise to different adducts at different reaction temperatures and reaction times, resulting in multiple peaks. Hence, after reaction, methylamine-acetonitrile solu- tion was added to eliminate excess deriva- tizing agent to prevent problems in evalu- ating temperature and time effects on ami- kacin derivative formation. The effect of reaction time at 50 ~ and 70 ~ on deri- vatization of amikacin from spiked plas- ma ( 5 2 n m o l m L 1) is shown in Figure 4. At 70 ~ the formation of the amikacin derivative reached equilibrium in 1 h; whereas at 50 ~ equilibrium was not at- tained in 2 h and resulted in lower yield of derivative. A more complicated chroma- togram was observed at a higher reaction temperature: 80 ~ but the yield of ami- kacin derivative was the same as at 70 ~ in 1 h. Therefore, the reaction time and temperature were set at 1 h and 70 ~ re- spectively.
Elimination of Excess
Derivatizing Agent
A large excess of derivatizing agent was usually used to speed up the derivatization reaction and achieve maximum formation of the derivative. A broad and tailing peak from the excess NITC, with a retention time > 40min, was found. Methylamine
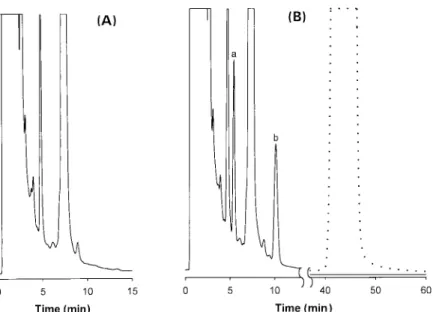
and dimethylamine were examined to eliminate excess reagent after derivatiza- tion. Methylamine was better than di- methylamine. A more polar compound than the derivatizing agent was formed during treatment of the reaction mixture with methylamine acetonitrile solution at the end of derivatization. The resulting product of N I T C and methylamine, with its more polar character, was eluted com- pletely from a reversed-phase column prior to the amikacin derivative. There- fore, methylamine can stop derivatization of amikacin with NITC, simplify the chro- matogram and shorten analysis time. Chromatograms obtained for amikacin extracted from plasma under optimized conditions are shown in Figure 5 (A) and (B). Figure 5 (A) shows the chromato- gram of the plasma blank and Figure 5 (B) shows the chromatogram of amikacin extracted from spiked human plasma, re- spectively, and then derivatized. Peak a and peak b in Figure 5 represent the ami- kacin derivative and the I. S., respectively. There was no interference from the re- agent blank with the resolution of the peak of amikacin derivative. The selectiv- ity of the method was studied by spiking standard amikacin with other aminogly- coside antibiotics, including kanamycin A and tobramycin. The spiked plasma was analyzed according to procedure de- scribed under Sample Preparation and Derivatization Procedures sections. The amikacin-naphthyl derivative could be re- solved from those of the other drugs, indi- cating that other aminoglycoside antibio- tics did not interfere with the H P L C ana- lysis of amikacin in this study. In conse- quence, the proposed method is specific and feasible for the analysis of amikacin in plasma for biological study or thera- peutic drug monitoring.
Stability of the Amikacin
Derivative
The relative stability of the amikacin deri- vative to I.S. at room temperature after derivatization and methylamine treatment was studied over a period of 14h. N o sig- nificant change in the ratio of the peak area of the derivative to I. S. was found, indicating that the derivative is stable en- ough in the reaction mixture for routine LC analysis. O r~ < n 1.2 0.8 0.4 I i i i i 0 6 12 18 24 30 36 1-naphthyl isothiocyanate (/1 mol)
Figure 3. Effect of amount of 1-naphthyl iso- thiocyanate on formation of amikacin deriva- tive. Reactions at 70 ~ for 1 h with 6 30 ~tmol 1-naphthyl isothiocyanate pyridine derivatiz- ing agent. 1.2 O t~ rv" 0.8 < -~ 0.4 01 m 12. i i i i 0 30 60 90 120 150 T i m e (min)
Figure 4. Effect of reaction temperature and re- action time on formation of amikacin deriva- tive. Reactions were at 50 ~ and 70 ~ at var- ied reaction times with 0.3mL 80mM (24 ~tmol) 1-naphthyl isothiocyanate pyridine solution derivatizing agent.
Analytical Calibration
On the basis of the optimized conditions, we formulated an analytical procedure for amikacin determination as described un- der Experimental. To validate quantita- tive application of the method, four differ- ent concentrations of amikacin over the range 10 5 2 n m o l m L 1 were evaluated. The calibration graph was established with the peak-area ratio of the derivative to I. S. as ordinate y; the amount of amika- cin in nmol as abscissa x and the correla- tion coefficient as r. The linear regression equations were obtained as follows: y = @0.0498 • 0.0055) + (0.0180 • 0.0002) x for intra-day assay (n = 6, r = 0.998) and y = @0.0404 • 0.0087) + (0.0177 • 0.0003) x for inter-day assay (n = 6, r = 0.998). The precision (relative standard deviation, R. S.D.) of the slope of the calibration graphs for intra-day and inter-day analysis is always < 1.7%. The data indicate good linearity of the method. The detection limit (signal to noise ratio = 3) of amikacin was 1 n m o l m L ] in a 10 ixL injection. The sta- bility of amikacin in spiked plasma under
(A)
l
(B)--It
) ) 10 40 Time (rain) 0 5 10 15 0 5 50 60 Time (rain)Figure 5. LC chromatograms for determination of amikacin in human plasma. (A) Plasma blank. (B) Amikacin in human plasma, with (solid line) and without (dashed line) methylamine treatment, after derivatization. Peaks: (a) amikacin derivative; (b) naphthalene (I. S.). LC conditions: LiChro- CART Rpl 8 column (55 x 4mm I.D.; 3~tm); mobile phase, water-acetonitrile (57:43,
v/v);
0.8 mL min 1 ; detection, 230 nm.
TaMe I. Precision and accuracy in analysis of amikacin-spiked human plasma.
Concentration Concentration R.S.D. R.E.
known (~tM) found (~tM) (%) (%) Intra-day* (n = 6) 52.00 52.68 • 0.94 1.78 + 1.31 30.00 28.87 • 0.68 2.37 3.76 20.00 19.55 • 0.38 1.96 2.28 10.00 10.96 • 0.11 1.04 + 9.55 Inter-day* (n = 6) 52.00 52.62 • 0.98 1.87 + 1.20 30.00 29.73 • 1.61 5.41 0.91 20.00 19.08 • 0.68 3.59 4.59 10.00 10.83 • 0.63 5.80 + 8.29
* Intra-day data based on six replicate analyses and inter-day from six consecutive days.
Table II. Relative recoveries of amikacin in human plasma.
Sample Amount spiked Amount foun& Recovery
(~M) (~M) (%) 0 N.D. N.D. 50.00 50.68 • 0.55 101.36 35.00 32.06 • 0.95 91.60 10.00 10.25 • 0.36 102.50 0 N.D. N.D. 50.00 48.59 • 0.87 97.18 35.00 34.56 • 0.85 98.74 10.00 9.80 • 0.52 98.00 0 N.D. N.D. 50.00 50.15 • 0.42 100.30 35.00 35.05 • 0.30 100.14 10.00 10.24 • 0.25 102.40 0 N.D. N.D. 50.00 51.88 • 0.64 103.76 35.00 36.35 • 0.53 103.86 10.00 10.76 • 0.08 107.60
a Mean • S.D. of triplicate analyses. N. D. = not detected.
storage was also examined. Three different c o n c e n t r a t i o n s of a m i k a c i n a t 10, 30 a n d 50 n m o l m L 1 in spiked p l a s m a were stu- died to assess the stability o f the a m i k a c i n at 40 ~ • 2 ~ F o r each sample, determi- n a t i o n of p l a s m a a m i k a c i n was p e r f o r m e d o n days 0, 7, a n d 21. Statistical analysis o f the results did n o t show any significant dif- ference; therefore, a m i k a c i n is stable in p l a s m a samples stored at 40 ~ • 2 ~ for periods u p to 21 days. This indicates favor- able stability of a m i k a c i n in h u m a n p l a s m a for analyses.
Precision and Accuracy
The reproducibility a n d reliability o f the p r o p o s e d m e t h o d were assessed a t three different c o n c e n t r a t i o n s of a m i k a c i n a n d e v a l u a t e d as relative s t a n d a r d d e v i a t i o n (R. S . D . ) a n d relative recovery, respec- tively. As s h o w n in T a b l e I, the precision o f the m e t h o d for a m i k a c i n spiked in hu- m a n p l a s m a was < 5.8% R. S. D. for b o t h i n t r a - d a y a n d i n t e r - d a y assays. T h e rela- tive recovery of a m i k a c i n , as s h o w n in Ta- ble II, is > 91%, o b t a i n e d f r o m the calibra- t i o n g r a p h of p l a s m a spiked w i t h different a m o u n t o f a m i k a c i n over the r a n g e 10 52 n m o l m L 1.
Conclusion
A simple a n d sensitive H P L C m e t h o d b a s e d o n the p r e - c o l u m n d e r i v a t i z a t i o n of a m i k a c i n in h u m a n p l a s m a w i t h 1-naph- thyl i s o t h i o c y a n a t e has b e e n established a n d optimized. V a l i d a t i o n of the m e t h o d for q u a n t i t a t i o n o f a m i k a c i n in spiked p l a s m a s h o w e d t h a t the m e t h o d h a s h i g h accuracy. T h e d e t e c t i o n limit (signal-to- noise ratio, 3; injection volume, 10 ixL) is ca. 1 n m o l m L 1. F u r t h e r a p p l i c a t i o n of the m e t h o d to the sensitive analysis of o t h e r aminoglycoside antibiotics is b e i n g carried out.Acknowledgement
The a u t h o r s are grateful to the N a t i o n a l Science Council, R O C , for financial sup- p o r t o f the w o r k ( N S C 89-2113-M-037- 032).
References
[1] Gilman, A.G.; Rail, T.W.; Nies, A.S.; Taylor, P. Goodman and Gilman's The
Pharmacological Basis o f Therapeutics,
8th Ed., Pergamon Press, New York, 1991.
[2] Kirby, W.M.M.; Clarke, J.T.; Libke, R.D.; Regamey, C. J. lnject. Dis. 1976,
134, s312 s315.
[3] Bodey, G.P.; Valdivieso, M.; Feld, R.; Ro- driguez, V. Antimicrob. Ag. Chemother. 1974,5, 508 512.
[4] Edson, R.S.; Terrell, C.L. Mayo Clin.
Proc. 1999, 74, 519 528.
[5] White, L.O. Trends Anal. Chem. 1986, 5, 29 31.
[6] Fukuchi, H.; Yoshida, M.; Tsukiai, S.; Ki- taura, T.; Konishi, T. Am. J. Hosp. Pharm. 1984, 41,690 693.
[7] Kurtz, M.J.; Billings, M.; Koh, T.; Olan- der, G.; Tyner, T.; Weaver, B.; Stone, L.
Clin. Chem. 1983,29, 1015 1019. [8] Li, T.M.; Robertson, S.P.; Crouch, T.H.;
Pahuski, E.E.; Bush G.A.; Hydo, S.J. Clin.
Chem. 1983,29, 1628 1634.
[9] Walter, B.; Greenquist, A.C.; Howard, W.E. Anal. Chem. 1983,55, 873 878. [10] Weber, A.; Smith, A.L.; Opheim, K.E.J.
Clin. Microbiol. 1985,21,419 424. [11] Woo, F.L.; Johnson, A.P.; Insler, M.S.;
George, W.J.; LaCorte, W.S. Arch.
Ophthalmol. 1985, 103,216 218. [12] Khabbaz, R.F.; Standiford, H.C.; Bern-
stein, D., Nipper, H.C.; Tatem, B.A.;
Smalls, U.; Drusano, G.L.; Caplan, E. J.
Clin. Microbiol. 1985, 22, 699 701. [13] Stevens, P.; Young, L.S.; Hewitt, W.L.J.
Antibiotics 1976,29, 829 832.
[14] White, L.O.; Holt, H.A.; Reeves, D.S.; MacGowan, A.P. Z Antimicrob. Che-
mother. 1997, 39, 355 361.
[15] Blaser,J.; K6nig, C.; Fatio, R.; Follath, F.; Cometta, A.; Glauser, M. members of the International Antimicrobial Therapy Cooperative Group of the European Or- ganization for Research and Treatment of Cancer, Ther. Drug Monit. 1995, 17, 133 136.
[16] Zaninotto, M.; Secchiero, S.; Paleari, C.D.; Burlina, A. Ther. Drug Monit. 1992,
14, 301 305.
[17] Nanji, A.A.; Filipenko, J.D.; Smith, J.A.; Ngui-Yen, J. Drug lntel. Clin. Pharm. 1984, 18, 738.
[18] Ngui-Yen, J.H.; Doyle, P.W.; Smith, J.A.
J. Clin. Microbiol. 1984, 20, 962 965. [19] Nilsson-Ehle, I. J. liq. Chromatogr. 1983,
6, 251 293.
[20] Isoherranen, N.; Soback, S. J. AOAC Int. 1999, 82, 1017 1045.
[21] Solt6s, L., Biomed. Chromatogr. 1999, 13, 3 10.
[22] Lung, K.R.; Kassal, K.R.; Green, J.S.; Hovsepian, P.K.J. Pharm. Biomed. Anal.
1998,16,905 910.
[23] Gambardella, P.; Punziano, R.; Gionti, M.; Guadalupi, C.; Mancini, G. J. Chro-
matogr. 1985, 348, 229 240.
[24] Kabra, P.M.; Bhatnager, P.K.; Nelson, M.A.J. Chromatogr. 1984, 307, 224 229. [25] Papp, E.A.; Knupp, C.A.; Barbhaiya,
R.H.J. Chromatogr. 1992, 574, 93 99. [26] Barends, D.M.; Blauw, J.S.; Smits, M.H.;
Hulshoff, A. J. Chromatogr. 1983, 276, 385 394.
[27] Wong, L.T.; Beaubien, A.R.; Pakuts, A.P.
J. Chromatogr. 1982,231, 145 154. [28] Sampath, S.S.; Robinson, D.H.J. Pharm.
Sci. 1990, 79, 428 431.
[29] Morovj/m, Gy.; Csok~n, P.P.; N6meth- Konda, L.; Chromatographia 1998, 48, 32 36.
[30] Izquierdo, P.; Pavdn, P.; Gbmez-Hens, A.; Pdrez-Bendito, D. Fresenius J. Anal.
Chem. 1994, 349, 820 823.
[31] Sar, F.; Leroy, P.; Nicolas, A. Anal. Lett. 1992,25, 1235 1250.
[32] Caturla, M.C.; Cusido, E. J. Chromatogr. 1992, 593, 69 72.
[33] Wichert, B.; Schreier, H.; Derendorf, H. J.
Pharm. Biomed. Anal. 1991,9, 251 254. [34] Essers, L. J. Chromatogr. 1984, 305, 345
352.
[35] Feng, C.H.; Lin, S.J.; Wu, H.L.; Chen, S.H.J. Liq. Chrom. & Rel. Technol., (in press).
Received: Oct 2, 2000 Accepted: Oct 18, 2000