行政院國家科學委員會專題研究計畫成果報告
利用高壓合成新材料及其特性分析(2/3)
Synthesis and Study of New Materials Using High Pressure
計畫編號:NSC 92-2113-M-002-018
執行期限:92 年 2 月 1 日至 93 年 1 月 31 日
主持人: 劉如熹 台灣大學化學系
共同主持人: 楊弘敦 中山大學物理系
波方主持人:Prof. S. M. Filipek 波蘭科學院物理化學研究所
計畫參與人員:王健源、康佳正、黃文彥 台灣大學化學系
黃家莞、康玉清 中山大學物理系
一、 中文摘要 AB2型儲氫合金(即Laves Phase 型合金) 為近十年儲氫材料之研發重點,與其他型儲 氫材料相較,AB2 型儲氫合金具較高之儲氫 量、較優異之抗氧化及抗腐蝕能力、較高之 體積電容量與製程較容易等優點,且無污染 環境之問題,故成為儲氫材料中之研究之重 點。 本計畫與波蘭科學院物理化學研究所 S.M. Filipek 教授合作,利用高壓設備成功地 合成YMn2Dx(x = 6)金屬氘化物。樣品分 析方面,利用X 光粉末繞射、元素分析儀、 X 光吸收近邊緣結構與 SQUID 磁性量測等方 法進行合成產物之分析。 關鍵詞:氫化物、氘化物、Laves 相、釔錳化 合物、高壓合成 Abstract:The AB2 Laves phase hydrogen storage alloys continuously attract interest of research in the field of hydrogen storage materials for their high ability of absorbing hydrogen, good stability, and high energy density. Moreover, AB2 alloys are environmental friendly compounds.
We cooperate with Prof. S.M. Filipek at the Institute of Physical Chemistry of the Polish Academy of Sciences to synthesize the intermetallic duteride YMn2Dx with x = 6 by
using the high pressure gaseous hydrogen and deuterium. X ray powder diffraction (XRD) has been used to characterize he crystal structure. Elemental analysis has been used to determine the quantity of deuterium in the alloy, and the valence of Mn was determined by X-ray absorption near edge structure (XANES). The magnetic properties of the synthesized products were analyzed by SQUID magnetometer.
Keywords: hydride, deuteride, Laves phase,
YMn2, high pressure synthesis 二、 緣由與目的 於60年代末與70年代初,國際間因石油 短缺而引發能源危機,故替代能源崛起,暫 時解決當時之能源危機,然替代能源無法完 全取代現有使用之能源,且隨著石油之大量 開採,據專家之保守估計,全球石油蘊藏量 約僅能再供應50年,屆時將引發更嚴重之能 源危機。再者,因石油之使用所引發之全球 環境污染問題日益嚴重,吾人於追求更高物 質生活水準之同時,應更注意其所夾帶對環 境造成之破壞。為儘早解決未來即將爆發之 種種危機,科學家們莫不積極地尋找既乾淨 又有效率的替代能源,此時科學家提出“氫能 經濟”,除氫氣之產生外,還要解決氫氣儲存 與運輸問題,各種氫氣儲存技術因而逐一發 展。 較常用的氫氣儲存方法有(1)以高壓氣態
的方式儲存於鋼瓶內,(2)將其液化或固化儲 存於容器內,以及(3)近幾年來已迅速發展之 固態金屬氫化物儲氫法。比較上述三種氫氣 儲存方法,其中氣態儲氫方式會造成金屬脆 化,有安全上之顧慮。而液態其固態儲氫法 則會耗費過多之能量而有經濟方面之顧慮。 固體氫化物所能溶入的氫氣高於液態氫(表 一)[1],因此固態金屬氫化物儲氫法及其儲氫 效率在研究漸漸成為占有一席之地。 1866 年,英國人Thomas Gramham 發現 金屬鈀(Pd)能大量吸收氫氣[2]。自此開始金屬 氫化物之研究。金屬氫化物一般稱為儲氫合 金。1914 年,Sieverts發現低濃度下金屬固 體中氫的濃度正比於P1/2 (P為氫的壓力),此 即為Sieverts規則。除鈀能吸收氫而成為金屬 氫化物之外,原則上所有過渡金屬皆可能形 成氫化物,特別是過渡金屬之化合物可以形 成各種性能不同之金屬氫化物。 60 年代,Zylstra 和Westendory 發現於 20 atm、室溫環境中,每莫耳SmCo5 能吸收 2.5克原子氫,並於壓力降至1 atm 後能釋放 吸入之氫氣[3]。荷蘭之Philips公司近一步發現 LaNi5儲氫合金於常溫下具良好之可逆吸放 氫性能[4,5]。美國Brookhaven國立研究所研發 TiFe系合金[6]。日本於1970年後,企業界及研 究 機 構 陸 續 投 入 儲 氫 合 金 之 開 發 與 生 產 [7,8],如松下電器產業中央研究所研發TiMn 系合金;大阪工業技術試驗所研發MmNi5摻 雜Al、Cr與Zr之合金;金屬材料研究所研發 之Fe-Ti-O系合金等。 自1950 年代起,儲氫合金被大量地發 掘,這些合金根據成分的不同可分為AB2、
AB5、AB、AB3、A2B7 及Mg-based 等[9]。
其中已被商業化之儲氫合金大多為AB5型與
AB2 Laves Phase 型兩種。其中A主要扮演強
吸氫元素,B則扮演觸媒之角色,以加速吸氫
與脫氫的反應。用於金屬氫化物的材料一般 可分為AB5及AB2型兩種。
(1) AB5型合金
AB5 型 合 金 以 LaNi5 及 其 改 良 型 LaNi5-xMx、MmNi5、La2-xZrxNi5為代表。LaNi5 為荷蘭Philips實驗室所研發成功之產品,最
初應用於鎳氫電池負極材料,因吸氫容量 少,於氧化及腐蝕之問題較嚴重,導致內部 氣體壓力高、壽命短與功率低等缺點,因而 於 商 業 化 方 面 遇 到 瓶 頸[10]。 直 至1984 年 Philips 實 驗 室 之 Willimes 於 LaNi5系 統 中 以 Co、Si與Mn等元素部分取代Ni,並用Nd取代 部分La[11],延長儲氫合金之壽命,於儲氫材 料發展方面取得重大突破,隨後以較便宜之 混合稀土金屬(Mm:Misch metal)取代昂貴之 鑭(La),使材料之價格大幅度降低,讓AB5 儲氫合金真正得以商業化。 表一、不同物質中氫的溶解度[1] (2) AB2型合金 AB2型Laves phase合金被認為比AB5型
具較高之理論儲氫量[12]。所謂Laves phase指
金屬化合物中,原子半徑dA及dB之比例為
1.225 或接近此比例之一連串最密堆積構造 之 合 金 。 第 一 個Laves phase 為 Pebler 及 Gulbransen 於1966年發現[13]。Laves phase 歸
類成三種結構,以鎂基合金為例[14],如圖一
所示:
圖一、AB2 Laves Phase 之結晶結構[14] (1) 六方堆積C14 結構:MgZn2 (hexagonal C14, ABABAB) (2) 立方堆積C15 結構:MgCu2 (cubic C15, ABCABC) (3) 六方堆積C36 結構:MgNi2 (hexagonal C14, ABACABAC) 通 常AB2型 的 合 金 其dA/dB於1.2 至 1.3 間,合金會形成六方堆積C14或立方堆積C15 之結構。關於AB2 Laves phase 合金,電子密 度 也 為 決 定 結 構 排 列 之 因 素 ,Elliot 與 Rostocker於1958年提出平均外層d 電子數 (Averaged number of outer electrons, ANOE) 與結構排列之關係[15,16]。以Ti-base 及Zr-base AB2 型合金為例:
(1) ANOE < 4.67 不會形成Laves phase (2) 4.67 < ANOE < 5.4 Zr-base 會形成
C15 Laves phase,Ti-base 無法形成Laves phase。 (3) 5.4 < ANOE < 7 Ti-base,Zr-base 會形 成C14 Laves phase。 (4) ANOE > 7 Ti-base,Zr-base 會形成C15 Laves phase。 此些結構每一層中原子以不同方式進行 堆積。合金吸收氫時會佔據Laves phase 之 sub-units 中 四 面 體 空 隙 位 置 (A2B2、AB3與 B4)[17],如圖二所示 。
圖二、Hexagonal C14 Laves phase 的結構。 黑球表示B原子,白球表示A原子。 (a)B4 sites,(b)AB3 sites與(c)A2B2 sites。 而氫化物的結構仍與原來合金相同,僅 晶格因氫原子填入而擴張。大部分之AB2合 金為多相組成,Rietveld method可有效用於辨 別 多 相 合 金 相 及 各 相 之 含 量 。C14 Laves phase 晶格常數(dhkl)及與單位晶格體積(V)可 利用下列之方程式計算[18]: l/dhkl = 4/3 [(h2 + hk + k2)/a2 + l2/c2 V = 0.866ac2 a與c為晶格常數,如圖三所示。 於金屬氫化物之熱力學性質方面,氫與 金屬平衡最好的描述是壓力、成分與等溫線 的 吸 放 氫 曲 線 圖[19](Pressure Composition Temperature, PCT),對金屬氫化物在不同溫 度與壓力所量測吸收氫氣的含量,如圖四所 示。
圖三、(a)六方堆積C14 晶體結構中菱形的晶 格單位。(b)菱形的晶格單位以(110)平面原子 A及原子B距離[18]。 圖四、三種溫度之理想PCT 曲線以及Van’t Hoff 圖。 金屬氫化物在氫氣存在之條件下會自發 性地產生吸氫反應形成氫化物。加熱或氫氣 減壓會使反應逆轉。Anani[20]提出,於第一階 段氫為金屬吸收形成固溶體稱為α相。而當氫 氣濃度很大時壓力增加,此區域是氫化物區 稱為β相。而中間的平坦區域是α和β兩相共存 區具一水平之氫平衡壓稱為平台壓(plateau pressure)。平台區之壓力值在吸氫與放氫時循 環 會 有 落 差 , 謂 之 為 吸 放 氫 遲 滯 曲 線 (hysteresis curve),如圖五所示。壓力與溫度 的關係式可以由Van’t Hoff方程式描述:
ln
PH2=
RT H ∆-
R S ∆ ΔH 及ΔS 是吸收氫氣時的焓與熵,R 是氣體常數,T 是絕對溫度。依據此方程式, 可以計算金屬氫化物反應在不同溫度下的焓 與熵的PCT 圖。 圖五、氣相吸氫—放氫的原理 本 計 畫 為 探 討AB2系 列 儲 氫 材 料 中 之 YMn2化合物其氫化物與氘化物之性質。藉高 壓合成技術合成YMn2Hx與YMn2Dx,尋求X 可能存在之最大值,於去年度計畫執行期間 已成功合成YMn2H4.5化合物,並進行結構判 定,Mn之價數鑑定與磁性量測等,於今年度 已成功合成YMn2D6氘化物,並進行結構、元 素含量及價數鑑定,計畫執行成果於國際會 議中發表三篇海報論文,一篇口頭報告論 文,研究結果並投稿至Chem. Mater. 期刊。 三、 研究方法製備起始物 YMn2 合金之方法為利用真 空電弧融鍊爐(Arc melting furnace)。將釔金屬
與錳金屬置於水冷式無氧銅座中,於Ar 氣氛 中反覆融鍊數次,即可得合成金屬氫化物之 前驅物 YMn2 合金。以 X 光粉末繞射技術 (XRD)鑑定所得之 YMn2合金,其為立方結構 (cubic),其空間群為Fd-3m,晶格常數為a = 7.681(1) Å。 合成金屬氫化物部分,先將 YMn2粉末 放入不銹鋼製之樣品槽中,再將樣品槽置入 鈹銅合金之反應槽內,溫度升高到 100℃以 幫浦抽真空至10-3 torr,將溫度降到室溫再通 入 5N 的氘氣(D2)緩慢加壓至 1atm,維持 此壓力約十九小時,期間每隔半小時觀察壓 力是否有降低,之後加壓至9atm 溫度升高至 200℃,維持此壓力與溫度約二小時,待溫度 降 至 室 溫 取 出 樣 品 , 此 時 所 得 之 樣 品 為 YMn2D4.5,經XRD 測試確定樣品純度後再將 樣品置入樣品槽中繼續進行第二階段之高壓 合成,此階段的所需之壓力極大,因此樣品 槽之材質置換為鈹銅合金,為將更多氘氣壓 入 YMn2 中,除重複第一階段的前段步驟, 再將壓力加大至 200 atm,而溫度則設定於 140℃,持續約三小時將溫度降至室溫得 YMn2D6化合物,取出樣品進行分析研究。 樣品分析方面,利用X 光粉末繞射儀鑑 定結構,光源為銅靶(Cu Kα,λ = 1.5406 Å), 以0.02°為間格收集 2θ 範圍為 10°至 120°之數 據,所得之結果再配合 Rietveld refinement 進 行 結 構 精 算 。 利 用 元 素 分 析 儀 (Perkia Elmer 240)測定碳、氫及氮之含量,此儀器 對氫之誤差為0.3%,乃用電導度之原理測定 之。其中乃將樣品置入白金坩鍋中,通入氧 氣,於 900℃、15 分鐘後即可量測。校正此 儀 器 之 化 合 物 一 般 常 用 C8H9NO (acetanilid)。利用同步輻射光源進行金屬 氫化物中 Mn 金屬價數之判定。並利用超導 量 子 干 涉 儀 (superconducting quantum interference device, SQUID)量測磁性特性。 四、實驗結果與討論 圖六為YMn2D6化合物之X光粉末繞射 圖譜,圖中黑色十字表示實驗值,淺色實線 代表理論計算值,下方之短直線表示理論計 算之繞射峰所應出現之位置,最下方之曲線 則 為 理 論 值 與 實 驗 值 之 誤 差 。 表 二 則 為 Rietveld refinement結構精算所得之結構參數 及可靠度係數。由圖六及表二觀之,所合成 之YMn2D6化合物其結構屬立方晶系,晶格常 數a = b = c = 6.7093(1) Å, α = β = γ = 90°。可 靠度參數Rp = 10.7 %、Rwp = 13.4 % 與 χ2 = 2.41均位於可接受之範圍內。於約30°出現 Y2O3化 合 物 之 繞 射 峰 , 且 仍 有 少 部 分 YMn2D4.5化合物之繞射峰存在,顯示仍有少 量非YMn2D6之雜質存在。 圖六、YMn2D6之X光粉末繞射圖 表二、YMn2D6結構精算之參數列表 圖七所示為配合結構精算之結果所得之 YMn2D6化合物原子座標圖。淺色為釔金屬,
Atoms x y z Wyckoff Uiso (Å2) Y Mn (1) Mn (2) 0.00 0.50 0.25 0.00 0.50 0.25 0.00 0.50 0.25 4a 4b 4c 3.131 1.436 1.436 Lattice parameters Reliability factors
space group: F-43m (216) a = b = c = 6.7093(1) Å α = β = γ = 90° Rp = 10.7% Rwp = 13.4% χ2 = 2.41 20 30 40 50 60 70 80 90 100 110 120 130 -1000 0 1000 2000 3000 4000 YMn2D6 Y2O3 YMn2D4.5 In te ns ity ( coun ts p er s eco nd) 2 theta
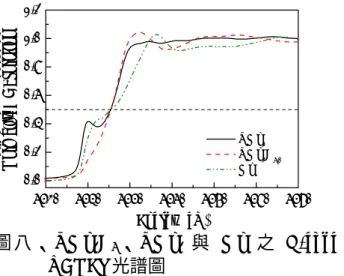
深色則為錳金屬。於 C15 型結構中,AB3、 A2B2 以及 B4 三種四面體位置可為氘原子所 占據。實驗上可證實A2B2位置乃為合金與氘 原子最利於鍵結產生之處,故其應最先被填 入氘原子。然而當氘含量增多時,填入另二 種位置之機率亦不能被排除。填入他種四面 體位置可能導致主體結構扭曲而變形為斜方 晶系之結構。然本研究之YMn2D6.0晶體對稱 性並非與C15 結構直接相關,亦即 YMn2D6.0 之形成乃伴隨著釔與錳原子大幅度地重新排 列。換言之,結構中氫或氘原子可占據之位 置可能與 C15 結構中之 AB3、A2B2與 B4等 位置有極大差異。由圖六觀之,YMn2D6化合 物並無 B4 四面體位置可供氫原子或氘原子 佔據,而於立方體之角落共有8 個 A3B 四面 體位置可吸附氫原子及氘原子。而於晶格中 另有 32 個可能提供氫原子與氘原子佔據之 A2B2四面體,其中僅 16 個符合所求,將配 合各原子半徑及四面體空隙大小,並考慮質 子排斥力等效應尋找適合氫原子及氘原子填 入之四面體空隙。 圖七、YMn2D6之原子座標圖 圖八為 YMn2D6 與 YMn2 化合物兩者之 Mn 金屬 K-edge X 光近邊緣吸收光譜,以純 Mn 金屬為標準樣品,測定形成 YMn2化合物 與形成金屬氫化物 YMn2D6其 Mn 金屬之特 性是否改變。
圖 八 、YMn2D6、YMn2 與 Mn 之 K-edge XANES 光譜圖 由圖八觀之,Mn 金屬之價數於三種不同 樣品中並無差異,然其吸收邊緣之形狀隨不 同對稱性及化學環境而有所變化。於K-edge XANES 光譜中,吸收邊緣出現為過渡金屬 1s 軌域之電子吸收能量激發至 4p 空軌域所 致,然於L-edge XANES 光譜則為 2p 軌域電 子躍遷至 3d 軌域造成吸收邊緣之出現。故 L-edge XANES 光譜較不易外在化學環境及 對稱性之影響。圖九所示為扣除背景值之 YMn2D6與YMn2之L-edge XANES 光譜。由
圖中下方兩光譜之差值曲線可得知 YMn2D6
與 YMn2中之 Mn 金屬並無明顯之差異性,
顯示 Mn 元素於此兩種化合物中均為金屬
態,並與Y 金屬形成合金。
圖九、YMn2D6與YMn2之L-edge XANES 光譜圖 圖十所示為 YMn2D6與 YMn2之磁化率 635 640 645 650 655 660 (b) (a) difference: (a)-(b) YMn2 YMn2D6.0 N orm aliz ed ab so rp tion , I s /I 0 ( ar b. u nit) Energy (eV)
Y
Mn
6530 6540 6550 6560 6570 6580 6590 0.0 0.2 0.4 0.6 0.8 1.0 1.2 YMn2 YMn2D6.0 Mn No rm ali ze d A bso rpt ion Energy (eV)隨溫度變化之趨勢圖,可知吸收氘氣可提高 化合物之磁化率,而自120 K 降至 5 K 之過 程中可觀察到 YMn2D6之磁化率迅速上升。 圖十一所示為 YMn2D6與 YMn2之磁化迴路 曲線圖,可知吸收氘氣之 YMn2D6具較高之 磁化程度,此外,此二種化合物之磁化作用 於外加磁場增為 ± 5.5 T 時皆未達其飽和磁 化狀態。於5 K 下之遲滯程度而言,YMn2D6 所測得之遲滯程度較YMn2高。 圖十、YMn2D6與YMn2之磁化率對溫度作圖 圖十一、YMn2D6與YMn2之磁化迴路曲線圖 五、結論 本研究所合成之新 AB2型氘化物,經查 證文獻之前並未有相關報導。以X 光繞射分 析配合結構精算得知其屬立方晶系(a = b = c = 6.7093(1) Å, α = β = γ = 90°)空間群為 F-43m,經元素分析確定化學式為YMn2D6。 氘化物中 Mn 之價數為金屬態,同時與釔元 素形成合金。並以超導量子干涉儀測樣品之 磁化率,發現吸收氘氣後之 YMn2D6具較高 之磁矩,且於5K 下具較高之遲滯程度。 六、計劃成果自評 已利用高壓合成設備成功合成新相之 YMn2D6高吸氫(氘)量之儲氫合金,經搜尋即 查證相關資料,此新相化合物為首度發現之 研究成果。並利用X 光粉末繞射技術配合結 構精算鑑定其結構,利用元素分析儀判定氘 含量,以X 光近邊緣吸收光譜鑑定化合物中 錳金屬之價數,並利用超導量子干涉儀研究 磁之特性,達成本年度計畫之目標。研究之 成果於2003 年 7 月 7 日~7 月 11 日法國波爾 多市(Bordeaux)舉行之國際高壓科技會議 (High pressure science and technology)中與 Filipek 教授合作發表 2 篇論文,其中一篇名 為“Characterization of Several Laves Phase Hydride Recently Synthesized under High Hydrogen Pressure”之口頭報告論文,另一篇 名 為 “ A New Hydride Phase of YMn2 Synthesized under High Hydrogen Pressure and Its Characterization”之壁報論文,如圖十二 所示。 圖十二、於法國波爾多市參加國際高壓會議 另外亦於台南崑山科技大學舉行之2003 材料年會(11 月 21 日~11 月 22 日)中 Filipek 教授合作發表一篇名為“A New Phase of
YMn2D6.0 Deuteride Synthesized by High Pressure Apparatus”之壁報論文,並獲得佳 作之獎項,如圖十三所示。同時亦投稿參加 於中壢中原大學舉行之2003 化學年會(11 月 -6 -4 -2 0 2 4 6 -5 -4 -3 -2 -1 0 1 2 3 4 Mome nt ( em u/ g) Magnetic field (T) YMn2 YMn2D6 (at 5 K) 0 50 100 150 200 250 300 2.2 2.3 2.4 2.5 6 8 10 12 14 16 18 20 χ mo l ( x10 -3 ) Temperature (K) YMn2 YMn2D6 (H = 1 T) -4 -2 0 2 4 -0.5 -0.4 -0.3 -0.2 -0.10.0 0.1 0.2 0.3 0.4 0.5 M ome n t ( emu /g )
Magnetic field (kOe)
Y Mn2
Y Mn2D6
28 日~11 月 30 日)之壁報論文,題目為” Studies on Synthesis and Characterization of New Intermetallic Deuteride YMn2D6.0
Synthesized by High Pressure Apparatus”。
圖十三、於台南崑山科技大學參加2003 材料年會 本計畫至目前為止之研究成果並已投稿 至Chemistry of Materials ( Chem. Mater. )題 目 為” New Phase of YMn2D6.0 Deuteride
Synthesized by High Pressure Apparatus” 作者 為 Chien-Yuan Wang, Valerie Paul-Boncour, Ru-Shi. Liu, Annick Percheron-Guegan, Maria Dorogova, Iryna Marchuk, Toshiya Hirata, Stanislaw M. Filipek, Hwo-Shuenn Sheu, Ling-Yun Jang, Jin-Ming Chen, and Hung-Duen Yang 等人。
七、參考文獻
[1] A. E. Dwight, Trans. Am. Soc. Met. 53, 479 (1961).
[2] H. Yamada and M. Shimizu, Phys. Lett. A 117, 313
(1986).
[3] Y. Nakamura, M. Shiga and S. Kawano, Physica B 120, 212 (1983).
[4] H. Wada, H. Nakamura, K. Yoshimura, M. Shiga and Y. Nakamura, J. Magn. Magn. Mater. 70, 134
(1987).
[5] T. Okamoto, H. Nagata, H. Fujii and Y. Makihara, J. Magn. Magn. Mater. 70, 139 (1987).
[6] M. Shiga, H. Wada, H. Nakamura, K. Yoshimura and Y. Nakamura, J. Phys. F 17, 1781 (1987).
[7] G. Oomi, T. Terada, M. Shiga and Y Nakamura, J. Magn. Magn. Mater. 70, 137 (1987).
[8] R. Ballou, J. Deportes, R. Lemaire, Y. Nakamura and B. Ouladdiaf, J. Magn. Magn. Mater. 70, 129 (1987).
[9] K. Yoshimura and Y. Nakamura, J. Magn. Magn. Mater. 40, 55 (1983).
[10] H. Nakamura, N. Metoki, S. Suzuki, F. Takayanagi and M. Shiga, J. Phys.: Condens. Matter 13, 475
(2001).
[11] S. Asano and S. Ishida, J. Magn. Magn. Mater. 70,
39 (1987).
[12] H. Yamada and M. Shimizu, J. Magn. Magn. Mater. 70, 47 (1987).
[13] H. Fuji, M. Saga and T. Okamoto, J. Less-Common Met. 130, 25 (1987).
[14] J. Przewoznik, V. Paul-Boncour, M. Latroche and A. Percheron-Guegan, J. Alloys Compds. 225, 436
(1995).
[15] M. Latroche, V. Paul-Boncour, J. Przewoznik, A. Percheron-Guegan and F. Bouree-Vigneron, J. Alloys Compds. 231, 99 (1995).
[16] M. Latroche, V. Paul-Boncour, A.
Percheron-Guegan and F. Bouree-Vigneron, J. Alloys Compds. 274, 59 (1998).
[17] H. Figiel, A. Lindbaum, Cs. Kapusta and E. Gratz, J. Alloys Compds. 217, 157 (1995).
[18] H. Figiel, J. Przewoznik, V. Paul-Boncour, A. Lindbaum, E. Gratz, M. Latroche, M. Escorne, A. Percheron-Guégan and P. Mietniowski, J. Alloys Compds. 274, 29 (1998).
[19] H. Fujii, M. Saga and T. Okamoto J. Less-Common Metals 130, 25 (1987).
[20] I. N. Goncharenko, I. Mirebeau, A. V. Irodova and E. Suard, Phys. Rev. B 56, 2580 (1997).