On: 26 April 2014, At: 02:36 Publisher: Taylor & Francis
Informa Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House, 37-41 Mortimer Street, London W1T 3JH, UK
Liquid Crystals
Publication details, including instructions for authors and subscription information: http://www.tandfonline.com/loi/tlct20
Synthesis and characterization of achiral
banana
‐shaped liquid crystalline molecules containing
bisnaphthyl moieties
Po‐Jen Yang a & Hong‐Cheu Lin aa
Department of Materials Science and Engineering , National Chiao Tung University , Hsinchu, Taiwan, ROC
Published online: 20 Feb 2007.
To cite this article: Po‐Jen Yang & Hong‐Cheu Lin (2006) Synthesis and characterization of achiral banana‐shaped liquid crystalline molecules containing bisnaphthyl moieties, Liquid Crystals, 33:5, 587-603, DOI: 10.1080/02678290600633394 To link to this article: http://dx.doi.org/10.1080/02678290600633394
PLEASE SCROLL DOWN FOR ARTICLE
Taylor & Francis makes every effort to ensure the accuracy of all the information (the “Content”) contained in the publications on our platform. However, Taylor & Francis, our agents, and our licensors make no
representations or warranties whatsoever as to the accuracy, completeness, or suitability for any purpose of the Content. Any opinions and views expressed in this publication are the opinions and views of the authors, and are not the views of or endorsed by Taylor & Francis. The accuracy of the Content should not be relied upon and should be independently verified with primary sources of information. Taylor and Francis shall not be liable for any losses, actions, claims, proceedings, demands, costs, expenses, damages, and other liabilities whatsoever or howsoever caused arising directly or indirectly in connection with, in relation to or arising out of the use of the Content.
This article may be used for research, teaching, and private study purposes. Any substantial or systematic reproduction, redistribution, reselling, loan, sub-licensing, systematic supply, or distribution in any
form to anyone is expressly forbidden. Terms & Conditions of access and use can be found at http:// www.tandfonline.com/page/terms-and-conditions
Synthesis and characterization of achiral banana-shaped liquid
crystalline molecules containing bisnaphthyl moieties
PO-JEN YANG and HONG-CHEU LIN*
Department of Materials Science and Engineering, National Chiao Tung University, Hsinchu, Taiwan, ROC
(Received 29 September 2005; accepted 6 January 2006 )
Three analogous series of symmetric banana-shaped liquid crystalline molecules containing bisnaphthyl units were synthesized and characterized. The effects of linking groups (on the side wings) and lateral meta-fluoro substitutions (on the middle outer rings) on the mesogenic properties were examined. The type of mesophase depends on the lengths of terminal alkoxy chains. Thus, achiral molecules with shorter flexible chains (n58) exhibit a rectangular columnar (B1) phase, while analogous derivatives with longer flexible chains (n512) display
the B2 phase. All lateral meta-fluoro substituted analogues (series II) possess the lowest
isotropization temperatures and the narrowest mesophasic ranges of the B1and B2phases.
The B1and B2phases were confirmed by X-ray diffraction, polarizing optical microscopy
(POM) and electro-optical (EO) switching experiments. An electric field-induced transition from an antiferroelectric (tristable) state to a ferroelectric (bistable) state was observed in the EO measurements. Spontaneous polarization (by switching current response), tilt angle of chiral domains (by POM), and transmittance–voltage measurements of the B2 phase in
related compounds have been surveyed in this study.
1. Introduction
Since the first ferroelectric liquid crystalline (FLC) material was reported by Meyer et al. in 1975 [1], FLCs were characterized as tilted arrangements of smectic liquid crystalline phases in materials composed of chiral molecules, allowing the occurrence of spontaneous polarizations. Later, antiferroelectric liquid crystals (AFLCs) were discovered and finally identified in 1989 [2]; subsequently, AFLCs were developed into fast electro-optic devices due to the quick response of these materials used for flat-panel displays [3].
In 1996, Niori et al. [4] first demonstrated that smectic phases of achiral molecules with bent cores can exhibit ferroelectric switching properties. These achiral molecules have a sterically induced polar packing character, and are arranged in smectic layers in such a way that the dipoles point along a common direction within the layer, and tilt with respect to the layer normal. These factors can give rise to chiral layer symmetry, although the molecules themselves are achiral. These liquid crystalline molecules with a bent molecular shape, so-called banana-shaped liquid crys-tals, have attracted much attention owing to their novel ferroelectric properties. Although eight different banana phases (B1 to B8) of achiral molecules have been
identified [5, 6] electro-optical switching behaviour is observed only in the smectic B2, B5 and B7 phases. Among these phases with switching properties, the B2 phase is the most frequently investigated. According to the report of Link et al. [7], the spontaneous formation of macroscopic chiral domains in a fluid smectic is derived from achiral molecules with antiferroelectric switching characteristics. The B2 phase exhibit four different supramolecular architectures, denoted SmCAPA, SmCSPA, SmCAPF and SmCSPF; here syn-clinic (S) or antisyn-clinic (A) represents molecular tilt correlation, and ferroelectric (F) or antiferroelectric (A) specifies polar correlation between adjacent layers. When the layer chiralities in adjacent layers have the same handedness, such as those in SmCAPA and SmCSPF, the structures are called homochiral; whereas, when the chiralities in adjacent layers alternate, such as those in SmCAPFand SmCSPA, the structures are called racemic. More recently, different parts of the achiral molecular structures, such as the central parts, lateral substituents, linking groups, terminal chains, and the number of rings, have been varied to study the influence of molecular design on the mesomorphic behaviour [5, 8–14].
In this study, three analogous series of symmetric banana-shaped liquid crystalline molecules containing bisnaphthyl units were synthesized and characterized. All these compounds are derived from central resorcinol *Corresponding author. Email: [email protected]
Liquid Crystals
ISSN 0267-8292 print/ISSN 1366-5855 online # 2006 Taylor & Francis http://www.tandf.co.uk/journals
DOI: 10.1080/02678290600633394
cores and connected by ester linking groups, and the side wings of the molecules are connected via ester or Schiff base linkages symmetrically, with different lengths (n58, 12 and 16) of flexible alkoxy chains. Their molecular structures are shown in figure 1, which includes three variable parts; i.e. with/without lateral substitutions (X5F or H), different linking groups (Y5–COO– or –CH5N–), and alkoxy flexible tails with three different lengths of n.
2. Experimental 2.1. Characterization 1
H NMR spectra were recorded on a Varian unity 300 MHz spectrometer using CDCl3 and DMSO-d6as solvents. Elemental analyses were performed on a Heraeus CHN-OS RAPID elemental analyser. Mesophase textures were characterized by a polarizing optical microscopy (POM) using a Leica DMLP equipped with a hot stage. Temperatures and enthalpies of transition were determined by differential scanning calorimetry (DSC) using a Perkin-Elmer Pyris 7, under N2at a heating and cooling rate of 5uC min21.
Synchrotron powder X-ray diffraction (XRD) mea-surements were performed at beamline BL17A of the National Synchrotron Radiation Research Center (NSRRC), Taiwan, where the X-ray wavelength was 1.32633 A˚ . The XRD data were collected using imaging plates (IP, of an area520640 cm2and a pixel resolution of 100) curved with a radius equivalent to a sample-to-image plate distance of 280 mm, and the diffraction signals were accumulated for 3 min. The powder samples were packed into a capillary tube and heated by a heat gun, whose temperature controller is programmable by a PC with a PID feedback system. The scattering angle theta was calibrated by a mixture of silver behenate and silicon.
The electro-optical properties were determined in commercially available ITO cells (from Mesostate
Corp., thickness59mm, active area50.25 cm2) with rubbed polyimide alignment coatings (antiparallel rub-bing direction). The optical transmission experiments used cells of active area51 cm2, 6mm thick (antiparallel rubbing direction), with monochromatic light from a He-Ne laser beam (632.8 nm). The cell was placed between crossed polarizers and its orientation was adjusted to obtain the minimum transmission of light (detected by a photodiode) without an electric field. The photodiode detector was set about 20 cm away from the cell sample, with the incident laser beam normal to the cell. A digital oscilloscope (Tektronix TDS-3012B) was used in these measurements, and a high power amplifier connected to a function generator (GW Model GFG-813) with a d.c. power supply (Keithley 2400) was utilized in the d.c. field experiments. During electro-optical measurements, texture modulations on applying an electric field were observed by POM.
2.2. Synthesis
Chemicals and solvents were reagent grades and purchased from Aldrich, ACROS, TCI and Lancaster Chemical Co. Dichloromethane (CH2Cl2) was redis-tilled under nitrogen and dried over calcium hydride before use. The other chemicals were used without further purification.
The synthetic route to the banana-shaped compounds of this study is shown in scheme 1 (series I and II) and scheme 2 (series III). 6-n-Alkoxynaphthalene-2-car-boxylic acids were prepared according to literature procedures. 6-Hydroxynaphthalene-2-carboxylic acid was converted to methyl 6-hydroxynaphthalene-2-car-boxylate using the modified procedure described by Gu¨ller et al. [15]; this ester was treated with the appropriate n-alkyl bromides followed by hydrolysis [16]. 6-n-Alkoxynaphthalene-2-carbaldehyde [17] and 4-benzyloxybenzoic acid [18] were synthesized according to the respective literature procedures.
Figure 1. Molecular structures of the synthesized series I, II and III compounds.
3-Fluoro-4-benzyloxybenzoic acid was prepared as described in [19], followed by a similar procedure to that for 4-benzyloxybenzoic acid.
2.2.1. 1,3-Phenylene bis(4-benzyloxybenzoate), 2a (X5H). To a suspension of resorcinol (1.5 g, 13.6 mmol) in a solution of 4-benzyloxybenzoic acid 1a (6.2 g, 27.2 mmol) in dry dichloromethane (60 ml), were added N,N-dicyclohexylcarbodiimide (DCC) (5.9 g, 28.6 mmol) and a catalytic amount of 4-(N,N-dimethylamino)pyridine (DMAP). The reaction mixture was stirred under nitrogen for 15 h at room temperature.
The precipitated dicyclohexylurea (DCU) was filtered off and washed with an excess of CHCl3(100 ml). The filtrate was washed with water and dried over anhydrous magnesium sulphate. After removal of the solvent by evaporation under reduced pressure, the residue was purified by column chromatography (silica gel, CHCl3). The collected product was crystallized from a mixture of dichloromethane and 2-propanol to give a white solid; yield 5.1 g (71%). 1H NMR (ppm, CDCl3): 8.16 (d, J58.9 Hz, 4H, Ar–H), 7.4927.35 (m, 10H, Ar–H), 7.1627.13 (m, 4H, Ar–H), 7.07 (d, J58.9 Hz, 4H, Ar– H), 5.16 (s, 4H, ArCH2O–).
Scheme 1. Synthetic route for series I and II compounds.
2.2.2. 1,3-Phenylene bis(3-fluoro-4-benzyloxybenzoate), 2b (X5F). This compound was obtained from the reaction of resorcinol and 3-fluoro-4-benzyloxybenzoic acid 1b, following a similar procedure to that described for compound 2a. The product was isolated as a white solid; yield 69%.1H NMR (ppm, CDCl3): 7.9427.88 (m, 4H, Ar–H), 7.4727.35 (m, 11H, Ar–H), 7.1427.06 (m, 5H, Ar–H), 5.25 (s, 4H, ArCH2O–).
2.2.3. 1,3-Phenylene bis(4-hydroxybenzoate), 3a (X5H). Compound 2a (5 g, 9.4 mmol), was dissolved in a mixture of 1,4-dioxane (200 ml) and THF (100 ml) containing a suspension of 10% Pd-C catalyst (1 g). The mixture was
stirred under hydrogen at room temperature until no further hydrogen was taken up (c. 10 h). The catalyst was removed by filtration through Celite and washed with THF (100 ml). The solvent was removed by evaporation under reduced pressure and the crude product purified by column chromatography (silica gel, CHCl3/EtOAc 60/1) to give a white solid; yield 90%.1H NMR (ppm, DMSO-d6): 10.54 (s, 2H, 26Ar–OH), 7.98 (d, J58.7 Hz, 4H, Ar– H), 7.49 (t, J58.7 Hz, 1H, Ar–H), 7.2027.17 (m, 3H, Ar– H), 6.92 (d, J58.7 Hz, 4H, Ar–H).
2.2.4. 1,3-Phenylene bis(3-fluoro-4-hydroxybenzoate), 3b (X5F). This compound was obtained from Scheme 2. Synthetic route for series III compounds.
compound 2b, following a similar procedure to that described for compound 3a. The product was isolated as a white solid; yield 92%.1H NMR (ppm, DMSO-d6): 11.09 (br s, 2H, 26Ar–OH), 7.8627.80 (m, 4H, Ar–H), 7.52 (t, J58.4 Hz, 1H, Ar–H), 7.2727.15 (m, 3H, Ar– H), 7.1227.09 (m, 2H, Ar–H).
Compounds of series I and series II were prepared by similar methods to that described for compound 2a. After work-up, the crude products were purified by column chromatography (silica gel, CHCl3) and twice recrystallized from a mixture of chloroform and ethanol. The yields of these products after purification were c. 50–65%.
2.2.5. 1,3-Phenylene bis[4-(6-n-octyloxynaphthalene-2-carbonyloxy)benzoate], Ia (n58). The crude product was isolated as a white solid from the reaction of compound 3a with 6-n-octyloxynaphalene-2-carboxylic acid. 1H NMR (ppm, CDCl3): 8.72 (s, 2H, Ar–H), 8.31 (d, J58.7 Hz, 4H, Ar–H), 8.15 (d, J58.7 Hz, 2H, Ar–H), 7.90 (d, J59.0 Hz, 2H, Ar–H), 7.82 (d, J59.0 Hz, 2H, Ar–H), 7.51 (t, J58.4 Hz, 1H, Ar–H), 7.44 (d, J58.7 Hz, 4H, Ar–H), 7.2627.19 (m, 7H, Ar–H), 4.12 (t, J56.6 Hz, 4H, 26OCH2), 1.9321.86 (m, 4H, 26OCH2CH2), 1.5521.31 (m, 20H, 106CH2), 0.90 (t, J56.6 Hz, 6H, 26CH3). Anal: calc. for C58H58O10, C 76.13, H 6.39; found, C 76.04, H 6.60%.
2.2.6. 1,3-Phenylene bis[4-(6-n-dodecyloxynaphthalene-2-carbonyloxy)benzoate], Ib (n512). The crude product was isolated as a white solid from the reaction of compound 3a with 6-n-dodecyloxynaphalene-2-carboxylic acid. 1H NMR (ppm, CDCl3): 8.72 (s, 2H, Ar–H), 8.31 (d, J58.7 Hz, 4H, Ar–H), 8.15 (d, J58.7 Hz, 2H, Ar–H), 7.90 (d, J59.0 Hz, 2H, Ar–H), 7.82 (d, J59.0 Hz, 2H, Ar–H), 7.53 (t, J58.4 Hz, 1H, Ar–H), 7.44 (d, J58.7 Hz, 4H, Ar–H), 7.2627.19 (m, 7H, Ar–H), 4.12 (t, J56.6 Hz, 4H, 26OCH2), 1.9021.83 (m, 4H, 26OCH2CH2), 1.5621.28 (m, 36H, 186CH2), 0.90 (t, J56.6 Hz, 6H, 26CH3). Anal: calc. for C66H74O10, C 77.16, H 7.26; found C 77.07, H 7.28%.
2.2.7. 1,3-Phenylene bis[4-(6-n-hexadecyloxy-naphthalene-2-carbonyloxy)benzoate], Ic (n516). The crude product was isolated as a white solid from the reaction of compound 3a with 6-n-hexadecyloxy-naphalene-2-carboxylic acid. 1H NMR (ppm, CDCl3): 8.72 (s, 2H, Ar–H), 8.31 (d, J58.7 Hz, 4H, Ar–H), 8.15 (d, J58.7 Hz, 2H, Ar–H), 7.90 (d, J59.0 Hz, 2H, Ar– H), 7.82 (d, J59.0 Hz, 2H, Ar–H), 7.53 (t, J58.4 Hz, 1H, Ar–H), 7.44 (d, J58.7 Hz, 4H, Ar–H), 7.2627.19 (m, 7H, Ar–H), 4.12 (t, J56.6 Hz, 4H, 26OCH2), 1.9021.82 (m, 4H, 26OCH2CH2), 1.5521.31 (m, 52H, 266CH2), 0.90 (t, J56.6 Hz, 6H, 26CH3). Anal: calc. for C74H90O10, C 78.00, H 7.96; found, C 78.23, H 7.97%. 2.2.8. 1,3-Phenylene bis[4-(6-n-octyloxynaphthalene-2-carbonyloxy)-3-fluorobenzoate], IIa (n58). The crude product was isolated as a white solid from the reaction of compound 3b with 6-n-octyloxynaphalene-2-carboxylic acid. 1H NMR (ppm, CDCl3): 8.73 (s, 2H, Ar–H), 8.1728.06 (m, 6H, Ar–H), 7.90 (d, J59.0 Hz, 2H, Ar–H), 7.82 (d, J58.7 Hz, 2H, Ar–H), 7.5127.47 (m, 3H, Ar–H), 7.2627.19 (m, 7H, Ar–H), 4.12 (t, J56.6 Hz, 4H, 26OCH2), 1.9221.83 (m, 4H, 26OCH2CH2), 1.5421.31 (m, 20H, 106CH2), 0.90 (t, J56.6 Hz, 6H, 26CH3). Anal: calc. for C58H56F2O10, C 73.25, H 5.93; found, C 73.28, H 6.06%.
2.2.9. 1,3-Phenylene bis[4-(6-n-dodecyloxynaphthalene-2-carbonyloxy)-3-fluorobenzoate], IIb (n512). The crude product was isolated as a white solid from the reaction of compound 3b with 6-n-dodecyloxynaphalene-2-carboxylic acid. 1H NMR (ppm, CDCl3): 8.73 (s, 2H, Ar–H), 8.1728.06 (m, 6H, Ar–H), 7.90 (d, J59.0 Hz, 2H, Ar–H), 7.82 (d, J58.7 Hz, 2H, Ar–H), 7.5527.49 (m, 3H, Ar–H), 7.2627.19 (m, 7H, Ar–H), 4.12 (t, J56.6 Hz, 4H, 26OCH2), 1.9021.85 (m, 4H, 26OCH2CH2), 1.5421.28 (m, 36H, 186CH2), 0.88 (t, J56.6 Hz, 6H, 26CH3). Anal: calc. for C66H72F2O10, C 74.55, H 6.83; found, C 74.45, H 6.94%.
2.2.10. 1,3-Phenylene bis[4-(6-n-hexadecyloxynaphtha-lene-2-carbonyloxy)-3-fluorobenzoate], IIc (n516). The crude product was isolated as a white solid from the reaction of compound 3b with 6-n-hexadecyloxynaphalene-2-carboxylic acid. 1H NMR (ppm, CDCl3): 8.73 (s, 2H, Ar–H), 8.1728.06 (m, 6H, Ar–H), 7.90 (d, J59.0 Hz, 2H, Ar–H), 7.82 (d, J58.7 Hz, 2H, Ar–H), 7.5527.49 (m, 3H, Ar–H), 7.2627.19 (m, 7H, Ar–H), 4.12 (t, J56.6 Hz, 4H, 26OCH2), 1.8821.83 (m, 4H, 26OCH2CH2), 1.5421.28 (m, 52H, 266CH2), 0.90 (t, J56.6 Hz, 6H, 26CH3). Anal: calc. for C74H88F2O10, C 75.61, H 7.55; found C 75.20, H 7.53%.
2.2.11. 3-(4-Nitrobenoyloxy)phenyl 4-nitrobenzoate, 4. This compound was obtained from resorcinol and 4-nitrobenzoic acid, following a similar procedure to that described for compound 2a. After work up, the crude product was purified by column chromatography (silica gel, CHCl3/EtOAc 20/1), and recrystallized from a mixture of dichloromethane and ethanol. The product
was isolated as a yellow solid; yield 80%. 1H NMR (ppm, CDCl3): 8.38 (s, 8H, Ar–H), 7.54 (t, J58.1 Hz, 1H, Ar–H), 7.2527.22 (m, 3H, Ar–H).
2.2.12. 3-(4-Aminobenzoyloxy)phenyl 4-aminobenzoate, 5. This compound was obtained from compound 4, following a similar procedure to that described for compound 3a. After work-up, the crude product was purified by column chromatography (silica gel, CHCl3/ EtOAc 2/1). The product was isolated as a white solid; yield 89%.1H NMR (ppm, CDCl3): 7.78 (d, J58.7 Hz, 4H, Ar–H), 7.45 (t, J57.8 Hz, 1H, Ar–H), 7.1227.08 (m, 3H, Ar–H), 6.62 (d, J58.7 Hz, 4H, Ar–H), 4.16 (s, 4H, Ar–NH2).
2.2.13. 1,3-Phenylene bis[4-(6-n-octyloxynaphthalen-2-ylideneamino)benzoate], IIIa (n58). A few drops of acetic acid (glacial) were added to a solution of compound 5 (0.4 g, 1.15 mmol) and 6-n-octyloxynaphthalene-2-carbaldehyde (0.69 g, 2.42 mmol) dissolved in absolute ethanol (50 ml). The mixture was heated at reflux for 4 h, then cooled to room temperature. The precipitated product was filtered off and, washed with hot absolute ethanol. It was purified by several recrystallizations from a chloroform and ethanol mixture to give a yellow solid; yield 0.35 g (35%).1H NMR (ppm, CDCl3): 8.57 (s, 2H, –CH5N–), 8.25 (d, J58.4 Hz, 4H, Ar–H), 8.16 (s, 2H, Ar–H), 8.12 (d, J58.7 Hz, 2H, Ar–H), 7.82 (t, J59.6 Hz, 4H, Ar–H), 7.48 (t, J58.4 Hz, 1H, Ar–H), 7.27 (d, J58.4 Hz, 4H, Ar–H), 7.2427.18 (m, 7H, Ar– H), 4.01 (t, J56.6 Hz, 4H, 26OCH2), 1.9221.82 (m, 4H, 26OCH2CH2), 1.5721.26 (m, 20H, 106CH2), 0.90 (t, J56.6 Hz, 6H, 26CH3). Anal: calc. for C58H60N2O6, C 79.06, H 6.86, N 3.18; found, C 79.06, H 7.03, N 3.43%.
2.2.14. 1,3-Phenylene bis[4-(6-n-dodecyloxynaphthalen-2-ylideneamino)benzoate], IIIb (n512). The crude product was isolated as a yellow solid from the reaction of compound 6 with 6-n-dodecyloxynaphthalene-2-carbaldehyde, following a similar procedure to that described for compound IIIa; yield 40%. 1H NMR (ppm, CDCl3): 8.57 (s, 2H, 2CH5N2), 8.25 (d, J58.4 Hz, 4H, Ar–H), 8.17 (s, 2H, Ar–H), 8.12 (d, J58.4 Hz, 2H, Ar–H), 7.85 (t, J59.3 Hz, 4H, Ar–H), 7.52 (t, J58.4 Hz, 1H, Ar–H), 7.32 (d, J58.7 Hz, 4H, Ar–H), 7.2627.18 (m, 7H, Ar– H), 4.11 (t, J56.6 Hz, 4H, 26OCH2), 1.9221.83 (m, 4H, 26OCH2CH2), 1.5821.28 (m, 36H, 186CH2), 0.88 (t, J56.6 Hz, 6H, 26CH3). Anal: calc. for C66H76N2O6, C 79.80, H 7.71, N 2.82; found, C 79.46, H 7.64, N 2.81%.
2.2.15. 1,3 - Phenylene bis [ 4 - ( 6 - n -hexadecyloxy-naphtha1len-2-ylideneamino)-benzoate], IIIc (n516). The crude product was isolated as a yellow solid from the reaction of compound 6 with 6-n-hexadecyloxynaphthalene-2-carbaldehyde, following a similar procedure to that described for compound IIIa; yield 42%. 1H NMR (ppm, CDCl3): 8.57 (s, 2H, 2CH5N2), 8.25 (d, J58.4 Hz, 4H, Ar–H), 8.17 (s, 2H, Ar–H), 8.12 (d, J58.4 Hz, 2H, Ar–H), 7.85 (t, J59.3 Hz, 4H, Ar–H), 7.52 (t, J58.4 Hz, 1H, Ar–H), 7.32 (d, J58.7 Hz, 4H, Ar–H), 7.2627.18 (m, 7H, Ar– H), 4.11 (t, J56.6 Hz, 4H, 26OCH2), 1.9021.82 (m, 4H, 26OCH2CH2), 1.5421.28 (m, 52H, 266CH2), 0.90 (t, J56.6 Hz, 6H, 26CH3). Anal: calc. for C74H92N2O6, C 80.39, H 8.39, N 2.53; found, C 79.87, H 8.35, N 2.46%.
3. Results and discussion
3.1. Mesophases and thermal properties
The phase transition temperatures and corresponding enthalpy changes of all the compounds obtained from differential scanning calorimetry (DSC) are summarized in table 1. Only one mesophase (either B1 or B2) was observed for each compound in both series; then exhibited monotropic or enantiotropic phase behaviour. The mesomorphic behaviour is significantly influenced by the terminal alkoxy flexible chains. All compounds bearing the longest chain length (n516) in the three analogous series show the B2 phase (Ic and IIIc are enantiotropic but IIc is monotropic). In contrast, the analogous compounds bearing the shortest chain length (n58) show the B1phase (Ia and IIa are enantiotropic but IIIa is monotropic).
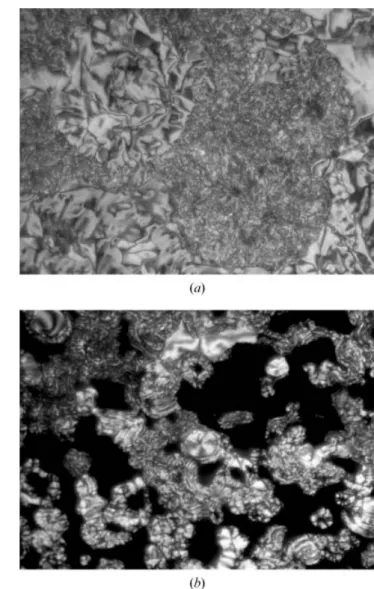
The B1phase exhibits both mosaic-like and dendritic nuclei textures as it grows from the isotropic phase [5]; the dendritic nuclei texture of the B1phase in compound Ia at 157uC (cooling) is shown in figure 2 (a). As the temperature was further lowered, the dendritic nuclei rapidly coalesced into a structured mosaic-like texture [20]. After further cooling of compound IIIa, a typical mosaic-like texture of the B1phase was seen at 150uC, as shown in figure 2 (b). Similar textural features of the B1phase were observed in compounds IIa and IIb. The transition enthalpies of the B1–isotropic transition are in the range 12.6–14.9 kJ mol21.
For analogous compounds with the medium chain length (n512) the B2phase was dominant, except that a monotropic B1 phase was observed in compound IIb (X5F and Y5–COO–) due to the lateral fluorine. Hence, based on the same length of flexible chain (n512), the B2phase is not favoured by the lateral meta-fluoro substitution of compound IIb compared with
series Ib (X5H and Y5–COO–). An oval nuclei texture of the B2 phase cooling from the isotropic liquid is observed to coalesce gradually to form a non-uniform texture. Compound Ic reveals schlieren and non-specific grainy texture regions at 128uC as shown in figure 3 (a), and compound IIIb displays spherulite domains and stripe textures coexisting with schlieren textures in some
parts of small regions at 138uC, figure 3 (b), which demonstrate the characteristics of typical texture features observed in the B2 phase [21–23]. Similar textural features of the B2phase were also observed in compounds Ib, IIc and IIIc. The transition enthalpies of the B2–isotropic transition are in the range 13.3– 24.1 kJ mol21.
Table 1. The transition temperatures (uC) and enthalpies (in parentheses, kJ mol21
)aof all compounds in series I–III.
Comparing analogous compounds among these three series, the lateral meta-fluoro-substituted derivatives of series II have the lowest isotropization temperatures and the Schiff base linkage derivatives of series III have the highest isotropization temperatures. Moreover, the lateral meta-fluoro-substituted derivatives of series II possess the narrowest mesophasic ranges of the B1and B2 phases. This suggests that the dipolar effects and steric factors of lateral fluorine are detrimental to the formation of the B1 and B2 phases, especially the B2 phase. Although the B2 phase can be obtained by increasing the flexible chain length to n516 in series II, compound IIc only displays a short range of a monotropic B2 phase. Thereby, in contrast to the
non-substituted series I, the lateral meta-fluoro sub-stitution in series II destabilizes the B2phase.
3.2. X-ray investigation
In order to determine the structure of the mesophases by X-Ray diffraction (XRD), measurements were carried out in the mesophase temperature ranges of the compounds Ic, IIa and IIIc (non-oriented samples). The XRD pattern of the mesophase obtained from compound IIa at 125uC is shown in figure 4 (a). In the small angle region, two sharp reflection peaks are obtained at d1526.2 A˚ and d2522.7 A˚ ; these reflections can be indexed as (1 1) and (0 2), respectively, for a Figure 2. Polarizing optical micrographs of the B1 phase
obtained on cooling from the isotropic phase: (a) growth of dendritic nuclei of compound Ia at 157uC, (b) mosaic-like texture of compound IIIa at 150uC.
Figure 3. Polarizing optical micrographs of the B2 phase
obtained on cooling from the isotropic phase: (a) schlieren and non-specific grainy texture of compound Ic at 128uC, (b) spherulite domains and stripe textures of compound IIIb at 138uC.
centre-rectangular lattice with two-dimensional lattice parameters calculated as a532.1 A˚ and b545.4 A˚, which can be identified as a B1 phase (rectangular columnar mesophase, Colr) as described in the literature [20, 24]. The molecular length (L) values of the banana-shaped molecular structures can be calculated by molecular modelling, on the assumtion that all molecular struc-tures are coplanar, the bent rigid cores have a central bending angle of 120u [20, 25] and the alkyl chains are in the fully extended all-trans conformation. The value of the lattice parameter b corresponds approximately to the fully extended molecular length of 46.2 A˚ by molecular modelling calculation.
The XRD patterns of the mesophases obtained from compounds Ic and IIIc at 122 and 120uC, respectively, are shown in figure 4 (b). Compound Ic in the meso-phase showed three sharp reflection peaks at d1542.9 A˚ , d2521.5 A˚ and d3514.4 A˚ , which can be indexed as (0 1), (0 2), and (0 3), in the small angle region. The layer (d ) spacing values are in the ratio of 1 : 1/2 : 1/3, indicating that a lamellar order exists in the mesophase. Similar diffraction patterns for IIIc in the small angle region, viz. three sharp reflection peaks at d1545.5 A˚ , d2522.6 A˚ , and d3515.1 A˚ , again indicate lamellar order. This is clearly indicative of a two-dimensional structure of the B2phase, which has been confirmed in previous reports [26]. The d values determined from the XRD measurements are smaller than the calculated molecular length (L) values from molecular modelling, i.e. L558.2 and 59.2 A˚ for compounds Ic and IIIc, respectively, indicating tilted smectic phases. From the values of d spacing (XRD) and L (molecular modelling), the tilt angle (a) can be calculated to be around 42.5u and 39.8u for compounds Ic and IIIc, respectively. In addition, wide angle diffuse peaks corresponding to a spacing value of 4.6 A˚ in figures 4 (a) and 4 (b) indicate that similar liquid-like in-plane orders with average intermolecular distances around 4.6 A˚ are prevalent inside the smectic layers of compounds Ic, IIc and IIIc.
3.3. Electro-optical behaviour
Spontaneous polarization (Ps) experiments were carried out on LC cells of 9mm thickness (antiparallel rubbing direction) by the triangular wave voltage method [27], the current response being measured across a 1 kV resistance. Regarding the electro-optical measurements, the most interesting compounds are Ib, Ic, IIIb and IIIc, because they exhibit the best mesomorphic properties and switching behaviour (i.e. B2phase). Since both B1rev and B1rev,tilt phases exhibit switching phenomena on applying electric fields [14, 28], the B1 phase is further confirmed by the electro-optical observation that the B1 phase exhibits no switching phenomena or texture variation under polarizing microscopy, on applying an electric field. For instance, the spontaneous polarization of compounds Ib and IIIb were measured representa-tively (because both compounds exhibit the B2phase). The samples were slowly cooled (1uC min21
) from the isotropic phase to the mesophase under a triangular wave voltage. If a lower triangular wave voltage (E,2 Vmm21) was applied, only one sharp peak was obtained in the Ps experiments. However, when a sufficiently higher triangular voltage (E,7 Vmm21) was applied, a second peak became clearly visible and sharper. Thereafter, the current response peak became saturated above certain critical values of external fields. Figure 4. X-ray diffraction intensity against angle profiles
obtained upon cooling from the isotropic phase: (a) in the B1
phase of compound IIa at 125uC; (b) in the B2 phase of
compound Ic at 122uC and compound IIIc at 120uC (top and bottom, respectively).
Two current peaks per half-period of an applied triangular voltage are obtained in the switching current response curve of compounds Ib and IIIb, as shown in figures 5 (a) and 5 (b), respectively. The two-peak response is the characteristic behaviour of a sequential electric response due to a ferroelectric state switched into an antiferroelectric ground state and back to the opposite ferroelectric state; so this result confirms the SmCPAstructure of the B2phase in compounds Ib and IIIb [29]. The Ps values calculated by integrating the area under the switching current peaks are estimated to
be c. 940 nC cm22 for compound Ib at 132uC, and 820 nC cm22for compound IIIb at 134uC.
Figure 6 shows the values of spontaneous polariza-tion plotted as a funcpolariza-tion of the temperature for compounds Ic and IIIc. On cooling from the isotropic phase, the spontaneous polarizations increase initially with decreasing temperature, then level off. The abrupt increase of Ps around the isotropization temperature indicates that the I-B2phase transition is of first order. The Ps value exhibits maxima of 895 nC cm22 for compound Ic at 127uC and 680 nC cm22
for IIIc at Figure 5. Switching current response on applying the triangular wave method (antiparallel rubbed 9mm thick LC cells): (a) the B2
phase of compound Ib at 132uC (Vpp5100 V, f5100 Hz); (b) the B2phase of compound IIIb at 134uC (Vpp580 V, f550 Hz).
122uC. The Psmeasurements for compound IIc are not shown in figure 6 because of its narrow mesophase range; its maximum Ps value is about 368 nC cm22 at 104uC. In comparison, the Ps value of compound IIc (368 nC cm22) is much smaller than that of the analogous compound Ic (895 nC cm22) without the lateral meta-fluoro-substitution.
Figures 5 and 6 suggest that the Ps values of compounds Ib and Ic (940 nC cm22 and 895 nC cm22, respectively) are larger than those of the analogous compounds IIIb and IIIc (820 nC cm22 and 680 nC cm22, respectively), by reason of the different linking groups (on the side wings) in series I (Y5 –COO–) and series III (Y5–CH5N–). In addition, compounds Ib and IIIb have larger Psvalues than the respective compounds Ic and IIIc owing to different lengths of the flexible terminal chains. Thus, the order of Psvalues in relation to molecular design is as follows: Ps(without fluoro-substitution).Ps (with lateral meta-fluoro-substitution on the middle outer rings); Ps (Y5 –COO–, linking group on the side wings).Ps (Y5 –CH5N–); Ps (n512, shorter flexible chains).Ps (n516, longer flexible chains).
In order to investigate the dynamics of the polariza-tion switching behaviour, the switching currents of compound Ib in 9mm thick LC cells (antiparallel rubbed) were measured with an applied square wave field (Vpp580 V, f510 Hz) in the B2phase, at 121uC on cooling from the isotropic phase. The resulting switch-ing current curves are shown in figure 7 (a) as a function of time. The switching time (t) values were determined
from the time elapsed between the appearance of the maximum of the current signal and the field reversal [30]. The switching time as a function of temperature is shown in figure 7 (b) with the same field and frequency. A decrease in the switching time with increasing temperature for both compounds Ib and IIIc (in the range 10–40ms) was observed. The switching time of compound Ib (n512, Y5–COO–, Ps5940 nC cm22) is a little larger than that of compound IIIc (n516, Y5 –CH5N–, Ps5680 nC cm22), with less difference at higher temperatures. The switching time is influenced by viscosity but is unrelated to the Psvalue.
On slow cooling of compound Ib from the isotropic phase, the mesophase gradually appeared and coalesced as small fractal domains; non-specific grainy domains along with small fan-like domains developed at 125uC on further cooling. By applying a sufficiently high electric field (E,7 Vmm21
), the texture was transformed into a fan-like texture. As the electric field was removed, the switched off state relaxed back to a state with non-specific texture, possessing only very small irregularly distributed bright spots, which was nearly dark between crossed polarizers, see figure 8 (a). When one polarizer was rotated clockwise by a small angle (c. 10u) from the crossed position, dark and bright domains become clearly distinguishable. On rotating the polarizer counterclockwise by the same angle from the crossed position, the previously dark domains became bright and vice versa, see figures 8 (b) and 8 (c). This observa-tion is indicative of the occurrence of chiral domains with opposite handednesses. Thus, two types of Figure 6. Temperature dependence of spontaneous polarization in the B2phase of compounds Ic and IIIc (antiparallel rubbed
9mm thick LC cells).
optically active domain with opposite chiralities were formed; this phenomenon of exchanging brightness with rotation of the polarizer against the analyser, counter-clockwise or counter-clockwise, has been reported previously [13].
In order to investigate further the switching process of the B2 phase, a d.c. electric field was applied to antiparallel rubbed 9mm thick LC cells between crossed polarizers [29]. When the isotropic phase was slowly
cooled (0.5uC min21
) in the presence of a high d.c. field (about 60 V), large and intact circular domains were induced. Moreover, many circular domains were formed in the B2 phase in which the smectic layers were circularly arranged around the centres of the domains. The layer structure arrangement corresponds to the domain models proposed by Link et al. [7]. After removing the electric fields, the extinction crosses were reoriented along the crossed polarizer position in the off Figure 7. (a) Time dependences of the switching current under a square wave field for compound Ib at 121uC; (b) switching time as a function of temperature for compounds Ib and IIIc (antiparallel rubbed 9mm thick LC cells, Vpp580 V, f510 Hz).
state, see figure 9 (a), indicating an anticlinic tilt in the antiferroelectric ground state (SmCAPA). As shown in figures 9 (b) and 9 (c), on applying electric fields with reverse polarities, the extinction crosses rotated coun-terclockwise (positive field) and clockwise (negative field). The extinction crosses in the switched on state did not coincide with the crossed polarizer positions in the off-state. The angles between the extinction directions and the crossed polarizer positions are about 41u for ¡60 V at 124uC, which corresponds to the optical tilt angle. The switching behaviour of the extinction crosses of domains rotated in opposite directions, depending on the sign of the field, is characteristic of field-responsive textures for chiral domains of the B2 phase [31]. However, the extinction crosses should rotate in only
one direction in the racemic domains, independent of the sign of the field, which is characteristic of field-responsive textures for racemic domains of the B2 phase (see figure 10). The angles between the extinction directions and the crossed polarizer positions are about 37u for ¡60 V at 127uC [32].
The switching process can also be studied by measuring the tilt angle as a function of applied d.c. electric fields in the B2phase of compounds Ic and IIIc (at 124 and 130uC, respectively), as shown in figure 11. The tilt angles are estimated from the angles of rotation between the extinction brushes and crossed polarizers on applying an electric field, which indicates that a rotated phase structure from the anticlinic-antiferro-electric (SmCAPA) state to the synclinic-ferrelectric Figure 8. Polarizing optical microscopic texture of the B2 phase in compound Ib after removing the electric field at 125uC
(cooling): (a) crossed position, (b) and (c) rotation of the polarizer counterclockwise and clockwise, respectively, from the crossed polarizer position. (Arrows are the directions of polarizers and analysers.)
(SmCSPF) state has occurred. The tilt angles of both compounds increase with increasing applied voltage, and saturate at fields of 28 V and 17 V for compounds Ic and IIIc, respectively. Moreover, both compounds Ic and IIIc have similar tilt angles at lower applied voltages and similar saturated tilt angles around 40u.
Optical transmission studies were carried out on the B2 phase of compound Ic in 6mm thick LC cells (antiparallel rubbed) using a triangular wave voltage (Vpp560 V, f51 Hz) at 129uC. The electric field dependence of the transmitted light intensity is shown in figure 12. The switching process exhibits a thresh-oldless V-shaped behaviour, with highest transmittance at around 30 V; a similar phenomenon has been observed in another bent-core structure [33]. This result
should coincide with the previous title angle experiment of compound Ic, where the highest transmittance at around 30 V is due to the saturated title angle at around 28 V (see figure 11); the turn on voltage is below 2 V. The V-shaped or pseudo-V-shaped electro-optical response in transmittance is abruptly increased on increasing the applied voltage from below 7 V, then is gradually relaxed near saturation at around 30 V. The shape of the electro-optical response curve and turn on voltage in transmittance may be further improved by reducing the thickness of the LC cell.
4. Conclusions
Three analogous series of achiral banana-shaped liquid crystalline molecules containing bisnaphthyl units and Figure 9. Polarizing optical micrographs of the circular domains in the B2phase for compound Ic (antiparallel rubbed 9mm thick
LC cells). Chiral domains obtained by various d.c. fields: (a) 0 V, SmCAPA, (b) 260 V, SmCSPF, (c)+60 V, SmCSPFat 124uC.
(Arrows are the directions of polarizers and analysers.)
derived from resorcinol cores have been prepared. The formation of the B2phase with antiferroelectric switch-ing behaviour is favoured in the achiral bisnaphthyl derivatives with longer flexible chains; however, the B1 phase is favoured in corresponding derivatives with shorter flexible chains. The B1 and B2phases are less favoured in compounds of series II with lateral meta-fluoro-substitution on the middle outer phenyl rings, in comparison with series I without fluoro-substitution. The B1or B2phases have been confirmed by XRD and POM measurements. The electro-optical switching behaviour of two current peaks found during a triangular voltage half-period has also confirmed the B2phase. This process exhibits an electric field-induced transition from an antiferroelectric (tristable) state to a
ferroelectric (bistable) state. The spontaneous polariza-tion (by switching current response), tilt angle of chiral domains (by POM), and transmittance–voltage mea-surements of the B2phase in related compounds have been surveyed in this study.
Acknowledgements
The powder XRD measurements were supplied by beamline BL17A (operated by Dr J.-J. Lee) of the National Synchrotron Radiation Research Center (NSRRC), Taiwan. Financial support provided by the National Science Council of Taiwan, ROC through NSC 92-2113-M-009-016, and AUO (Taiwan) through NCTU-94C060 is acknowledged.
Figure 10. Polarizing optical micrographs of the circular domains in the B2phase for compound Ic (antiparallel rubbed 9mm thick
LC cells). Racemic domains obtained by various d.c. fields: (a) 0 V, SmCSPA, (b) 260 V, SmCAPF, (c)+60 V, SmCAPFat 127uC.
(Arrows are the directions of polarizers and analysers.)
References
[1] R.B. Meyer, L. Liebert, L. Strzelecki, P. Keller. J. Phys. (Paris) Lett., 36, L69 (1975).
[2] A.D.L. Chandani, Y. Ouchi, H. Takezoe, A. Fukuda, K. Terashima, K. Furukawa, A. Kishi. Jpn. J. appl. Phys., 28, L1261 (1989).
[3] A. Fukuda, Y. Takanishi, T. Isokaki, K. Ishikawa, H. Takezoe. J. mater. Chem., 4, 997 (1994).
[4] T. Niori, T. Sekine, J. Watanabe, T. Furukawa, H. Takezoe. J. mater. Chem., 6, 1231 (1996).
[5] G. Pelzl, S. Diele, W. Weissflog. Adv. Mater., 11, 707 (1999). [6] J.P. Bedel, J.C. Rouillon, J.P. Marcerou, M. Laguerre, H.T. Nguyen, M.F. Achard. Liq. Cryst., 28, 1285 (2001). Figure 11. Tilt angle of the B2phase as a function of d.c. electric field in compounds Ic and IIIc (antiparallel rubbed 9mm thick
LC cells) at 124 and 130uC, respectively.
Figure 12. Transmittance versus applied voltage (triangular wave, Vpp560 V, f51 Hz) in the B2 phase of compound Ic
(antiparallel rubbed 6mm thick LC cells) at 129uC.
[7] D.R. Link, G. Natale, R. Shao, J.E. Maclennan, N.A. Clark, E. Ko¨rblova, D.M. Walba. Science, 278, 1924 (1997).
[8] W. Weissflog, H. Na`dasi, U. Dunemann, G. Pelzl, S. Diele, A. Eremin, H. Kresse. J. mater. Chem., 11, 2748 (2001).
[9] M.W. Schro¨der, S. Diele, G. Pelzl, U. Dunemann, H. Kresse, W. Weissflog. J. mater. Chem., 13, 1877 (2003). [10] R.A. Reddy, B.K. Sadashiva, V.A. Raghunathan. Chem.
Mater., 16, 4050 (2004).
[11] C. Keith, R.A. Reddy, C. Tschierske. Chem. Commun., 7, 871 (2005).
[12] C. Achten, R. Cuypers, M. Giesbers, A. Koundijs, A.T.M. Marcelis, E.J.R. Sudho¨lter. Liq. Cryst., 31, 1167 (2004).
[13] M.W. Schro¨der, S. Diele, G. Pelzl, W. Weissflog. ChemPhysChem, 5, 99 (2004).
[14] D. Kardas, M. Prehm, U. Baumeister, D. Pociecha, R.A. Reddy, G.H. Mehl, C. Tschierske. J. mater. Chem., 15, 1722 (2005).
[15] R. Gu¨ller, A. Binggeli, V. Breu, D. Bur, W. Fischi, G. Hirth, C. Jenny, M. Kansy, F. Montavon, M. Mu¨ller, C. Oefner, H. Vieira, M. Wilhelm, W. Wostl, H.P. Ma˘tki. Bioorg. med. Chem. Lett., 9, 1403 (1999).
[16] M. Debono, W.W. Turner, L. LaGrandeur, F.J. Burkhadt, J.S. Nissen., et al. J. med. Chem., 38, 3271 (1995). [17] R. Gomez, J.L. Segura, N. Martin. J. org. Chem., 65,
7501 (2000).
[18] C. Cativiela, J.L. Serrano, M.M. Zurbano. J. org. Chem., 60, 3074 (1995).
[19] A. Svensson, T. Fex, J. Kihlberg. Tetrahedron Lett., 37, 7649 (1996).
[20] D. Shen, S. Diele, G. Pelzl, I. With, C. Tschierske. J. mater. Chem., 9, 661 (1999).
[21] T. Sekine, T. Niori, M. Sone, J. Watanabe, S.W. Choi, Y. Takanishi, H. Takezoe. Jpn.J. appl. Phys., 36, 6455 (1997).
[22] H. Dehne, M. Po¨tter, S. Sokolowski, W. Weissflog, S. Diele, G. Pelzl, I. Wirth, H. Kresse, H. Schmalfuss, S. Grande. Liq. Cryst., 28, 1269 (2001).
[23] R.A. Reddy, B.K. Sadashiva. Liq. Cryst., 30, 1031 (2003). [24] G. Dantlgraber, D. Shen, S. Diele, C. Tschierske. Chem.
Mater., 14, 1149 (2002).
[25] J. Ortega, M.R. de la Fuente, J. Etxebarria, C.L. Folcia, S. Dı´ez, J.A. Gallastegui, N. Gimeno, M.B. Ros, M.A. Pe´rez-Jubindo. Phys. Rev. E., 69, 011703 (2004). [26] V. (a) Prasad, S.W. Kang, S. Kumar. J. mater. Chem. 13,
1259 (2003); (b) H.N. Shreenivasa Murthy, B.K. Sadashiva. Liq. Cryst., 31, 1337 (2004).
[27] K. Miyasato, S. Abe, H. Takezoe, A. Fukuda, E. Kuze. Jpn. J. appl. Phys., 22, L661 (1983).
[28] J. (a) Mieczkowski, K. Gomola, J. Koseska, D. Pociecha, J. Szydlowska, E. Gorecka. J. mater. Chem. 13, 2132 (2003); (b) E. Gorecka, N. Vaupoticˇ, D. Pociecha, M. Cˇ epicˇ, J. Mieczkowski. ChemPhysChem, 6, 1087 (2005); (c) K. Pelz, W. Weissflog, U. Baumeister, S. Diele. Liq. Cryst., 30, 1151 (2003).
[29] M. Zenyoji, Y. Takanishi, K. Ishikawa, J. Thisayukta, H. Watanabe, J. Takezoe. J. mater. Chem., 9, 2775 (1999). [30] C.V. Yelamaggad, U.S. Hiremath, S. Anitha Nagamani,
D.S. Shankar Rao, S. Krishna Prasad. J. mater. Chem., 11, 1818 (2001).
[31] D. Shen, A. Pegenau, S. Diele, I. Wirth, C. Tschierske. J. Am. chem. Soc., 122, 1593 (2000).
[32] S. Shubashree, B.K. Sadashiva, S. Dhara. Liq. Cryst., 29, 789 (2002).
[33] A. Ja´kli, Y.M. Huang, K. Fodor-Csorba, A. Vajda, G. Galli, S. Diele, G. Pelzl. Adv. Mater., 15, 1606 (2003).