This article was downloaded by: [China Medical University], [Calvin Yu-Chian Chen] On: 05 September 2011, At: 07:58
Publisher: Taylor & Francis
Informa Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House, 37-41 Mortimer Street, London W1T 3JH, UK
Molecular Simulation
Publication details, including instructions for authors and subscription information: http://www.tandfonline.com/loi/gmos20
Treat Alzheimer's disease by traditional Chinese
medicine?
Mao-Feng Sun ab , Tung-Ti Chang ab , Kuan-Chung Chen a , Shun-Chieh Yang a , Kai-Wei Chang a , Tsung-Ying Tsai a , Hsin-Yi Chen c , Fuu-Jen Tsai cd , Jaung-Geng Lin a & Calvin Yu-Chian Chen acef
a
Laboratory of Computational and Systems Biology, School of Chinese Medicine, China Medical University, Taichung, 40402, Taiwan, ROC
b
Department of Acupuncture, China Medical University Hospital, Taichung, Taiwan, ROC c
Department of Bioinformatics, Asia University, Taichung, 41354, Taiwan, ROC d
Department of Medical Genetics, China Medical University, Taichung, 40402, Taiwan, ROC e
Department of Systems Biology, Harvard Medical School, Boston, MA, 02115, USA f
Computational and Systems Biology, Massachusetts Institute of Technology, Cambridge, MA, 02139, USA
Available online: 26 Aug 2011
To cite this article: Mao-Feng Sun, Tung-Ti Chang, Kuan-Chung Chen, Shun-Chieh Yang, Kai-Wei Chang, Tsung-Ying Tsai, Hsin-Yi Chen, Fuu-Jen Tsai, Jaung-Geng Lin & Calvin Yu-Chian Chen (2011): Treat Alzheimer's disease by traditional Chinese medicine?, Molecular Simulation, 37:11, 923-931
To link to this article: http://dx.doi.org/10.1080/08927022.2011.577074
PLEASE SCROLL DOWN FOR ARTICLE
Full terms and conditions of use: http://www.tandfonline.com/page/terms-and-conditions
This article may be used for research, teaching and private study purposes. Any substantial or systematic reproduction, re-distribution, re-selling, loan, sub-licensing, systematic supply or distribution in any form to anyone is expressly forbidden.
The publisher does not give any warranty express or implied or make any representation that the contents will be complete or accurate or up to date. The accuracy of any instructions, formulae and drug doses should be independently verified with primary sources. The publisher shall not be liable for any loss, actions, claims, proceedings, demand or costs or damages whatsoever or howsoever caused arising directly or indirectly in connection with or arising out of the use of this material.
Treat Alzheimer’s disease by traditional Chinese medicine?
Mao-Feng Suna,b, Tung-Ti Changa,b, Kuan-Chung Chena, Shun-Chieh Yanga, Kai-Wei Changa, Tsung-Ying Tsaia, Hsin-Yi Chenc, Fuu-Jen Tsaic,d, Jaung-Geng Linaand Calvin Yu-Chian Chena,c,e,f*
a
Laboratory of Computational and Systems Biology, School of Chinese Medicine, China Medical University, Taichung 40402, Taiwan, ROC;bDepartment of Acupuncture, China Medical University Hospital, Taichung, Taiwan, ROC;cDepartment of Bioinformatics, Asia University, Taichung 41354, Taiwan, ROC;dDepartment of Medical Genetics, China Medical University, Taichung 40402, Taiwan,
ROC;eDepartment of Systems Biology, Harvard Medical School, Boston, MA 02115, USA;fComputational and Systems Biology,
Massachusetts Institute of Technology, Cambridge, MA 02139, USA (Received 28 December 2010; final version received 20 March 2011)
Upregulated phosphodiesterase 4D (PDE4D) disrupts the regulation of calcium ion channel in the central nerve system, and hence it is considered as one of the causes of Alzheimer’s disease. We employed structure-based drug design techniques and the world’s largest traditional Chinese medicine (TCM) database for identifying potential TCM-based PDE4D inhibitors. We then applied multiple linear regression (MLR) and support vector machine (SVM) for quantitative structure – activity relationship model, as well as for molecular dynamics simulation analysis. Screening results suggested that metal cations, Zn2þand Mg2þ, played key roles in mediating stable protein – ligand interactions with the ligand-binding residues, Asp367 and Asp484. In addition, each ligand was shown to interfere with the active residue His326 that suggested inhibitory effects. The MLR and SVM prediction models further implied the PDE4D inhibitory effect of each TCM compound. The molecular simulation further suggested the binding stability of each compound in the PDE4D binding site. We identified three TCM compounds, such as mumefural, 2-O-feruloyl tartaric acid and kainic acid, as potential PDE4D inhibitors. In addition, we further identified the key interaction features associated with the protein – ligand-binding stabilities.
Keywords: traditional Chinese medicine; docking; support vector machine; molecular dynamics; Alzheimer’s disease
1. Introduction
Phosphodiesterase 4D (PDE4D) is a key regulator of cell growth, differentiation, survival and inflammatory pro-cesses [1]. The protein hydrolyses intracellular cyclic adenosine monophosphate (cAMP) which is a key second messenger involved in numerous hormones and neuro-transmitter signalling cascades [2]. The cAMP is an important second messenger which, in response to various hormone stimuli, regulates the protein kinase A-dependent signalling pathways as well as the Ca2þ ion channel. Hence, the inhibition of PDE4D affects cAMP concen-tration, which subsequently affects the downstream biological pathways such as proliferation, metabolism, signalling transduction and neurotransmission. PDE4D inhibitors have been developed in an attempt to resolve inflammatory diseases such as chronic obstructive pulmonary disease, bronchopulmonary dysplasia and asthma, as well as neurodegenerative diseases such as Alzheimer’s disease [3,4]. This study focuses on the discovery and potential application of novel PDE4D inhibitors as a cure for Alzheimer’s disease.
Computer-aided drug design (CADD) has been used to screen, design and analyse various drugs or drug leads from a selected compound database. In this study, we
investigated the drug research potentials of traditional Chinese medicine (TCM) using the CADD methods. Although TCM has been recognized as an alternative treatment, individual TCM compounds were less studied. Nevertheless, previous reports further supported the TCM drug research potentials on viral, anti-inflammatory and anti-tumour [5 – 16]. Hence, we employed CADD and the world’s largest TCM database [17] to investigate TCM-based PDE4D inhibitors for Alzheimer’s disease treatment caused by an abnormal PDE4D upregulation. Hence, we employed the com-pounds from the TCM Database@Taiwan [17], the current world’s largest small molecule database on TCM, for the identification of potential PDE4D inhibitor compounds. The activities of each compound were validated with two quantitative structure – activity relationship (QSAR) models and supported by molecular dynamics (MD) simulations.
2. Materials and methods 2.1 Data collection
The X-ray structure of the PDE4D complex (PDB ID: 3G4G) [4] and more than 20,000 TCM compounds
ISSN 0892-7022 print/ISSN 1029-0435 online
q2011 Taylor & Francis
DOI: 10.1080/08927022.2011.577074 http://www.informaworld.com
*Corresponding author. [email protected]; [email protected] Vol. 37, No. 11, September 2011, 923–931
downloaded from the TCM database [17] were used for virtual screening. The predictive models were established with a total of 16 compounds from Card’s study [18]. The ionisation state of the ionising functional groups for all compounds was modified using Accelrys Discovery Studio 2.5 (DS 2.5) [19].
2.2 Docking
The docking simulation with the CHARMm force field [20] was performed by the Flexible Docking module [21] of DS 2.5, and the cAMP binding site was defined by the volume of co-crystallised compound, RS-25344. The natural substrate of the PDE4D, cAMP, was also used as control. Binding energy was calculated by the Calculate Binding Energies module [22] of DS 2.5. Table 1. Docking results and predicted pIC50for the top TCM
compounds and cAMP.
Predicted pIC50
Name Binding energy SVMa MLR
Mumefural 2 460.19 Active 5.92
2-O-Feruloyl tartaric acid 2 443.67 Active 4.25
Kainic acid 2 393.02 Active 7.76
cAMPb 2 347.35 Active 8.62
4-O-Caffeoylquinic acid 2 335.37 Active 4.64
4-O-Feruloylquinic acid 2 330.58 Active 2.01
Digallic acid 2 328.18 Active 8.49
a
Active when predicted pIC50. 4. bControl. O O –O –O OH Mumefural (a) (b) (c) (d) (e) (g) (f) Kainic acid 4-O-Caffeoylquinic acid Cyclic AMP 4-O-Feruloylquinic acid Digallic acid 2-O-Feruloyl tartaric acid O O O O O OH O O O –O –O O OH +H2N –O O O O– OH OH O –O O O OH OH OH OH OH O O– HO O O OH OH O OH O O O –O HO HO OH N N N N NH2 O OH O P O O –O
Figure 1. The scaffold of (a) mumefural, (b) 2-O-feruloyl tartaric acid, (c) kainic acid, (c) digallic acid, (e) 4-O-caffeoylquinic acid, (f) 4-O-feruloylquinic acid and control and (g) cAMP.
M.-F. Sun et al. 924
2.3 QSAR modelling
The QSAR prediction models were established using both support vector machine (SVM) and multiple linear regressions (MLRs) to predict the bioactivity of TCM compounds. The SVM model was established using LibSVM constructed by Chang and Lin [23], and the MLR function was established using MATLAB [24].
Sixteen compounds from Card’s study [18] were used as a training set of prediction models with activity descriptor of pIC50(log(1/IC50). The genetic function approximation
(GFA) module [25] of DS 2.5 was used to select the suitable molecular descriptors for the prediction models with the training set of 16 molecules. The fitness of individual model for all the possible QSAR models was estimated by the squared correlation coefficient (R2). Cross-validation test was used to validate the prediction model.
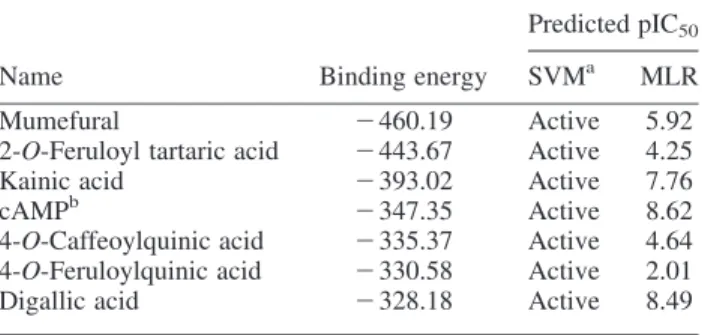
The support vector regression (SVR) model was established using the regression of continuous data with SVM and the gridregression.py program in the libsvm-2.91 package based on the radial basis function [23]. Figure 2. Docking poses of (a) mumefural (b) 2-O-feruloyl tartaric acid (c), kainic acid and (d) cAMP in the PDE4D cAMP binding site. The residues for ligand-binding site (blue) and active site (orange), as well as the electrostatic interactions (light blue line), pi – pi interactions (red line) and hydrogen bond interactions (purple dashed line), were illustrated (colour online).
Table 2. Predicted pIC50of the MLR and SVM models.
MLR SVM
Compound pIC50 Prediction Residual Prediction Residual
1 4.09 4.36 0.27 4.34 0.25 2 4.72 4.71 2 0.01 4.97 0.25 3 6.57 6.75 0.18 6.82 0.25 4 6.06 6.33 0.27 5.81 2 0.25 5 4.72 5.92 1.20 4.91 0.19 6 4.3 4.24 2 0.06 4.55 0.25 7 4.01 3.69 2 0.32 4.26 0.25 8 5.34 5.58 0.24 5.09 2 0.25 9 4.68 4.98 0.30 4.93 0.25 10 4.85 4.65 2 0.20 4.60 2 0.25 11 5.04 4.97 2 0.07 5.28 0.24 12 5.7 5.32 2 0.38 5.93 0.23 13 6 6.85 0.85 6.25 0.25 14 6.8 6.28 2 0.52 6.55 2 0.25 15 7.72 7.10 2 0.62 7.47 2 0.25 16 7.68 6.55 2 1.13 6.30 2 1.38
2.4 MD simulation
The CHARMM force field [20] was applied for the MD simulation using the Simulation package of DS 2.5 [19]. A 7 A˚ solvation shell was created with TIP3P water and neutralised by the additional sodium cations using the Solvation module in DS 2.5. Two minimisation steps were performed with restrained and flexible proteins in sequence. A maximum of 6000 cycles with Steepest Descent [26] and then with Conjugate Gradient [27] was performed in each minimisation step. The time steps for all MD stages were set to 0.002 ps. The SHAKE algorithm was used to fix all bonds involving hydrogen atoms. The long-range electrostatics throughout the MD simu-lation was treated by the PME method. In the heating procedure, the system was gradually heated from 51 to 310 K within 50 ps and the followed by a 200 ps
equilibration phase. In the production procedure, the NVT canonical ensemble was performed with 0.4 ps of temperature coupling decay time for the Berendsen thermal coupling method for 20 ns. The post-processing of the trajectory after MD was analysed using the Analyze Trajectory module in DS 2.5.
3. Results and discussions 3.1 Docking
Table 1 shows the docking results and the predicted activity of top six compounds and cAMP ranked by the binding energy. The scaffold of top six compounds such as mumefural, 2-O-feruloyl tartaric acid, kainic acid, 4-O-caffeoylquinic acid, 4-O-feruloylquinic acid, digallic acid and control cAMP is shown in Figure 1. From the docking pose as shown in Figure 2, the scaffolds of top compounds showed that each compound had at least one carboxyl group that had electrostatic interaction with the two divalent metal cations, Zn2þ and Mg2þ, which mediated the binding to key residues, Asp367 and Asp484. For mumefural, a hydrogen bond (H-bond) with His326 Table 3. Squared correlation coefficients (R2) for each cross-validation test in the MLR model.
Group All 1 2 3 4 5
Squared correlation coefficient (R2) 0.7801 0.7567 0.7619 0.8268 0.8538 0.766
0.0 0.4 0.8 1.2 1.6 2.0 2.4 2.8 3.2 Ligand RMSD 0.0 0.4 0.8 1.2 1.6 2.0 (b) (c)
Mumefural 2-O-Feruloyl tartaric acid
Kainic acid cAMP
Whole molecule RMSD (a) 0 2 4 6 8 10 12 14 16 18 20 –18800 –18600 –18400 –18200 –18000 –17800 –17600 –17400 Mumefural
2-O-Feruloyl tartaric acid Kainic acid cAMP T otal ener gy (kcal/mol) Time (ns)
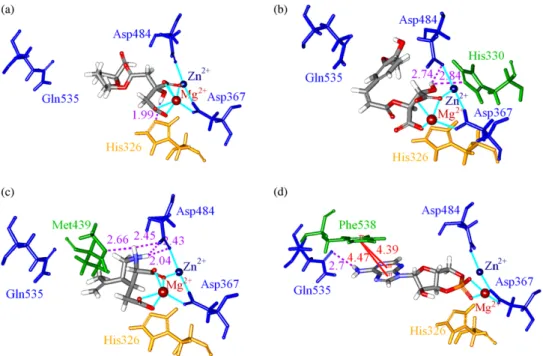
Figure 4. Molecular simulation trajectories of (a) RMSD of PDE4D in complex with mumefural, 2-O-feruloyl tartaric acid, kainic acid and cAMP; (b) only ligand RMSD and (c) total energy of each complex.
3 4 5 6 7 8 3 4 5 6 7 8 3 4 5 6 7 8 95% Prediction bands Predicted activity (MLR) Observed activity R2 = 0.7801 3 4 5 6 7 8 95% Prediction bands Predicted activity (SVM) Observed activity R2 = 0.8860
Figure 3. Comparative plots of observed activity vs. predicted activity (pIC50) of the MLR (top) and SVM (bottom) models.
The black line and magenta lines represent correlation trend and 95% prediction boundaries, respectively.
M.-F. Sun et al. 926
increased the inhibition potential by blocking the access to this active residue (Figure 2(a)). For 2-O-feruloyl tartaric acid and kainic acid, the H-bonds with Asp484 and other residues in the binding domain further stabilised the binding with PDE4D (Figure 2(b), (c)). For the control cAMP-binding conformation, the key binding residue, Gln535, has an H-bond with cAMP. The two divalent metal cations, Zn2þ and Mg2þ, mediated the binding between cAMP and the other two key binding residues, Asp367 and Asp484, through electrostatic interactions. In addition, the cAMP positioned right by the key active residue, His326, suggesting the compound was ready for hydrolysis (Figure 2(d)). This implied that the docking algorithm was reliable by identifying the actual cAMP-binding conformation. Although all compounds did not
interact with the key binding residue, Gln535, the metal ion mediated interactions with the residues Asp367 and Asp484, as well as the interaction with the active residue His326, contributed to the binding stabilities.
3.2 QSAR modelling
GFA was used to select the most representative descriptors to build QSAR models. The selected representative descriptors are Num_RingAssemblies, Jurs_FNSA_1, Jurs_WNSA_1 and Shadow_XY. Each predicted pIC50
and prediction residual are listed in Table 2. A training set of 16 compounds with the four aforementioned descriptors was used to establish an MLR model. The MLR model was cross-validated (Table 3) and the modelling formula was Figure 5. The docking pose snapshots at the 20-ns MD simulation for PDE4D with (a) mumefural, (b) 2-O-feruloyl tartaric acid, (c) kainic acid and (d) cAMP. Residues for binding site (blue) and active site (orange), as well as electrostatic interactions (cyan line) and hydrogen bond interactions (purple dashed line), were illustrated (colour online).
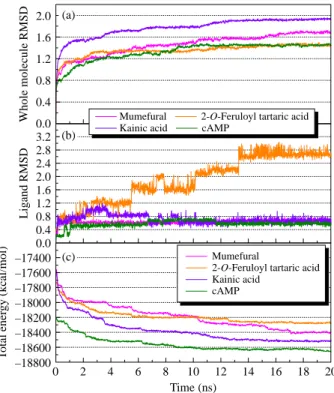
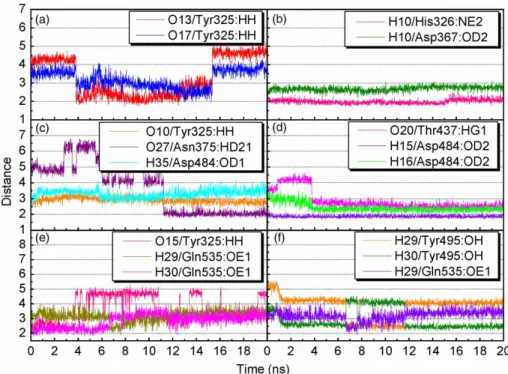
Figure 6. Distance (A˚ ) of hydrogen bonds between PDE4D and (a,b) mumefural, (c) 2-O-feruloyl tartaric acid, (d) kainic acid and (e,f) cAMP.
Figure 7. RMSF (A˚ ) of PDE4D with mumefural, 2-O-feruloyl tartaric acid, kainic acid and cAMP during the 18–20-ns MD simulation for residue numbers (a) 256 – 570, (b) 315 – 380, (c) 455 – 505 and (d) 505 – 550.
M.-F. Sun et al. 928
obtained as follows: Predicted pIC50 ¼ 213:4556 þ 2:6445 £ Num_RingAssemblies þ 51:0269 £ Jurs_FNSA_1 2 0:1611 £ Jurs_WNSA_1 þ 0:1371 £ Shadow_XY:
Same training set of 16 compounds was also used to establish the SVM model. The squared correlation coefficients (R2) were 0.78 and 0.89 for the MLR and SVM models, respectively. Figure 3 shows the statistical significance of each model. The predicted pIC50 using
MLR and SVM models showed that all compounds except 4-O-feruloylquinic acid were potent inhibitors (Table 1).
3.3 MD simulation
The MD simulation was performed for further analysis on each protein – ligand interaction. The whole molecule RMSD and ligand RMSD trajectories displayed the atomic fluctuations during the MD simulation (Figure 4). All ligands tended to stabilise after 14 ns of MD. The trend was also observed in the total energy deviation, in which each complex tended to stabilise after 14 ns of MD (Figure 4(c)). An exception was observed on the top one ligand, mumefural, which was stabilised after 17 ns of simulation. Nevertheless, the trajectory changes between the 14 and 17 ns time points could be neglected in terms of binding stability. In addition, the snapshot of each ligand at 20 ns of MD (Figure 5) indicated that the two divalent metal cations, Zn2þand Mg2þ, acted as a bridge for coordinating ligand interactions to Asp367 and Asp484. Consequently, the electrostatic interactions shown in the docking pose of each complex became more stabilised. For all compounds, stabilised H-bonds were observed during the 20-ns MD simulation (Figure 6). In addition to the common metal cation-mediated bonding, mumefural had an additional stable H-bond with His326 during the MD simulation. Similarly, an H-bond with Asp367 was observed, which further stabilise the binding pose (Figure 6(b)). 2-O-Feruloyl tartaric acid showed H-bonds with Tyr325 and Asn375 during the MD simulation, which indicated the increase in stable position that may interfere the activity of active residue, His326 (Figure 6(c)). Kainic acid had stabilised H-bonds with Asp484 as well as Thr437 during the MD simulation (Figure 6(d)). For cAMP, the H-bonds were stabilised during the MD simulation (Table 4 and Figure 6(e), (f)). In the same way, cAMP gained H-bond interaction with Tyr495 during the MD simulation, suggesting a stabilising docking pose. According to the
Table 4. H-bond distances and occupancies of PDE4D with each of the top three compounds and the control cAMP. Ligand H-bond Ligand atom Amino acid Maximum distance Average distance Minimum distance H-bond occupancy (%) Mumefural 1 O13 Tyr325:HH 5.42 1.75 3.30 37.06 2 O17 Tyr325:HH 4.34 1.92 3.20 8.39 3 H10 His326:NE2 2.63 1.73 1.99 99.85 4 H10 Asp367:OD2 3.11 2.20 2.68 11.24 2-O -Feruloyl tartaric acid 1 O10 Tyr325:HH 3.57 2.91 2.36 1.20 2 O27 Asn375:HD21 6.81 3.72 1.74 43.26 3 H35 Asp484:OD1 4.17 3.30 2.45 0.15 Kainic acid 1 O20 Thr437:HG1 3.74 2.43 1.96 37.51 2 H15 Asp484:OD2 4.64 2.85 2.05 99.90 3 H16 Asp484:OD2 2.31 1.87 1.68 75.42 cAMP 1 O15 Tyr325:HH 5.28 1.94 3.63 11.74 2 N 8 Tyr495:HH 3.29 2.16 2.75 10.84 3 H29 Tyr495:OH 5.67 2.20 3.81 11.79 4 H30 Tyr495:OH 4.54 2.15 2.96 34.17 5 H29 Gln535:OE1 4.20 1.90 3.20 5.99 6 H30 Gln535:OE1 4.04 1.79 2.86 29.72 N ote: H-bond occupan cy cuto ff is 2.5 A˚ .
RMSF in 18 – 20 ns (Figure 7), Asp367, Asp484 and Gln535 were stabilised after binding with the compounds as well as with the cAMP; each had a value of RMSF under 0.2 A˚ . In addition, three compounds also reacted with His326, in which the value of RMSF was reduced from 0.44 A˚ (for cAMP) to under 0.2 A˚.
4. Conclusion
Nowadays, molecular simulation is widely used in material science and drug design [28 – 44]. We applied this technology in this study. The residues Asp367, Asp484 and Gln535 were the key binding residues for the cAMP-binding domain, and the residue His326 was the active residue for PDE4D. According to the analysis each of the TCM compounds, a binding pattern was demonstrated that involved with metal ions, Zn2þand Mg2þ, observed from the docking pose of each TCM compound as well as the corresponding MD simulation. The divalent metal cations were the interaction bridges between compounds and residues, Asp367 and Asp484. The carboxyl groups on each ligand formed stable electrostatic interactions with the divalent metal cations. These stable interactions held the compounds in close approximation to Asp367 and Asp484 and suggested potential PDE4D inhibition. Additionally, all three TCM compounds were bound close to the active residue, His326, for cAMP hydrolysis and consequently interrupted the catalytic process of PDE4D. The inhibition efficacy of TCM compounds was supported by the predicted pIC50of MLR and SVM models. Hence, we suggested these
TCM compounds, mumefural, 2-O-feruloyl tartaric acid, and kainic acid, as potential PDE4D inhibitors.
Acknowledgements
The research was supported by the grants from the National Science Council of Taiwan (NSC 99-2221-E-039-013-), China Medical University (CMU98-TCM, CMU99-TCM, CMU99-S-02) and Asia University (CMU98-ASIA-09). This study was also supported, in part, by Taiwan Department of Health Clinical Trial and Research Center of Excellence (DOH100-TD-B-111-004) and Taiwan Department of Health Cancer Research Center of Excellence (DOH100-TD-C-111-005). We were grateful to the National Center of High-performance Computing for computer time and facilities.
References
[1] M.D. Houslay, M. Sullivan, and G.B. Bolger, The multienzyme PDE4 cyclic adenosine monophosphate-specific phosphodiesterase family: Intracellular targeting, regulation, and selective inhibition by compounds exerting anti-inflammatory and antidepressant actions, Adv. Pharmacol. 44 (1998), pp. 225 – 342.
[2] C. Mehats, C.B. Andersen, M. Filopanti, S.L.C. Jin, and M. Conti, Cyclic nucleotide phosphodiesterases and their role in endocrine cell signaling, Trends Endocrinol. Metab. 13 (2002), pp. 29 – 35. [3] D. Spina, PDE4 inhibitors: Current status, Br. J. Pharmacol. 155
(2008), pp. 308 – 315.
[4] A.B. Burgin, O.T. Magnusson, J. Singh, P. Witte, B.L. Staker, J.M. Bjornsson, M. Thorsteinsdottir, S. Hrafnsdottir, T. Hagen, A.S. Kiselyov, L.J. Stewart, and M.E. Gurney, Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhan-cing cognition with improved safety, Nat. Biotechnol. 28 (2010), pp. 63 – 70.
[5] H.F. Lu, Y.J. Chie, M.S. Yang, C.S. Lee, J.J. Fu, J.S. Yang, T.W. Tan, S.H. Wu, Y.S. Ma, S.W. Ip, and J.G. Chung, Apigenin induces caspase-dependent apoptosis in human lung cancer A549 cells through Bax- and Bcl-2-triggered mitochondrial pathway, Int. J. Oncol. 36 (2010), pp. 1477 – 1484.
[6] M.L. Lin, Y.C. Lu, J.G. Chung, Y.C. Li, S.G. Wang, S.H. Ng, C.Y. Wu, H.L. Su, and S.S. Chen, Aloe-emodin induces apoptosis of human nasopharyngeal carcinoma cells via caspase-8-mediated activation of the mitochondrial death pathway, Cancer Lett. 291 (2009), pp. 46 – 58.
[7] C.L. Chang, L.J. Zhang, R.Y. Chen, C.C. Wu, H.C. Huang, M.C. Roy, J.P. Huang, Y.C. Wu, and Y.H. Kuo, Quiquelignan A-H, eight new lignoids from the rattan palm Calamus quiquesetinervius and their antiradical, anti-inflammatory and antiplatelet aggrega-tion activities, Bioorg. Med. Chem. 18 (2010), pp. 518 – 525. [8] T.T. Chang, H.J. Huang, K.J. Lee, H.W. Yu, H.Y. Chen, F.J. Tsai,
M.F. Sun, and C.Y. Chen, Key features for designing phospho-diesterase-5 inhibitors, J. Biomol. Struct. Dyn. 28 (2010), pp. 309 – 321.
[9] H.J. Huang, K.J. Lee, H.W. Yu, H.Y. Chen, F.J. Tsai, and C.Y. Chen, A novel strategy for designing the selective PPAR agonist by the “sum of activity” model, J. Biomol. Struct. Dyn. 28 (2010), pp. 187 – 200.
[10] H.J. Huang, K.J. Lee, H.W. Yu, C.Y. Chen, C.H. Hsu, H.Y. Chen, and F.J. Tsai, Structure-based and ligand-based drug design for HER 2 receptor, J. Biomol. Struct. Dyn. 28 (2010), pp. 23 – 37. [11] C.Y. Chen and C.Y.C. Chen, Insights into designing the
dual-targeted HER2/HSP90 inhibitors, J. Mol. Graph. Model. 29 (2010), pp. 21 – 31.
[12] C.Y.C. Chen, Virtual screening and drug design for PDE-5 receptor from traditional Chinese medicine database, J. Biomol. Struct. Dyn. 27 (2010), pp. 627 – 640.
[13] C.Y.C. Chen, Bioinformatics, chemoinformatics, and pharmainfor-matics analysis of HER2/HSP90 dual-targeted inhibitors, J. Taiwan Inst. Chem. Eng. 41 (2010), pp. 143 – 149.
[14] C.Y. Chen, Weighted equation and rules – a novel concept for evaluating protein – ligand interaction, J. Biomol. Struct. Dyn. 27 (2009), pp. 271 – 282.
[15] C.Y. Chen, Computational screening and design of traditional Chinese medicine (TCM) to block phosphodiesterase-5, J. Mol. Graph. Model. 28 (2009), pp. 261 – 269.
[16] C.Y. Chen, Y.H. Chang, D.T. Bau, H.J. Huang, F.J. Tsai, and C.H. Tsai, Ligand-based dual target drug design for H1N1: Swine flu – a preliminary first study, J. Biomol. Struct. Dyn. 27 (2009), pp. 171 – 178.
[17] C.Y.-C. Chen, TCM database@Taiwan: The world’s largest traditional Chinese medicine database for drug screening in silico, PLoS ONE 6 (2011), p. e15939.
[18] G.L. Card, L. Blasdel, B.P. England, C. Zhang, Y. Suzuki, S. Gillette, D. Fong, P.N. Ibrahim, D.R. Artis, G. Bollag, M.V. Milburn, S.H. Kim, J. Schlessinger, and K.Y.J. Zhang, A family of phosphodiesterase inhibitors discovered by cocrystallo-graphy and scaffold-based drug design, Nat. Biotechnol. 23 (2005), pp. 201 – 207.
[19] Accelrys Software, Inc. Discovery Studio Modeling Environment, Accelrys Software, Inc., San Diego, CA, 2009.
[20] B.R. Brooks, R.E. Bruccoleri, B.D. Olafson, D.J. States, S. Swaminathan, and M. Karplus, CHARMM: A program for macromolecular energy minimization and dynamics calculations, J. Comp. Chem. 4 (1983), pp. 187 – 217.
[21] J. Koska, V.Z. Spassov, A.J. Maynard, L. Yan, N. Austin, P.K. Flook, and C.M. Venkatachalam, Fully automated molecular mechanics based induced fit protein – ligand docking method, J. Chem. Inf. Model. 48 (2008), pp. 1965 – 1973.
[22] J. Tirado-Rives and W.L. Jorgensen, Contribution of conformer focusing to the uncertainty in predicting free energies for protein – ligand binding, J. Med. Chem. 49 (2006), pp. 5880 – 5884.
M.-F. Sun et al. 930
[23] R.E. Fan, P.H. Chen, and C.J. Lin, Working set selection using second order information for training support vector machines, J. Mach. Learn. Res. 6 (2005), pp. 1889 – 1918.
[24] A.K. Saxena and P. Prathipati, Comparison of MLR, PLS and GA-MLR in QSAR analysis, SAR QSAR Environ. Res. 14 (2003), pp. 433 – 445.
[25] D. Rogers and A.J. Hopfinger, Application of genetic function approximation to quantitative structure – activity relationships and quantitative structure – property relationships, J. Chem. Inf. Comput. Sci. 34 (1994), p. 854.
[26] R. Fletcher, Optimization, Academic Press, New York and London, 1969.
[27] R. Fletcher and C.M. Reeves, Function minimization by conjugate gradients, Comput. J. 7 (1964), pp. 148 – 154.
[28] M.L. da Silva, A.D. Goncalves, P.R. Batista, J.D. Figueroa-Villar, P.G. Pascutti, and T.C.C. Franca, Design, docking studies and molecular dynamics of new potential selective inhibitors of Plasmodium falciparum serine hydroxymethyltransferase, Mol. Simul. 36 (2010), pp. 5 – 14.
[29] J. Xu, L.X. Wang, H. Zhang, C.H. Yi, and W.L. Xu, Accurate quantitative structure – property relationship analysis for prediction of nematic transition temperatures in thermotropic liquid crystals, Mol. Simul. 36 (2010), pp. 26 – 34.
[30] J. Wang, M.C. Yan, D.M. Zhao, Y. Sha, F. Li, and M.S. Cheng, Pharmacophore identification of PAK4 inhibitors, Mol. Simul. 36 (2010), pp. 53 – 57.
[31] R.W. Wang, L. Zhou, Z.L. Zuo, X. Ma, and M. Yang, 3D-QSAR studies of checkpoint kinase 1 inhibitors based on molecular docking and CoMFA, Mol. Simul. 36 (2010), pp. 87 – 110. [32] A. Stavrakoudis, Computational modelling and molecular dynamics
simulations of a cyclic peptide mimotope of the CD52 antigen complexed with CAMPATH-1H antibody, Mol. Simul. 36 (2010), pp. 127 – 137.
[33] R. Zhang, Z.G. Wang, B.P. Ling, Y.J. Liu, and C.B. Liu, Docking and molecular dynamics studies on the interaction of four imidazoline derivatives with potassium ion channel (Kir6.2), Mol. Simul. 36 (2010), pp. 166 – 174.
[34] H. Dalir, T. Nisisako, Y. Yanagida, and T. Hatsuzawa, DNA force – extension curve under uniaxial stretching, Mol. Simul. 36 (2010), pp. 221 – 228.
[35] Y. Iwai, H. Nakamura, Y. Arai, and Y. Shimoyama, Analysis of dissociation process for gas hydrates by molecular dynamics simulation, Mol. Simul. 36 (2010), pp. 246 – 253.
[36] C.D. Yoo, S.C. Kim, and S.H. Lee, Molecular dynamics simulation study of probe diffusion in liquid n-alkanes, Mol. Simul. 35 (2009), pp. 241 – 247.
[37] D.X. Li, G.L. Chen, B.L. Liu, and Y.S. Liu, Molecular simulation of b-cyclodextrin inclusion complex with 2-phenylethyl alcohol, Mol. Simul. 35 (2009), pp. 199 – 204.
[38] F. Luan, H.T. Liu, Y. Gao, and X.Y. Zhang, QSPR model to predict the thermal stabilities of second-order nonlinear optical (NLO) chromophore molecules, Mol. Simul. 35 (2009), pp. 248 – 257. [39] J.H. Jing, G.Z. Liang, H. Mei, S.Y. Xiao, Z.N. Xia, and Z.L. Li,
Quantitative structure – mobility relationship studies of dipeptides in capillary zone electrophoresis using three-dimensional holographic vector of atomic interaction field, Mol. Simul. 35 (2009), pp. 263 – 269.
[40] A.M. Al-Mekhnaqi, M.S. Mayeed, and G.M. Newaz, Prediction of protein conformation in water and on surfaces by Monte Carlo simulations using united-atom method, Mol. Simul. 35 (2009), pp. 292 – 300.
[41] M.L. Mihajlovic and P.M. Mitrasinovic, Applications of the ArgusLab4/AScore protocol in the structure-based binding affinity prediction of various inhibitors of group-1 and group-2 influenza virus neuraminidases (NAs), Mol. Simul. 35 (2009), pp. 311 – 324. [42] K. Roy and G. Ghosh, QSTR with extended topochemical atom (ETA) indices. 11. Comparative QSAR of acute NSAID cytotoxicity in rat hepatocytes using chemometric tools, Mol. Simul. 35 (2009), pp. 648 – 659.
[43] P. Nimmanpipug, V.S. Lee, P. Wolschann, and S. Hannongbua, Litchi chinensis-derived terpenoid as anti-HIV-1 protease agent: Structural design from molecular dynamics simulations, Mol. Simul. 35 (2009), pp. 673 – 680.
[44] P. Ghosh and M.C. Bagchi, Comparative QSAR studies of nitrofuranyl amide derivatives using theoretical structural proper-ties, Mol. Simul. 35 (2009), pp. 1185 – 1200.