Novel distyrylcarbazole derivatives as hole-transporting blue emitters for
electroluminescent devices{
Fang-Iy Wu, Ping-I Shih, Mao-Chuan Yuan, Ajay Kumar Dixit, Ching-Fong Shu,* Zhu-Ming Chung and
Eric Wei-Guang Diau*
Received 14th July 2005, Accepted 13th September 2005
First published as an Advance Article on the web 30th September 2005 DOI: 10.1039/b510035f
We have synthesized three novel distyrylcarbazole derivatives for use as simultaneously hole-transporting and light-emitting layers in blue light-emitting diodes. Each compound, which contains a rigid carbazole core and two 2,2-diphenylvinyl end groups substituted through either the 3,6- or the 2,7- position, forms films satisfactorily and exhibits a blue emission with its PL maximum in the range 459–470 nm. Photophysical measurements indicate that twisting of the adjacent C–C bonds in the 3,6-position of the carbazole core in dilute solutions causes an efficient nonradiative relaxation to occur, yielding a much smaller quantum yield for fluorescence in 3,6-linked carbazoles. As intense emissions of 2,7-linked carbazoles are observed, such deactivation from an excited-state is inefficient therein. Electrochemical studies revealed that incorporation of the carbazole core increased the HOMO energy effectively; this feature facilitates hole injection. These distyrylcarbazole derivatives are promising bifunctional, blue-emitting, hole-transporting molecules for use in simple double-layer devices of a general structure ITO/emitting layer/TPBI/Mg : Ag, in which TPBI—1,3,5-tris(N-phenylbenzimidazol-2-yl)benzene—serves as a hole-blocking and electron-transporting material. The devices prepared using
2,7-distyrylcarbazole as the emitter produced bright blue emissions having activating voltages below
3.0 V. A DPVTCz-based device attained a luminance efficiency of 3.11 cd A21at 5 V, a brightness
of 3062 cd m22, and CIE color coordinates of (0.14, 0.22).
Introduction
Conjugated organic materials that function as both efficient light emitters and/or charge transporters have attracted much interest because of their prospective applications in organic
light-emitting diodes (OLED).1 Organic materials that emit
light and have large-energy band gaps—so that they emit blue light efficiently—have, in particular, stimulated a wide range of interest because they might be applicable as sources of blue light in full-color display applications or as hosts for
exothermic energy transfer to lower-energy fluorophores.2
Such materials’ large band gaps lead to an inherent difficulty when attempting to inject charges into the blue emitters; this phenomenon hampers the performance of devices fabricated from these materials, especially for single- or double-layer
devices.3
Among the various known blue emitters, the most promising are distyrylarylene derivatives (DSA) because their neat solids
fluoresce intensely and because they form satisfactory films.4
Efficient organic diodes that emit blue and white light have been prepared using blue-emitting 4,49-distyrylbiphenyl
derivatives—e.g., 4,49-bis(2,2-diphenylvinyl)-1,19-biphenyl
(DPVBi)—that function as the electroluminescent layer.5The
most salient feature of DPVBi is that two nonplanar phenyl rings are located at each end of the molecule; this arrangement prevents exciplex formation at the interface between the
DPVBi layer and its neighboring charge-transporting layer.4c
Furthermore, distyrylarylene derivatives that incorporate arylamine moieties can exhibit both intense fluorescence and great mobility for the transport of holes. For instance, the hole mobility of BCzVBi, a DSA derivative possessing carbazoryl groups at both ends of the molecule, was determined to be as
large as 1023cm2V21s21(1–3 MV cm21) through
measure-ment of the transient behavior of the EL of ITO/BCzVBi
(120 nm)/Mg : Ag.4a In a double-layer device with the
configuration ITO/BCzVBi/PBD/Mg : Ag, a large power
efficiency (2.1 lm W21) was obtained at 5 V, with a brightness
of 135 cd m22.4bAs the emission of this BCzVBi-based device
occurred in the blue-green region, its use as a blue emitter is restricted in OLED applications.
Our objective in this study was to prepare distyryl derivatives that display both hole-transporting ability and highly efficient fluorescence in the blue region. Polymers and small molecules containing carbazole units are often effective hole-transporting materials because the nitrogen atom in
the carbazole ring bestows an electron-donating ability.6 We
designed three analogues of DPVBi in which we retained the 2,2-diphenylvinyl end groups and replaced the biphenyl core with a carbazole ring, substituted through either the 3,6- or the 2,7- position, in an attempt to realize blue distyrylcarbazole derivatives that might serve as both the hole-transporting layer
(HTL) and the emitting layer (EML).3b,4b,7We examined the
Department of Applied Chemistry, Institute of Molecular Science and Center for Interdisciplinary Molecular Science, National Chiao Tung University, Hsinchu, Taiwan, 30010
{Electronic supplementary information (ESI) available: details of experimental procedures and fabrication of light-emitting devices. See DOI: 10.1039/b510035f
thermal, optical, and electrochemical properties of these distyrylcarbazole derivatives, and fabricated double-layer devices to demonstrate the potential of these distyrylcarbazole compounds as blue light-emitting and hole-transporting materials for use in EL device applications.
Results and discussion
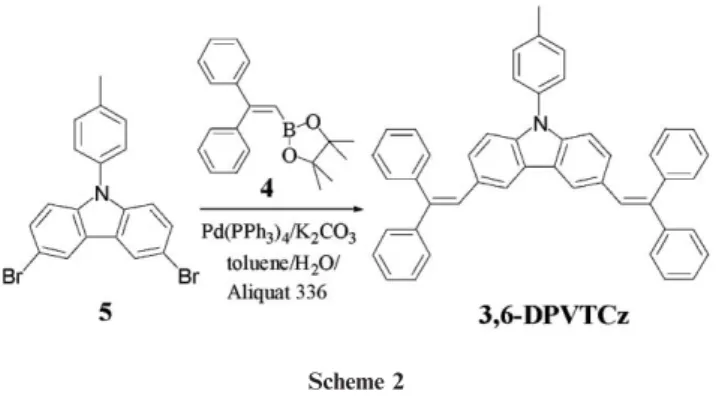
SynthesisScheme 1 illustrates the synthetic procedures used to prepare the 2,7-linked carbazole derivatives. The key intermediate, 2,7-dibromo-9H-carbazole (1), was synthesized according to a procedure described in the literature: nitration of 4,49-dibro-mobiphenyl with concentrated nitric acid followed by a
reductive Cadogan ring-closure.8 N-Alkylation of 1 with
isopropyl bromide in NMP–K2CO3 gave 2. The
copper-catalyzed Ullman coupling of 1 with 4-iodotoluene yielded 3. We prepared the borolane 4 through lithiation of
2,2-diphenylvinyl bromide9 with excess n-BuLi and subsequent
treatment with butyl borate and hydrolysis in aqueous HCl to give 2,2-diphenylvinyl boronic acid, which we then converted to the boronic ester through its reaction with pinacol. The Pd-catalyzed Suzuki coupling of borolane 4 with the 2,7-dibromocarbazole derivatives 2 and 3 afforded target compounds DPVICz and DPVTCz, respectively. Similarly, we prepared 3,6-DPVTCz through Suzuki coupling of 4 with the
3,6-dibromocarbazole compound10 5 (Scheme 2). DPVICz,
DPVTCz, and 3,6-DPVTCz, which we characterized using1H
and 13C NMR spectroscopy, were sublimable in a vacuum
chamber without their thermal decomposition. High-resolu-tion mass spectrometric and elemental analyses provided additional verification of the proposed structures.
Photophysical properties
We measured the absorption and fluorescence spectra of the three carbazole derivatives in dilute toluene solutions and as neat films on a quartz substrate (Fig. 1); Table 1 summarizes the corresponding spectral data. We observed no significant difference between the absorption spectra of DPVICz and DPVTCz in toluene; their absorption maxima occur at 376–378 nm. This finding indicates that the p-tolyl group in DPVTCz does not contribute significantly to extending the
length of p conjugation.11 In contrast, the absorption
maximum of 3,6-DPVTCz is located at 325 nm, i.e., it is blue-shifted by ca. 50 nm with respect to that of DPVTCz. This shift presumably originates from the disparate lengths of conjugation that result from the varied substitution patterns of 2,2-diphenylvinyl groups on the carbazole cores. In 2,7-linked carbazoles, conjugation can extend over the entire molecule, whereas in the 3,6-linked carbazole there is little conjugation
over large distances.12
Scheme 1
Scheme 2
Fig. 1 UV-Vis absorption and PL spectra of (a) DPVICz, (b) DPVTCz, and (c) 3,6-DPVTCz in dilute toluene solutions (solid lines) and in the solid state (dashed lines).
Both 2,7-disubstituted carbazoles display intense photo-luminescence in the blue region, with their respective maxima
occurring at ca. 460 nm. The fluorescence quantum yields (Wf)
of DPVICz and DPVTCz in dilute toluene solution were 0.37 and 0.51, respectively; they are significantly greater than that
of DPVBi (Wf = 0.25) when measured under the same
conditions with reference to 9,10-diphenylanthracene in
cyclohexane (Wf= 0.90).
13
In contrast, 3,6-DPVTCz exhibits a weak emission in toluene solution, with the maximum intensity occurring at ca. 460 nm; its fluorescence quantum yield is only ca. 0.01. The observed disparate magnitudes of
Wf—a factor of ca. 50—between 3,6-DPVTCz and DPVTCz
indicates the existence of an efficient channel for nonradiative relaxation that is activated in the 3,6-linked carbazole but inhibited in the 2,7-linked carbazole. Our control experiments indicate that the carbazole core itself displays an intense emission in dilute solution. Moreover, we observed an enhanced emission of the linked carbazole in a 3,6-DPVTCz/PMMA solid film when the nuclear motions of the molecule were substantially restricted in a PMMA matrix. Thus, we obtained strong evidence supporting the notion that deactivation of a carbazole derivative in its excited state is efficient when a key nuclear motion is activated for the system possessing p-conjugated substituents in the 3,6-positions, but not in the 2,7-positions.
We used G03 software to perform quantum chemical
calculations of the excited states of model systems14 in an
attempt to rationalize the observed large discrepancy in the
values of Wfbetween the 3,6- and 2,7-linked carbazoles. Our
preliminary calculations indicate that the phenyl groups are twisted substantially with respect to the carbazole ring. Therefore, we believe that the interactions between the phenyl groups and the carbazole center are weak. To diminish the computational cost and to allow a systematic comparison, we replaced all of the phenyl groups with saturated hydrocarbon units to serve as model systems in these calculations. As a result, the model systems contain one ethyl group connected to the central N atom and two dimethylvinyl groups connected to the carbazole core in either the 2,7- or 3,6-position. The geometries of both the 2,7-linked and 3,6-linked carbazoles in
the ground state (S0) were optimized based on density
functional theory (DFT) calculations at the B3LYP/6-31G(d) level. The optimized geometries of the 2,7- and 3,6-linked species are basically nonplanar with the two bridged C–C bonds being twisted substantially through a common torsion
angle (d) of ca. 37–38u for both molecules in their S0states.
In Fig. 2, we display a comparison of the corresponding HOMO-1, HOMO, and LUMO of the carbazole derivatives
optimized in the S0 state. Because electronic excitation from
the HOMO to the LUMO produces the first singlet excited
state (S1), the orbital features presented in Fig. 2 might provide
important clues toward understanding the distinct photo-physical properties of the two types of carbazole substituents. For the LUMO, the orbitals in the carbazole core are almost identical for both molecules, but an extension of the p-conjugation over the two dimethylvinyl groups occurs only for the substituents in the 2,7-positions, i.e., not in the 3,6-positions, which makes the bridged C–C bonds in the 2,7-linked carbazole display double-bond character. For the HOMO, p-conjugation of the orbitals is extended effectively to the dimethylvinyl groups in both the 2,7- and 3,6-linked carbazoles, but the bridged C–C bonds retain their single-bond character in both cases. Thus, we expected that a feasible twisting motion would occur in the ground-state surface for both molecules. According to the LUMO presented in Fig. 2, we expect that rotation about the bridged C–C bonds is more difficult to perform in the 2,7-linked carbazole than it is in the
3,6-linked carbazole in the S1state.
Rotation about the bridged C–C bonds toward a twisted geometry would involve an energy barrier for both model systems because of the lack of extended p-conjugation in the LUMO for the 2,7-linked carbazole and in the HOMOs of both molecules. To confirm this hypothesis, we performed geometry optimization calculations at the CIS/6-31G(d) level
of theory in the S1state. We found that the S1minimum (min)
of the 2,7-linked carbazole is located at d = ca. 28u, whereas the
S1 (min) of the 3,6-linked carbazole has an asymmetric
geometry with the two bridged C–C bonds being twisted by
35 and 49u, respectively. Along the single C–C bond twist
reaction coordinate (RC), the corresponding transition struc-tures are located at the perpendicular geometry (the single C–C
bond twist angle is 90u); note that d = ca. 37–38u at the
Franck–Condon (FC) geometry in both cases. To obtain the energy levels in an effort to improve our understanding of the interactions between excited-state surfaces, we performed single-point energy calculations at the TD B3LYP/6-31+G(d)
level of theory for three key geometries: the FC, the S1(min),
Table 1 Optical properties of distyrylcarbazole derivatives
Quantum yields (Wf)a,b Absorption, lmax/nm Photoluminescence,c lmax/nm Solution
(log e)a Filmd Solutiona Filmd
DPVICz 0.37 378 (4.71) 378 457 470
DPVTCz 0.51 376 (4.71) 378 456 469
3,6-DPVTCz 0.01 325 (4.70) 327 456 459 aMeasured in toluene.bThe relative quantum yield was measured in toluene with reference to that of 9,10-diphenylanthracene in cyclohexane.cThe photoluminescence maximum was recorded upon irradiation at the absorption maximum. dPrepared by spin-coating from their CHCl3solutions.
Fig. 2 Calculated HOMO-1, HOMOs, and LUMOs of dimethyl-vinylcarbazole derivatives; the geometries were optimized at the B3LYP/6-31G(d) level of theory.
and the perpendicular species (P1 or P2). According to our calculations, efficient nonradiative electronic relaxation might occur through twisting of the bridged C–C bond(s) along
either the single C–C twist RC (via P1species) or the concerted
C–C twist RC (via P2species) leading to a much smaller value
of Wfbeing observed for the 3,6-linked carbazole than for the
2,7-linked carbazole.15 The larger Stokes shift observed for
3,6-DPVTCz than for DPVTCz (Fig. 1) is consistent with our calculations that the relative energy between the FC geometry
(S3 for the 3,6-linked carbazole; S2 for the 2,7-linked
carbazole) and the S1minimum (given the maximum emission
intensity) of the 3,6-linked carbazole (ca. 0.5 eV) is larger than that of the 2,7-linked carbazole (ca. 0.35 eV).
According to these quantum chemical calculations, we provide a schematic illustration (Scheme 3) to rationalize the
observed discrepancy in Wf between the 2,7- and 3,6-linked
carbazoles. Because the singlet p,p*excited state is zwitterionic
in nature,16 its positive and negative charges are completely
separable through electronic resonance in the 2,7-linked molecule, whereas charge separation is localized in only the carbazole core of the 3,6-linked molecule (Scheme 3). As a result, electron redistribution in the 2,7-linked carbazole causes the original flexible C–C single bond between the diphenyl-vinyl moiety and the carbazole core to become a rigid bridge, having double-bond character, because rotation about the
C–C double-bond axis becomes unfavorable in the 1(p,p*)
excited state. Localization of the p-conjugation in the
carbazole core of the 3,6-linked molecule in the 1(p,p*) state
does not affect its feasible C–C single bond twisting motion, which, thus, enables efficient nonradiative relaxation. Hence, we conclude that localization of the p conjugation and the bridged C–C bond twisting motion in the excited-state surface
are responsible for the much smaller values of Wfobserved for
the 3,6-linked carbazole.
The absorption spectra of thin films prepared through spin-coating of the carbazole derivatives from chloroform solutions onto quartz plates are almost identical to those obtained in dilute solutions (see the dashed curves in Fig. 1). The emission spectra of the 2,7-linked carbazoles in thin films were red-shifted by ca. 10 nm, but the spectrum of the 3,6-linked carbazole is similar to that obtained in solution, reflecting the fact that molecular packing of the 2,7- and 3,6-linked carbazoles is different in their respective solid states.
Thermal properties
We investigated the thermal properties of these distyryl-carbazole derivatives using differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA); Table 2 pre-sents the results. Fig. 3 displays DSC curves recorded over a
temperature range 30–320uC. DPVICz and DPVTCz exhibit
distinct glass transition temperatures (Tg) at 80 and 92 uC,
respectively, followed by broad crystallization features at 121
and 142uC, respectively, and well-defined melting points at
193 and 244uC, respectively. For 3,6-DPVTCz, we observed a
glass transition at 100uC, but no crystallization or melting of
the sample at temperatures up to 320 uC. This result might
reflect the different symmetry in the molecular structure:6cthe
2,7-linked carbazoles have a rod-like architecture, whereas the 3,6-linked carbazole has a L-shaped structure, which might hinder close packing and diminish its tendency to crystallize. For comparison with these carbazole-based compounds, the
related analogue DPVBi has a glass transition at 64uC prior to
crystallizing at 106uC.17
Consequently, these distyrylcarbazole derivatives form more-stable amorphous glasses than does DPVBi and are, therefore, more promising—in terms of their thermal stability—for their application into OLEDs. We attribute the enhanced morphological stability of the carba-zole-based materials to the presence of their rigid carbazole
linkages.6c These distyrylcarbazole derivatives also exhibit
satisfactory thermochemical stability; as evident from the thermogravimetric analyses performed under a nitrogen atmosphere, their 5%-weight-loss temperatures reach as high
as 350uC.
Electrochemistry
We performed cyclic voltammetry (CV) measurements of the carbazole-containing compounds near 295 K using ferrocene as an internal standard. Cyclic voltammetry is a simple method
Scheme 3
Table 2 Thermal properties of distyrylcarbazole derivatives
DSCa TGAb
Tg(uC) Tc(uC) Tm(uC)
5% weight loss 10% weight loss DPVICz 80 121 193 351 368 DPVTCz 92 142 244 380 395 3,6-DPVTCz 100 n.a. n.a. 376 394 a
Measured under nitrogen at heating rates of 20 uC min21 and cooling rates of 40uC min21. n.a.: not available.bMeasured under nitrogen at a heating rate of 20uC min21
.
Fig. 3 DSC traces of distyrylcarbazole derivatives, recorded at a heating rate of 20uC min21
.
that allows the HOMO/LUMO energy levels of these materials to be determined; in this case, we calculated them with regard to the energy level of the ferrocene reference (4.8 eV below the
vacuum level).18Fig. 4 displays cyclic voltammograms of these
carbazole derivatives. Both of the 2,7-distyrylcarbazoles exhibit irreversible redox behavior. The onset potentials for the oxidations of DPVICz and DPVTCz are 0.58 and 0.62 V, respectively, and those for the reductions are 22.44 and
22.41 V, respectively, vs. Fc/Fc+. For 3,6-DPVTCz, we
observed a reversible oxidation at 0.55 V, with the onset potential at 0.48 V, and an irreversible reduction with its onset potential at 22.64 V. Similar phenomena have been reported previously for the 2,7- and 3,6-linked carbazole trimers in their cyclic voltammograms, with the reversible oxidation process
occurring only for the latter.12Hence, protection of the 3 and 6
positions through substitution prevents C–C coupling at these positions; such chemical reactions are the cause of irreversible
oxidation.11,12Table 3 summarizes electrochemical properties
of the three carbazole derivatives. From the electrochemistry data we estimate that the band gaps of DPVICz, DPVTCz, and 3,6-DPVTCz are 3.02, 3.03, and 3.12 eV, respectively; these values agree satisfactorily with the optical band gaps calculated from the absorption edges of the corresponding UV-Vis spectra. Again, the larger band gap of 3,6-DPVTCz is related to its smaller conjugation length.
Electroluminescence properties
To evaluate the utility of distyrylcarbazole derivatives as simultaneously emitting and hole-transporting layers, we fabricated double-layer EL devices with the configuration ITO/EML/TPBI/Mg : Ag. The emitting layer was deposited on ITO glass and a layer of TPBI was then deposited to function as the electron-transporting and hole-blocking layer. We co-evaporated a Mg–Ag alloy as the cathode, which we then protected with an additional layer of Ag. For comparison, we also constructed a control device using DPVBi as the emitting layer. Fig. 5 provides the energy levels of the materials used for the double-layer devices. According to this diagram, DPVICz, DPVTCz, and 3,6-DPVTCz have ionization energies of 5.38, 5.42, and 5.28 eV, respectively; these values are considerably smaller than that of DPVBi (5.59 eV, obtained through CV measurement), which indicates that holes can be injected from
the ITO electrode (WITO= 4.8 eV) into the HOMO levels of the
distyrylcarbazole derivatives more readily than into the HOMO of DPVBi. In contrast, the LUMO level of the TPBI
layer is 2.7 eV;19thus, electrons can readily cross the internal
heterojunction into the LUMO levels of the emitters by conquering a small energy barrier of 0.3–0.5 eV (Fig. 5). Fig. 6 displays plots of current density vs. applied voltage for the devices based on these distyryl derivatives. The I–V curve of the DPVBi-based device clearly differs from those of the other Fig. 4 Cyclic voltammograms of (a) DPVICz, (b) DPVTCz, and (c)
3,6-DPVTCz.
Table 3 Electrochemical properties of distyrylcarbazole derivatives Eox onset/ Va Ered onset/ Va HOMO/ eVb LUMO/ eVc Eel g/ eVd Eopt g / eVe DPVICz 0.58 22.44 25.38 22.36 3.02 2.90 DPVTCz 0.62 22.41 25.42 22.39 3.03 2.92 3,6-DPVTCz 0.48 22.64 25.28 22.16 3.12 3.12 aPotential values are listed vs. Fc/Fc+.bDetermined from the onset oxidation potential.cDetermined from the onset reduction potential. dElectrochemical band gap, estimated with a relation Eel
g = Eoxonset2 Ered
onset. e
Optical band gap, calculated from the absorption edge of the UV-Vis spectrum.
Fig. 5 Energy levels of materials used to prepare the double-layer devices.
Fig. 6 Current density-voltage (I–V) curves of devices with the configuration ITO/EML/TPBI/Mg : Ag.
three, and it is shifted to a greater voltage. We ascribe this increased operating voltage for the DPVBi-based device to its larger injection barrier for holes; the similar I–V characteristics that we observe among the three distyrylcarbazole compounds reflect their similar HOMO levels. Fig. 7 displays EL spectra of devices based on the distyrylcarbazole derivatives and DPVBi. These devices exhibit blue emissions that nearly coincide with their corresponding fluorescence spectra in the solid state; this observation implies that only the emitting layer con-tributes to the light emission and that no exciplexes form at the EML/TPBI interface. Moreover, the appearance of the EL spectra of each device that we investigated is independent of the applied voltage.
In this bilayer device geometry, blocked holes accumulate at the internal heterojunction, which results in a positive space– charge interfacial zone. This accumulated positive charge in turn enhances electron injection to neutralize the accumulated
holes, which improves the charge recombination.20 With the
facilitation of both hole and electron injections, the
distyryl-carbazole-based devices become activated at potentials
below 4.0 V and emit brighter light and have more efficient performance than does the DPVBi-based device (cf. Fig. 8 and 9). We obtained the maximum external quantum efficiency
(1.94%) at 5.0 V with a brightness of 3062 cd m22 for the
device based on DPVTCz; for the device based on DPVICz,
these values were 0.92%, 6.0 V, and 3059 cd m22, respectively.
Because of its small quantum yield for fluorescence, the maximum external quantum efficiency of the 3,6-DPVTCz-based device was only 0.11%; this performance occurred at an applied voltage 6.0 V and was accompanied by a brightness
of 80 cd m22. For the DPVBi-based device, the maximum
external quantum efficiency was only 0.03% at 9.5 V and it decreased rapidly upon increasing the current (Fig. 9). From our investigations of these EL devices, we find that the incorporation of the p-type carbazole core, rather than a biphenyl one, can significantly decrease the operating voltage and improve the device performance in a low-voltage region. Furthermore, taking the DPVTCz-based device as an example,
the maximum power efficiency was 2.18 lm W21, which
occurred at 4.0 V, with a brightness of 708 cd m22. This power
efficiency is comparable to that of other multi-layer devices
that exhibit efficient blue fluorescence.21 Furthermore, the
DPVTCz-based device can attain a brightness greater than
104 cd m22 at a potential below 7.5 V. Table 4 summarizes
the characteristics of the devices based on the four DPVBi analogues.
Conclusions
We have synthesized novel distyrylcarbazole derivatives, each of which contains part of the well-known DPVBi functionality, but with the biphenyl core replaced by a carbazole ring; we characterized their photophysical and electrochemical pro-perties with respect to their functions in OLEDs. In dilute solutions the 2,7-linked carbazole exhibits an intense emission
(Wf = 0.4–0.5), whereas the 3,6-linked carbazole displays a
weak emission (Wf = ca. 0.01). According to our theoretical
calculations, ISC deactivation in the 3,6-linked carbazole is activated efficiently through the twisting motion of the bridged C–C single bond(s), whereas this electronic relaxation is inefficient in the 2,7-linked carbazole because the extensive Fig. 7 EL spectra of devices having the configuration ITO/EML/
TPBI/Mg : Ag, recorded at an applied voltage of 7 V. Fig. 8configuration ITO/EML/TPBI/Mg : Ag.Luminescence–voltage (L–V) curves of devices having the
Fig. 9 Plots of luminance efficiency vs. current density for the distyryl derivative-based devices.
degree of p-conjugation of the orbitals provided the bridged C–C bond with double-bond character. These compounds form satisfactory films that possess the dual functions of emitting blue light and transporting holes. Double-layer devices prepared using 2,7-distyrylcarbazole derivatives—as the simultaneously emitting and hole-transporting layer—in combination with TPBI—as the hole-blocking and electron-transporting material—produced bright blue emissions and had activating voltages below 3.0 V. The luminance efficiency
of the DPVTCz-based device reached 3.11 cd A21at 5 V, a
brightness of 3062 cd m22, and CIE color coordinates of (0.14,
0.22). In contrast, the device based on pristine DPVBi displayed poor performance and required a larger operating voltage.
Acknowledgements
We thank the National Science Council for financial support. Our special thanks go to Professor C.-H. Cheng, Dr J.-P. Duan, and Dr H.-T. Shih for their support and cooperation during the preparation and characterization of the light-emitting devices.
References
1 (a) C. H. Chen, J. Shi and C. W. Tang, Coord. Chem. Rev., 1998, 171, 161; (b) U. Mitschke and P. Ba¨uerle, J. Mater. Chem., 2000, 10, 1471; (c) L. S. Hung and C. H. Chen, Mater. Sci. Eng., R, 2002, 39, 143.
2 (a) J. Kido, K. Hongawa, K. Okuyama and K. Nagai, Appl. Phys. Lett., 1994, 64, 815; (b) J. Kido, H. Shionoya and K. Nagai, Appl. Phys. Lett., 1995, 67, 2281; (c) F. C. Chen, Y. Yang, M. E. Thompson and J. Kido, Appl. Phys. Lett., 2002, 80, 2308. 3 (a) C. Adachi, T. Tsutsui and S. Saito, Appl. Phys. Lett., 1990, 57,
531; (b) K. R. J. Thomas, M. Velusamy, J. T. Lin, Y.-T. Tao and C.-H. Chuen, Adv. Funct. Mater., 2004, 14, 387; (c) W. L. Jia, X. D. Feng, D. R. Bai, Z. H. Lu, S. Wang and G. Vamvounis, Chem. Mater., 2005, 17, 164.
4 (a) C. Hosokawa, H. Tokailin, H. Higashi and T. Kusumoto, Appl. Phys. Lett., 1993, 63, 1322; (b) C. Hosokawa, H. Tokailin, H. Higashi and T. Kusumoto, J. Appl. Phys., 1995, 78, 5831; (c) C. Hosokawa, H. Higashi, H. Nakamura and T. Kusumoto, Appl. Phys. Lett., 1995, 67, 3853; (d) S. E. Shaheen, G. E. Jabbour, M. M. Morrell, Y. Kawabe, B. Kippelen, N. Peyghambarian, M.-F. Nabor, R. Schlaf, E. A. Mash and N. R. Armstrong, J. Appl. Phys., 1998, 84, 2324; (e) Y. Z. Wu, X. Y. Zheng, W. Q. Zhu, R. G. Sun, X. Y. Jiang, Z. L. Zhang and S. H. Xu, Appl. Phys. Lett., 2003, 83, 5077.
5 (a) Y.-S. Huang, J.-H. Jou, W.-K. Weng and J.-M. Liu, Appl. Phys. Lett., 2002, 80, 2782; (b) K. O. Cheon and J. Shinar, Appl. Phys. Lett., 2002, 81, 1738; (c) G. Li and J. Shinar, Appl. Phys. Lett., 2003, 83, 5359; (d) X. Y. Zheng, W. Q. Zhu, Y. Z. Wu, X. Y. Jiang,
R. G. Sun, Z. L. Zhang and S. H. Xu, Displays, 2003, 24, 121; (e) W. Xie, J. Hou and S. Liu, Semicond. Sci. Technol., 2003, 18, L42; (f) W. Xie, S. Liu and Y. Zhao, J. Phys. D: Appl. Phys., 2003, 36, 1246.
6 (a) O. Paliulis, J. Ostrauskaite, V. Gaidelis, V. Jankauskas and P. Strohriegl, Macromol. Chem. Phys., 2003, 204, 1706; (b) Y. Li, J. Ding, M. Day, Y. Tao, J. Lu and M. D’iorio, Chem. Mater., 2004, 16, 2165; (c) Q. Zhang, J. Chen, Y. Chen, L. Wang, D. Ma, X. Jing and F. Wang, J. Mater. Chem., 2004, 14, 895.
7 (a) W. Ni, J. Su, K. Chen and H. Tian, Chem. Lett., 1997, 101; (b) W. Zhu, M. Hu, R. Yao and H. Tian, J. Photochem. Photobiol., A, 2003, 154, 169; (c) P. Kundu, K. R. J. Thomas, J. T. Lin, Y.-T. Tao and C.-H. Chien, Adv. Funct. Mater., 2003, 13, 445; (d) C.-T. Chen, C.-L. Chiang, Y.-C. Lin, L.-H. Chan, C.-H. Huang, Z.-W. Tsai and C.-T. Chen, Org. Lett., 2003, 5, 1261; (e) K. R. J. Thomas, M. Velusamy, J. T. Lin, C.-H. Chuen and Y.-T. Tao, Chem. Mater., 2005, 17, 1860.
8 F. Dierschke, A. C. Grimsdale and K. Mu¨llen, Synthesis, 2003, 2470.
9 S. M. Korneev and D. E. Kaufmann, Synthesis, 2002, 491. 10 S. W. Cha and J.-I. Jin, J. Mater. Chem., 2003, 13, 479. 11 K. Brunner, A. van Dijken, H. Bo¨rner, J. J. A. M. Bastiaansen,
N. M. M. Kiggen and B. M. W. Langeveld, J. Am. Chem. Soc., 2004, 126, 6035.
12 M. Sonntag and P. Strohriegl, Chem. Mater., 2004, 16, 4736. 13 D. Eaton, Pure Appl. Chem., 1998, 60, 1107.
14 M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Jhonson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision B.5, Gaussian, Inc., Wallingford CT, 2004.
15 T.-T. Wang, S.-M. Chung, F.-I. Wu, C.-F. Shu and E. W.-G. Diau, manuscript submitted for publication.
16 M. Klessinger and J. Michl, Excited States and Photochemistry of Organic Molecules, VCH Publishers, New York, 1995.
17 S. Wang, W. J. Oldham, Jr., R. A. Hudack, Jr. and G. C. Bazan, J. Am. Chem. Soc., 2000, 122, 5695.
18 J. Pommerehne, H. Vestweber, W. Guss, R. F. Mahrt, H. Ba¨ssler, M. Porsch and J. Daub, Adv. Mater., 1995, 7, 551.
19 B. Chen, X. H. Zhang, X. Q. Lin, H. L. Kwong, N. B. Wong, C. S. Lee, W. A. Gambling and S. T. Lee, Synth. Met., 2001, 118, 193.
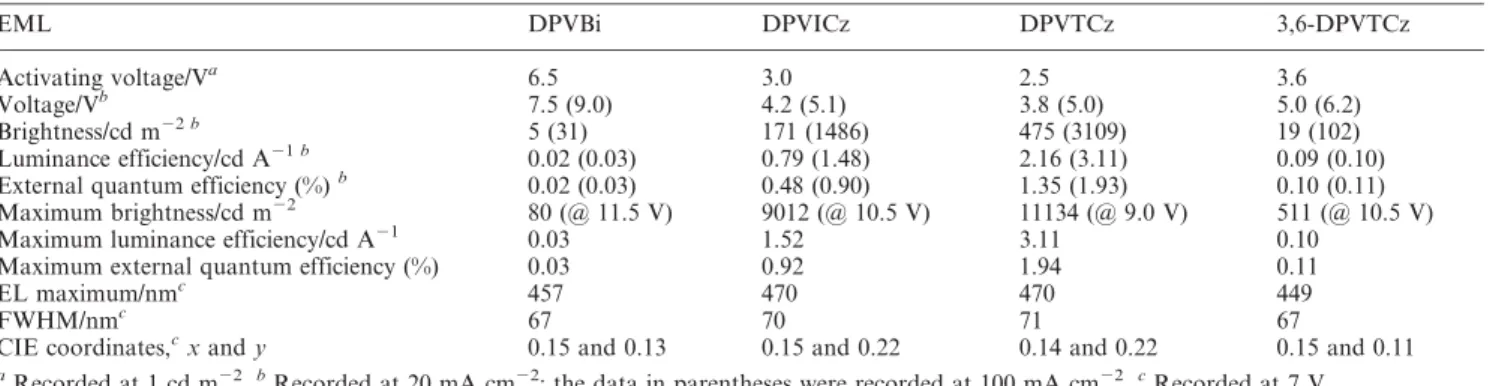
Table 4 Performance of ITO/EML/TPBI/Mg : Ag devices
EML DPVBi DPVICz DPVTCz 3,6-DPVTCz
Activating voltage/Va 6.5 3.0 2.5 3.6
Voltage/Vb 7.5 (9.0) 4.2 (5.1) 3.8 (5.0) 5.0 (6.2)
Brightness/cd m22 b 5 (31) 171 (1486) 475 (3109) 19 (102)
Luminance efficiency/cd A21 b 0.02 (0.03) 0.79 (1.48) 2.16 (3.11) 0.09 (0.10) External quantum efficiency (%)b 0.02 (0.03) 0.48 (0.90) 1.35 (1.93) 0.10 (0.11) Maximum brightness/cd m22 80 (@ 11.5 V) 9012 (@ 10.5 V) 11134 (@ 9.0 V) 511 (@ 10.5 V)
Maximum luminance efficiency/cd A21 0.03 1.52 3.11 0.10
Maximum external quantum efficiency (%) 0.03 0.92 1.94 0.11
EL maximum/nmc 457 470 470 449
FWHM/nmc 67 70 71 67
CIE coordinates,cx and y 0.15 and 0.13 0.15 and 0.22 0.14 and 0.22 0.15 and 0.11 aRecorded at 1 cd m22.bRecorded at 20 mA cm22; the data in parentheses were recorded at 100 mA cm22.cRecorded at 7 V.
20 (a) A. R. Brown, D. D. C. Bradley, J. H. Burroughes, R. H. Friend, N. C. Greenham, P. L. Burn, A. B. Holmes and A. Kraft, Appl. Phys. Lett., 1992, 61, 2793; (b) Y.-H. Tak and H. Ba¨ssler, J. Appl. Phys., 1997, 81, 6963; (c) B. K. Crone, P. S. Davids, I. H. Campbell and D. L. Smith, J. Appl. Phys., 2000, 87, 1947.
21 (a) K. Danel, T.-H. Huang, J. T. Lin, Y.-T. Tao and C.-H. Chuen, Chem. Mater., 2002, 14, 3860; (b) H.-T. Shih, C.-H. Lin, H.-H. Shih
and C.-H. Cheng, Adv. Mater., 2002, 14, 1409; (c) F. He, G. Cheng, H. Zhang, Y. Zheng, Z. Xie, B. Yang, Y. Ma, S. Liu and J. Shen, Chem. Commun., 2003, 2206; (d) Y. Kan, L. Wang, L. Duan, Y. Hu, G. Wu and Y. Qiu, Appl. Phys. Lett., 2004, 84, 1513; (e) W.-J. Shen, R. Dodda, C.-C. Wu, F.-I. Wu, T-H. Liu, H.-H. Chen, C.-H. Chen and C.-F. Shu, Chem. Mater., 2004, 16, 930.