Ab Initio Chemical Kinetics for SiH
2
+ Si
2
H
6
and SiH
3
+ Si
2
H
5
Reactions
and the Related Unimolecular Decomposition of Si
3
H
8
under a
‑Si/H
CVD Conditions
P. Raghunath and M. C. Lin*
Center for Interdisciplinary Molecular Science, Department of Applied Chemistry, National Chiao Tung University, Hsinchu 300, Taiwan
*
S Supporting InformationABSTRACT: The kinetics and mechanisms for SiH2+ Si2H6and SiH3+ Si2H5reactions and the related unimolecular decomposition of Si3H8have been investigated by ab initio
molecular orbital theory based on the QCISD(T)/CBS//QCISD/6-311++G(d,p) method in conjunction with quantum statistical variational Rice−Ramsperger−Kassel−Marcus (RRKM) calculations. For the barrierless radical association processes, their variational transition states have been characterized by the CASPT2//CASSCF method. The species involved in the study are known to coexist under CVD conditions. The results show that the association reaction of SiH2and Si2H6producing Si3H8occurs by insertion via its
lowest-energy path forming a loose hydrogen-bonding molecular complex with 8.3 kcal/mol binding energy; the reaction is exothermic by 55.0 kcal/mol. The chemically activated Si3H8adduct can fragment by several paths, producing SiH4+ SiH3SiH (−0.7 kcal/mol),
Si(SiH3)2+ H2(−1.4 kcal/mol), and SiH3SiH2SiH + H2(−1.4 kcal/mol). The predicted enthalpy changes as given agree well
with available thermochemical data. Three other decomposition channels of Si3H8occurring by Si−H or Si−Si breaking were found to be highly endothermic, and the reactions take place without a well-defined barrier. The heats of formation of Si3H8,
SiH2SiH, Si2H4, i-Si3H7, n-Si3H7, Si(SiH3)2, and SiH3SiH2SiH have been predicted and found to be in close agreement with those available data in the literature. The product branching rate constants for SiH2+ Si2H6and SiH3+ Si2H5reactions and the thermal
unimolecular decomposition of Si3H8for all low-energy paths have been calculated with multichannel variational RRKM theory covering varying P,T conditions typically employed in PECVD and Cat-CVD processes for hydrogenated amorphous silicon (a-Si/H)film growth. The results were also found to be in good agreement with available kinetic data. Our kinetic results may be employed to model and control very large-area a-Si/Hfilm growth for a new generation of solar cell applications.
■
INTRODUCTIONSilylene, SiH2, plays an important role in the reactions of silane or disilane forming higher silanes in the deposition of hydro-genated amorphous silicon (a-Si/H) thinfilms and polycrystal-line silicon (p-Si).1−6 For many decades, the technology has been used in the semiconductor industry for fabrication of solar cells, thin film transistors, and so on.7,8These films are prepared either by plasma-enhanced chemical vapor deposi-tion (PECVD) or, increasingly, by catalytic chemical vapor deposition (Cat-CVD).1−12 In both processes, silanes and hydrogen are employed as the source gases in a chamber. From the mechanistic point of view, the dissociation of a silane source gas, by collisions with electrons in a plasma or with a hot metal catalyst surface, leads to the generation of radicals, atoms, and ions. These species diffuse on to the substrate after the se-condary reactions in the gas phase, depositing a-Si/Hfilm through surface reactions. The chemical processes of the amorphous material formation thus involve gas-phase and surface reactions, which are affected by the system’s temperature and have a strong influence on the quality of the film. Roth et al. investigated the gas-phase parameters such as theflow, temperature, and pressure of participating reactants; the energy input for dissociation reactions, the size, and the structure of Si−H compounds have an important influence on the properties of the surface layer.13
Mechanistically, SiH2is known to be a primary decomposition product in the decomposition of silanes.1−19The interaction of the SiH2radical with Si2H6is a subject of interest in the present work because Si2H6 is one of the most commonly employed
reagents and silylene is known to be a very reactive diradical that inserts into Si−H bonds very efficiently, forming excited adduct Si3H8.
14−19The excited adduct can dissociate via intramolecular
1,2-H shifts, producing various products.20−22 Inoue et al.16 detected SiH2by laser-inducedfluorescence and found the rate
constants of the SiH2 reaction with Si2H6 to be 5.7 × 10−10 cm3molecule−1s−1in 1 Torr of helium at room temperature. Later, Jasinski et al.17 generated SiH2 by using laser absorption spec-troscopy and found the overall rate constant of the SiH2 with
disilane reaction from 1 to 10 Torr of He pressure at 298 K; the rate constant measured at 1 Torr of pressure was 1.5 × 10−10 cm3molecule−1s−1. Ditrich et al.4investigated the role of silylene in the laser-induced CVD of a-Si/H and the rate constant of SiH2with silane or disilane, forming higher silanes, Si2H6and Si3H8. The rate
constant for formation of the latter product was measured under the deposition condition to be 2.7± 0.4 × 10−10cm3molecule−1s−1.
Received: July 29, 2013
Revised: September 14, 2013
Published: September 23, 2013
Absolute rate constants for the reactions of SiH2 with Si2H6
have also been determined by Walsh and co-workers,18
covering the temperature range of 295−595 K by means of laserflash photolysis. Ring, O’Neal, and co-workers interpreted the mechanism for decomposition of Si3H8in the temperature range of 529−560 K.20,21 Arrhenius parameters for Si3H8
decomposition via 1,2-hydrogen migration reactions that lead to disilane and silylene at 530−570 K have been measured by Moffat et al.22
Various works have been carried out with Si2H6and Si3H8
as source gases for the preparation of a-Si/H thin films by low-temperature CVD.23,24 Kumata et al.23 investigated the deposition rate of a-Si/H films prepared from Si3H8 by the
direct photo-CVD method, which was found to befive times that from Si2H6. In our earlier studies, we used computational
tools to elucidate reaction mechanisms and provide accurate thermodynamic and kinetics data for gas-phase reactions of SiH3, SiH4, Si2H6, and Si3H8 with H and SiH3.25−28 These
species are known to coexist in media under CVD conditions. Under experimental conditions, the generated radicals and ions in the reaction chamber undergo a variety of primary and secondary reactions leading to reaction products, which are potential candidates for the layer-forming process at the substrate surface. It is well-known that SiH2is easily generated
in the electron-impact dissociation reaction of silane and hydrogen. Therefore, a detailed understanding of the gas-phase reactions involved may be useful to control the deposition parameters for optimization of the a-Si/H growth in a large substrate area under well-defined conditions. In this work, we focus our study on the effects of temperature and pressure on the association reactions of SiH2+ Si2H6and SiH3+ Si2H5via
the Si3H8intermediate for both forward and reverse processes,
which are critical to our ability in realistic simulations of a-Si−H
thinfilm growth by PECVD and Cat-CVD. We will study the
detailed mechanism for the title reactions by fully characterizing their common potential energy surface (PES), employing ab initio molecular orbital methods. For the SiH2 and Si2H6
reaction, it may take place by the following channels in the forward and reverse directions:
+ → * → + → + → + → + → + SiH Si H Si H Si H ( M) (1)
SiH SiH SiH (2)
H Si(SiH ) (3)
H SiH SiH SiH (4)
SiH Si H (5) 2 2 6 3 8 3 8 4 3 2 3 2 2 3 2 3 2 5
On the same PES, SiH3and Si2H5radicals produced from the
fragmentation of Si3H8* depicted above may react as follows in
a deposition medium: + → * → + → + → + → + → + SiH Si H Si H Si H ( M) (6) SiH Si H (7)
SiH SiH SiH (8)
H Si(SiH ) (9)
H SiH SiH SiH (10)
3 2 5 3 8 3 8
2 2 6
4 3
2 3 2
2 3 2
In the above reaction schemes, * denotes an internally
activated intermediate and M stands for a third body or
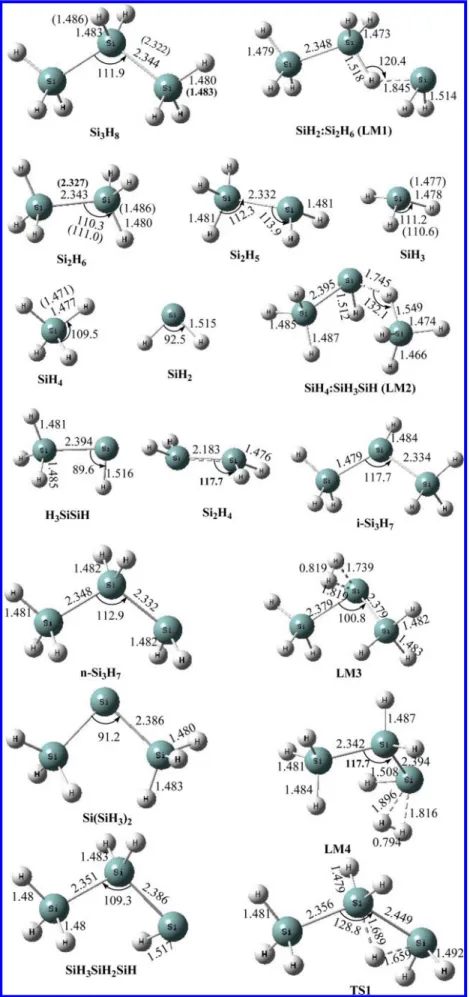
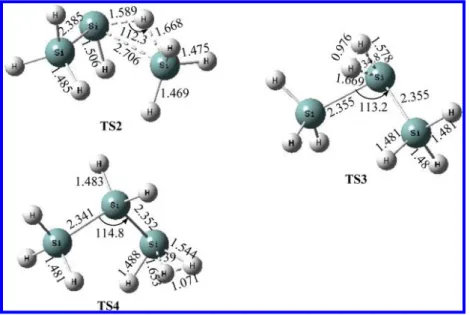
Figure 1.The optimized geometries of the reactants, intermediates, transition states, and products computed at the QCISD/6-311++G(d,p) level. The values in parentheses are the experimental values (refs 39 and 40). (Lengths in Å, and angles in degrees.)
Figure 2. Schematic energy diagram for the SiH2 + Si2H6 reaction computed at the QCISD(T)/CBS//QCISD/6-311++G(d,p) level with ZPE corrections. Relative energies are given in kcal/mol at 0 K.
quencher. The predicted geometries, vibrational frequencies, and heats of formation for new radical products at 0 K are given in the Results and Discussion section. The temperature and pressure dependences of the rate constants for the forward and reverse reactions and their related unimolecular decomposition processes have been derived using variational RRKM theory by solving the master equation covering the conditions commonly employed in industrial deposition of a-Si/Hfilms.
■
COMPUTATIONAL METHODSThe geometries of the reactants, products, intermediates, and transition states of the title reactions have been fully optimized by using the QCISD method29(the spin-unrestricted quadratic configuration interaction with single and double excitation) with the 6-311++G(d,p) basis set. Further improvement of energetics of the PES has been made with QCISD(T)/6-311+ +G(3df,2p) by single-point calculations. Vibrational frequencies
calculated at the QCISD/6-311++G(d,p) level have been used for characterization of stationary points, zero-point energy (ZPE) corrections, and reaction rate constants calculations. A recent study on silicon species has shown that the widely used QCISD method with split valence and Dunning correlation basis sets is quite suitable for geometry and property predic-tions.30,31 For a more accurate evaluation of the energetic parameters, single-point energy calculations of the stationary points were carried out by the QCISD(T)/CBS method,32in which the basis set extrapolation was based on the calculations with the cc-pVXZ (X = D, T, and Q) basis sets of Dunning.33 The CBS energies were estimated by the three-point extrapola-tion scheme.32All of the calculations were carried out using the Gaussian 03 program package.34
Rate constant calculations were carried out with the
VARIFLEX program35 based on the microcanonical RRKM
theory and variational transition-state theory (VTST).36,37The component rates were evaluated at the E/J-resolved level, and the pressure dependence was treated by one-dimensional master equation calculations using the Boltzmann probability of the intermediate (Si3H8*) for the J-distribution. For a
barrierless association/decomposition process, the variational TS36,37 was approximated with the Morse function, V(R) = De{1− exp[−β(R − Re)]}2, in conjunction with a potential
an-isotropy function to represent the minimum potential energy path (MEP), which will be discussed later. Here, De is the
binding energy excluding zero-point vibrational energy for an association reaction, R is the reaction coordinate (i.e., the distance between the two bonding atoms), and Reis the
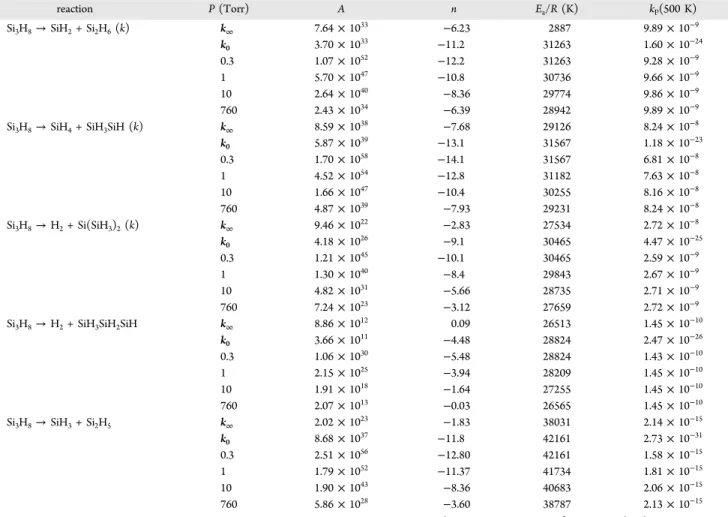
equilibrium value of R at the stable intermediate structure. Table 1. Calculated Relative Energies (kcal/mol, ZPE
corrections are included) for the SiH2Reaction with Si2H6at
Various Levels of Theory
QCISD/6-311+ +G(d,p) QCISD(T)/6-311+ +G(3df,2p)a QCISD(T)/ CBSa SiH2+ Si2H6 0.0 0.0 0.0 SiH2/Si2H6, LM1 −4.3 −7.0 −8.3 TS1 −1.5 −6.8 −9.3 Si3H8 −50.0 −52.7 −55.0 TS2 −4.8 −10.1 −12.2 SiH4/SiH3SiH, LM2 −4.8 −9.0 −10.6
SiH4+ SiH3SiH 0.2 −0.6 −0.7 TS3 3.4 −3.8 −6.2 H2/Si(SiH3)2, LM3 3.8 −3.9 −5.7
H2+ Si(SiH3)2 3.8 −1.7 −1.4
TS4 5.7 −0.9 −3.4 H2/SiH3SiH2SiH, LM4 3.0 −3.3 −4.7 H2+ SiH3SiH2SiH 2.5 −1.7 −1.2
SiH3+ Si2H5 17.7 19.0 19.0
H + i-Si3H7 31.7 29.9 30.0
H + n-Si3H7 33.6 31.8 31.9 aSingle-point calculations based on QCISD/6-311++G(d,p) optimized
geometries.
Table 2. Heats of Reaction (ΔrH0°) and Heats of Formation (ΔfH0°) of Species at 0 K Predicted at The QCISD/6-311+
+G(d,p)/CBS Level of Theory Given in kcal/mol
heat of reactionΔrH0° heat of formationΔfH0°
species reactionsa calculated literaturea calculated literaturea Si3H8 1SiH2+ Si2H6→ Si3H8 −55.0 −55.0 33.5 33.5± 1.0
SiH3 Si3H8→ SiH3+ Si2H5 74.0 73.4 48.3 47.7± 1.2
SiH3SiH Si3H8→ SiH4+ SiH3SiH 54.3 77.3
Si2H4 Si3H8→ SiH4+ Si2H4 45.0 47.0± 1.0 68.0 67.9± 0.9
i-Si3H7 Si3H8→ H + i-Si3H7 85.1 86 66.9± 1.0 67.8
n-Si3H7 Si3H8→ H + n-Si3H7 86.9 87.5 68.7± 1.0 69.3
Si(SiH3)2 Si3H8→ H2+ Si(SiH3)2 53.6 87.1
SiH3SiH2SiH Si3H8→ H2+ SiH3SiH2SiH 53.8 87.3
aThe experimental values employed in the calculations are obtained based on the enthalpies of formation at 0 K for H = 51.7 kcal/mol; H
2= 0.0 kcal/mol; SiH4= 10.5 kcal/mol (ref 42); SiH3= 47.7± 1.2 kcal/mol (ref 42);1SiH2= 65.6± 0.7 kcal/mol (ref 42); Si3H8= 33.5± 1.0 kcal/mol (ref 26); Si2H6= 22.9 kcal/mol (refs 41−43); Si2H5= 59.2 kcal/mol (ref 43); i-Si3H7= 67.8 kcal/mol (refs 41 and 26); n-Si3H7= 69.3 kcal/mol (refs 41 and 26); and Si2H4= 67.9± 0.9 (ref 43).
Table 3. Predicted Morse (β) and Exponential Coefficient Lennard-Jones Parameters Used in Rate Constant Calculations Lennard-Jones parameters47 reactions Morse (β) (Å) σ (Å) ε (cm−1) Si3H8→ SiH3+ Si2H5 1.68 Si3H8 5.563 230.05 LM1→ SiH2+ Si2H6 1.73 Ar 3.75 98.3
LM2→ SiH4+ SiH3SiH 1.66 He 2.55 10.22
LM3→ H2+ Si(SiH3)2 1.86
The Morse function was characterized variationally from the optimized bond length to separated radical pairs with an interval of 0.1 Å by second-order multireference perturba-tion theory (CASPT2//CASSCF(8,8)/6-311+G(3df,2p) level). Other geometric parameters were fully optimized. These calculations were performed with the MOLPRO code.38
■
RESULTS AND DISCUSSIONSPES and Reaction Mechanism. The optimized geometries of reactants, intermediates, transition states, and products are shown in Figure 1, along with the available experimental bond lengths.39 As aforementioned, the PES of the Si3H8system was predicted at the QCISD(T)/CBS//QCISD/6-311++G(d,p) + ZPVE level
Figure 3.Arrhenius plots of rate constants for the SiH2+ Si2H6reaction forming various products, Si3H8(a), SiH4+ SiH3SiH (b), H2+ Si(SiH3)2 (c), H2+ SiH3SiH2SiH (d), and SiH3+ Si2H5(e), at different pressures.
of theory, as shown in Figure 2. The corresponding energies estimated at different levels of the theory are summarized in Table 1. The moments of inertia and the vibrational frequencies of all of the species involved in these reactions are listed in the Supporting Information Table S1 for the kinetic calculations. The calculated heats of reaction and formation values are given in Table 2 and are compared with available experimental data at 0 K. Discussion of the thermochemical data will be made later.
The following discussion on the PES will be based on the energies of the TSs and intermediates relative to the reactants computed at the QCISD(T)/CBS//QCISD level.
SiH2 + Si2H6 Reaction. As shown in Figure 2, the initial
association reaction of SiH2and Si2H6, proceeding via the van der Waals complex, SiH2/Si2H6(LM1) with an 8.3 kcal/mol
binding energy, can readily occur by an insertion reaction into one of the Si−H bonds in Si2H6from the side with a somewhat
Table 4. Arrhenius Parametersafor the Bimolecular Reaction of SiH2with Si2H6Giving Different Products at Various Pressures
and Temperature Including High-Pressure (k∞) and Low-Pressure (k0) Limits
products P (Torr) A n Ea/R (K) kP(500 K) Si3H8(k1) k∞ 2.55× 107 −5.83 1141 4.64× 10−10 k0 5.51× 101 −9.2 2565 5.76× 10−26 0.3 1.60× 1020 −10.2 2565 3.34× 10−10 1 1.36× 1015 −8.4 2023 4.01× 10−10 10 2.82× 109 −6.5 1381 4.54× 10−10 760 3.54× 107 −5.9 1158 4.64× 10−10
SiH4+ SiH3SiH (k2) k0 9.11× 10−15 −3.6 2209 1.86× 10−26
1 2.97× 10−04 −2.0 1509 5.14× 10−11 10 1.28× 10−20 3.2 −180 7.92× 10−12 760 5.95× 10−33 6.7 −1409 1.18× 10−13 H2+ Si(SiH3)2(k3) k0 5.56× 10−22 −1.6 1866 8.53× 10−28 1 1.85× 10−11 0.1 1225 2.67× 10−12 10 3.98× 10−30 6.1 −649 4.97× 10−13 760 1.55× 10−47 11.3 −2460 8.09× 10−15 H2+ SiH3SiH2SiH (k4) k0 9.00× 10−27 −0.22 1678 8.09× 10−29
1 4.36× 10−15 1.05 1222 2.76× 10−13 10 1.15× 10−34 7.4 −705 5.84× 10−14 760 1.38× 10−54 13.5 −2789 1.02× 10−15 SiH3+ Si2H5(k5) k0 4.17× 10−28 1.18 9616 2.92× 10−33 1 2.39× 10−9 0.09 9667 1.69× 10−17 10 3.15× 10−5 −1.17 10529 1.56× 10−17 760 1.68× 10−27 6.13 8769 1.48× 10−18 ak(T) = ATnexp(−E
a/RT) predicted for various temperatures 300−600 K in units of cm3molecule−1s−1fork and k∞and cm6molecule−2s−1fork0.
Table 5. Arrhenius Parametersafor the Bimolecular Reaction of SiH3with Si2H5Giving Different Products at Various Pressures
and Temperatures Including High-Pressure (k∞) and Low-Pressure (k0) Limits
products P (Torr) temp (K) A n Ea/R (K) kP(500 K)
Si3H8(k6) k∞ 300−600 3.94× 10−10 0.11 51 6.93× 10−10 k0 300−600 6.36× 1010 −14.3 2634 6.78× 10−31 0.3 300−600 1.84× 1029 −15.3 2634 3.93× 10−15 1 300−600 4.45× 1032 −15.7 3237 2.24× 10−13 10 300−600 8.11× 1026 −12.7 3543 4.89× 10−11 SiH2+ Si2H6(k7) k0 300−600 7.23× 10−29 0.97 132 2.31× 10−26 1 300−600 2.68× 10−10 −0.06 156 1.34× 10−10 10 300−600 1.71× 10−6 −1.22 977 1.23× 10−10 760 300−600 1.53× 10−28 6.01 −755 1.18× 10−11 SiH4+ SiH3SiH2(k8) k0 300−600 3.48× 10−27 0.55 200 7.13× 10−26
1 300−500 7.48× 10−9 −0.4 190 4.14× 10−10 10 300−600 3.08× 10−4 −1.83 1102 3.90× 10−10 760 300−600 2.48× 10−26 5.41 −644 3.69× 10−11 H2+ Si(SiH3)2(k9) k0 300−600 3.03× 10−31 1.77 −50.7 2.0× 10−26 1 300−600 2.35× 10−12 0.64 27.2 1.16× 10−10 10 300−600 1.18× 10−8 −0.49 847 1.04× 10−10 760 300−600 7.62× 10−31 6.8 −888 1.02× 10−11 H2+ SiH3SiH2SiH (k10) k0 300−600 1.08× 10−33 2.41 −178.8 4.83× 10−27
1 300−600 1.03× 10−14 1.24 −81.6 2.78× 10−11 10 300−600 5.47× 10−11 0.11 753 2.48× 10−11 760 300−600 2.68× 10−33 7.45 −981.7 2.47× 10−12
ak(T) = ATnexp(−E
smaller activation barrier via TS1 forming Si3H8; the process is exothermic by 55.0 kcal/mol at the QCISD(T)/CBS//QCISD level. It should be noted that previous studies also reported a similar smaller activation energy.2,3 The ground electronic state of SiH2is singlet, not triplet as in the CH2case. We
con-sidered Si3H8and TS1 as a singlet state. The TS1 calculated at QCISD/6-311++G(d,p) and QCISD(T)/6-311++G(3df,2p)// QCISD/6-311++G(d,p) levels have small positive barriers of 2.8 and 0.2 kcal/mol, respectively, when compared to LM1 (see Table 1). The predicted Gibbs free energy of activation at 298 K from LM1 to TS1 was found to be positive also. Walsh’s
group theoretically explored the mechanism of the Si−H
insertion process by silylene. They explained that silylene has an empty p orbital and a lone pair of electrons. The electron density is transferred from the Si−H bond of silane to the empty p orbital of silylene, and simultaneously, the lone pair of electrons in silylene is donated to the Si of silane to produce the new Si−Si bond. As aforementioned, the two processes occur with the involvement of an intermediate complex.18,19 As shown in Figure 1, LM1 has C1symmetry, and SiH2is located with Si pointing toward a Si−H bond in the Si2H6molecule at a
distance of 1.845 Å. However, TS1 lies 9.3 kcal/mol below the reactants, and their structure has a tighter three-membered ring, in which the forming H−Si and Si−Si bond lengths are 1.659 and 2.449 Å, respectively, and the Si−H breaking bond length is elongated to 1.689 Å, which is 0.209 Å longer than that of Si2H6.
SiH3+ Si2H5Reaction. Both SiH3and Si2H5radicals formed
in the SiH2+ Si2H6reaction may coexit in the plasma-induced reactions of small silanes (SiH4, Si2H6, and Si3H8, for example).
The PES shown in Figure 2 suggests that all product pairs that lie below SiH3+ Si2H5can be accessed and produced in the
reaction, as listed in the Introduction (i.e., reactions 6−10). Decomposition of Si3H8. The initially formed chemically
activated Si3H8 adduct may eliminate several products, SiH4,
SiH3, H2, and H, directly, as shown in Figure 2. In the Si3H8 decomposition reactions, the lowest-energy channel producing SiH4+ H3SiSiH lies 54.3 kcal/mol above Si3H8. The 2,1-H shift occurs by migration of one of the H atoms in the secondary SiH2group to the SiH3group to form a van der Waals complex,
SiH4/SiH3SiH, LM2, via TS2. The barrier height at TS2 is 42.8 kcal/mol, and the complex is 44.4 kcal/mol above Si3H8.
The energies of TS2 predicted at the QCISD/6-311++G(d,p) and QCISD(T)/6-311++G(3df,2p)//QCISD/6-311++G(d,p) levels are 45.2 and 42.6 kcal/mol, respectively. The latter is close to the CBS limit, 42.8 kcal/mol. We also calculated the SiH4/SiH3SiH, LM2 energy with other methods; when
com-pared with TS2 at the QCISD/6-311++G(d,p) level, LM2 has the same energy while at the QCISD(T)/6-311++G(3df,2p)// QCISD/6-311++G(d,p) level, it is 1.1 kcal/mol above TS2. Here, although the transition state has a lower activation barrier compared with the complex, our result is consistent with pre-vious reports.2,3The predicted Gibbs free energy of activation at 298 K from LM2 to TS2 was found to be positive also. At TS2, the bond lengths of the breaking Si···H and Si···Si were predicted to be 1.589 and 2.706 Å, respectively, and the forming H···SiH3bond was predicted to be 1.668 Å calculated by the QCISD/6-311++G(d,p) method (Figure 1). In addition, the Si2H4+ SiH4 products may be formed by H3SiSiH
isom-erization with a small 1.6 kcal/mol barrier. Our result presented in Figure 2 shows that in the Si3H8dissociation reaction, the
secondary H-atom shifting to form SiH4has a lower activation energy when compared to that of the primary H-atom shifting
to form SiH2. This mechanism is in good agreement with
previous experimental work.1 Si2H4 is a stable and potential candidate for film formation.4 As shown in Figure 2, the interaction of the two H atoms at the secondary Si position in Si3H8 can eliminate H2 via a three-membered-ring transition
state TS3 (48.8 kcal/mol) to form a van der Waals complex, Si(SiH3)2/H2, LM3. At TS3, the two breaking Si−H bonds
lengthen unequally from 1.483 Å in Si3H8to 1.578 and 1.669 Å; the forming H−H bond length is 0.976 Å. Relative to SiH2+
Si2H6, the reaction product Si(SiH3)2+ H2is exothermic by 1.4 kcal/mol. Similarly, as shown in Figure 2, H2elimination can
also take place at a terminal Si atom of Si3H8 to form a SiH3SiH2SiH + H2 via TS4 and the SiH3SiH2SiH/H2, LM4
complex. The barrier height at TS4 is 51.6 kcal/mol, and the complex is 50.3 kcal/mol above Si3H8. The H2 elimination
barrier from the secondary Si atom in Si3H8is thus 2.8 kcal/mol lower compared with that from a terminal SiH3group.
On this PES, the next lowest-energy product channel is SiH3+ Si2H5, formed by direct cleaving of one of the Si−Si bonds in
Si3H8, requiring as much as 74.0 kcal/mol. The predicted heat of reaction is in good agreement with available experimental and computed values,25−28,39,40 as shown in Table 2. This process does not have a well-defined transition state. Due to the
Figure 4.(a) Pressure dependences of all product channels of SiH2+ Si2H6reaction rate constants at T = 500 K as functions of pressure. (b) Branching ratios of SiH2 + Si2H6 reaction products at 1 Torr of pressure.
absence of an intrinsic transition state for the fragmentation reaction, its dissociation potential function was computed variationally to cover a range of Si−Si separations from the equilibrium value 2.344 to 6.5 Å with an interval of 0.1 Å by second-order multireference perturbation theory (CASPT2)
based on the CASSCF optimized geometries with eight active electrons and eight active orbitals using the 6-311+G(3df,2p) basis set. Aforementioned in the above section, other geometric parameters were fully optimized. These calculations were
per-formed with the MOLPRO code.38 The computed potential
Figure 5.Arrhenius plots of rate constants for the SiH3+ Si2H5reaction forming various products Si3H8(a), SiH2+ Si2H6(b), SiH4+ SiH3SiH (c), H2+ Si(SiH3)2(d), and H2+ SiH3SiH2SiH (e) at different pressures.
energies could be fitted to the Morse function with the parameters of β = 1.68 Å−1 (SiH3 + Si2H5), for Si−Si bond
breaking. In the same manner, with much computational effort, we also determined the valuesβ = 1.73, 1.66, 1.86, and 1.89 Å−1 from LM1 to SiH2+ Si2H6, LM2 to SiH4+ SiH3SiH, LM3 to
H2+ Si(SiH3)2, and LM4 to H2+ SiH3SiH2SiH, respectively. These values will be used in the rate constant calculations to be discussed below. In the formulation of the Morse potential, r is the distance between the two bonding atoms for the separa-tion of two fragments. As shown in the PES of Figure 2, H2is
produced via LM3 and LM4. The reactions involved are reactions 3, 4, 9, and 10, shown in the reaction scheme. In these calculations, the separation “r” begins with the smaller of the two Si···H bonds from its equilibrium separation to about 4.5 Å at an interval of 0.1 Å. As the VTSs are typically located at 2.5− 3.5 Å, the values of r based on this method or the H2
center-of-mass separation from the Si involved are essentially the same; so are the values of the rate constant predicted. Finally, the cleavage of the secondary and primary Si−H bonds in the Si3H8
giving rise to the products H + i-Si3H7and H + n-Si3H7 are predicted to be endothermic by 82.6 and 84.5 kcal/mol, respectively, without intrinsic barriers (see Figure 2).
Enthalpies of Formation. The predicted heats of formation of all of the species related to the SiH2 reaction
with Si2H6 and the unimolecular decomposition of Si3H8 are
presented in Table 1 based on the energies computed at the QCISD/6-311++G(d,p), QCISD(T)/6-311++G(3df,2p)// QCISD/6-311++G(d,p) and QCISD(T)/CBS//QCISD/6-311++G(d,p) levels. As one would expect, the last two higher-level methods give rise to values closer to each other. The heats of formation were determined by combining the computed heats of reaction (ΔrH0°) based on the CBS limit
values and experimental heats of formation (ΔfH0°) of other
species involved in the reaction at 0 K. Theoretically, the heats of formation of Si3H8, n-Si3H7, and i-Si3H7are available in the
literature as 33.5± 1.0, 38.3, and 37.5 kcal/mol, respectively, at 0 K, as referenced in the footnote of Table 2. Thus, by using these values and experimental heats of formation, we obtained the values at 0 K for SiH3SiH, Si2H4, Si(SiH3)2, and SiH3SiH2SiH
to be 77.3, 68.0, 87.1, and 87.3 kcal/mol, respectively, with an estimated error of ±1.2 kcal/mol. Our predicted heats of formation of the species listed in Table 2 are in good agreement with the values derived from available experimental and theoretical data.25−28,41−43
Rate Constant Calculations. The rate constants for the bimolecular reactions SiH2+ Si2H6and SiH3+ Si2H5and the
related unimolecular decomposition processes can be com-puted with the predicted PES using energies obtained by QCISD(T)/CBS extrapolation and the QCISD/6-311++G-(d,p) molecular parameters of the reactants, intermediates, and transition states presented in Table S1 in the Supporting Information. The rate constants for the forward reactions of SiH2+ Si2H6via the low-energy channels have been computed
in the temperature range of 300−2000 K and the pressure range of 0.3−760 Torr with the Variflex code, whereas the higher-energy H-production channels are neglected. The VTST calculations were carried out with the unified statistical formulation of Miller44 including multiple reflection correc-tions45,46 above the shallow wells of the prereaction and postreaction complexes LM1, LM2, LM3, and LM4. The Lennard-Jones parameters for collision rate estimates are obtained by using σ and ε and are given in Table 3.47 The P,T conditions studied cover most of the conditions employed
in PECVD (typically 0.75−4 Torr, 373−723 K) and Cat-CVD (typically 0.1−10 Torr, 473−673 K)9−12
Rate Constant for SiH2+ Si2H6. The bimolecular reaction of
SiH2 and Si2H6 occurs exclusively by the insertion process forming the excited Si3H8 intermediate carrying as much as
55.0 kcal/mol of internal energy with 12.1 kcal/mol of excess energy above the transition state for 1,2-H elimination at TS2, giving the SiH4and SiH3SiH radical, as shown by the PES given in Figure 2. The predicted rate constants for all of the prod-uct channels represented by reactions 1−5 listed in the Introduction at various pressures between 0.3 and 760 Torr along with the high pressure limit in the temperature range of 300−2000 K are graphically presented in Figure 3 and are also listed in Table 4, covering the temperature range 300−600 K. In the rate constant calculation, the internal rotation of the SiH3group of Si3H8with the vibrational frequency of 77 cm−1 is hindered by a 1.0 kcal/mol barrier and thus also treated as a hindered rotor.
As shown in Figure 3a, under the high-pressure condition, the reaction occurs primary by a recombination/stabilization process producing Si3H8. Our predicted absolute rate constants
for Ar buffer gas agree quite well with experimental data.15−19 Walsh and co-workers18,19reported the values of 3.04× 10−10
Figure 6.(a) Pressure dependences of all product channels of SiH3+ Si2H5reaction rate constants at T = 500 K as functions of pressure. (b) Branching ratios of SiH3 + Si2H5 reaction products at 1 Torr of pressure.
exp(229/T) cm3molecule−1s−1for the temperature range of
295−595 K at 1.0−30.0 Torr in C8H8buffer gas. At 500 K, our predicted rate 4.54× 10−10cm3molecule−1s−1compares closely
with the experimental rate 4.87× 10−10cm3molecule−1s−1.18,19 White et al.15 theoretically estimated the rate constant at temperatures of 640−703 K with an order of magnitude lower value. At room temperature, the present predicted rate con-stants, 1.5× 10−9 at 1 Torr and 1.9× 10−9 at 10 Torr He pressure, are in reasonable agreement with the values of Inoue et al.,165.7× 10−10cm3molecule−1s−1, in 1 Torr of He gas and those of Jasinski et al.17 using laser absorption to obtain the 3.4× 10−10 cm3molecule−1s−1at 10 Torr in a He bath gas. When the pressure increases from 0.3 to 760 Torr, k1increases proportionally, as clearly illustrated in Figure 3a, reflecting the need for collisional deactivation of the excited Si3H8 adduct.
Under low-pressure conditions, the reaction may yield Si3H8
fragmentation products such as SiH4 + SiH3SiH, H2 +
Si(SiH3)2//SiH3SiH2SiH, and SiH3 + Si2H5; the predicted
rate constants for all product channels in the 300−2000 K temperature range including the high- and low-pressure limits are shown in the Figure 3b−e. The pattern of the pressure dependencies of the rate constants for formation of the product pairs revealed by thesefigures reflects the competitive nature of
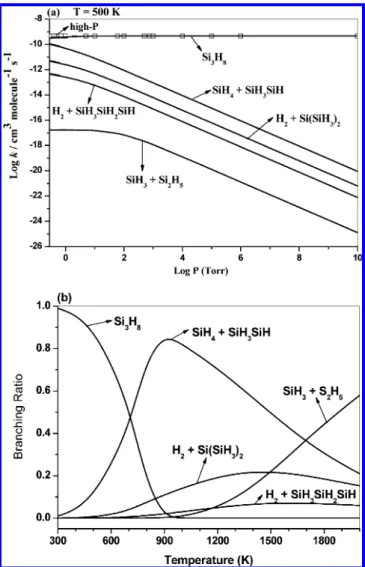
the collisional deactivation versus fragmentation of the excited Si3H8. The calculated pressure-dependent rate constants for com-peting product formation at 500 K are displayed in Figure 4a. The results clearly show that the formation of Si3H8 by
collisional deactivation is strongly P-dependent and dominant over the other production channels down to P < 1 Torr. The next dominant channel in the SiH2+ Si2H6reaction is SiH4+ SiH3SiH//Si2H4. Here, when the pressure increases, its rate also decreases due to the competition with Si3H8 formation.
Figure 4b displays the branching ratios of the individual product channels of the SiH2+ Si2H6reaction at 1 Torr in the
300−2000 K temperature range. The results show that at 300 K, formation of Si3H8 is dominant with 99% yield, and the
remaining 1% gives SiH4+ SiH3SiH. The production of Si3H8 up to 700 K temperature is dominant, above which SiH4 + SiH3SiH//Si2H4 becomes competitive. Other production
channels are not important until reaching a higher temperature of 1700 K, at which SiH3+ Si2H5becomes competitive.
Rate Constant for SiH3 + Si2H5. As aforementioned, the
association reaction of SiH3 with Si2H5 producing Si3H8with more than 74.0 kcal/mol of internal energy occurs without a well-defined transition state; the Si3H8 thus formed can
dis-sociate with 19 kcal/mol of excess energy above SiH2+ Si2H6
Figure 7.Arrhenius plots of rate constants for the Si3H8unimolecular dissociation forming various products, SiH2+ Si2H6(a), SiH4+ SiH3SiH (b), H2+ Si(SiH3)2(c), and SiH3+ Si2H5(d), at different pressures.
and even higher above other product pairs; thus, various prod-ucts, SiH2, SiH4, and H2, can be formed from the association/
decomposition reactions; see Figure 2 and reactions 6−10 presented in the Introduction section. The predicted values of k6forming Si3H8at various pressures between 0.3 and 760 Torr along with its high-pressure limit in the temperature range of 300−2000 K are graphically presented in Figure 5a and are also listed in Table 5. In the table, the value for the low-pressure limit is also given for kinetic modeling. The values of k6
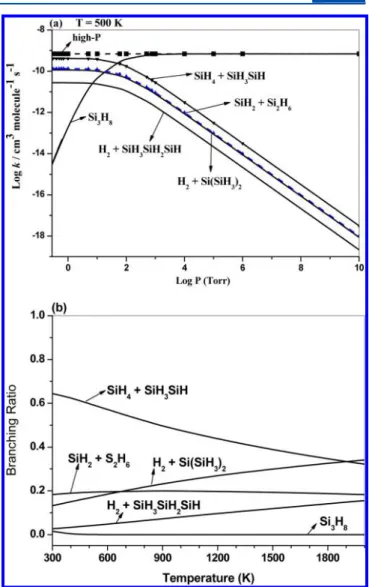
decrease as the temperature increases from 300 to 2000 K. When the pressure increases from 0.3 to 760 Torr, k6increases proportionally, as clearly illustrated in Figure 5a, reflecting the effect of collisional deactivation of the excited Si3H8. The predicted rate constants for the SiH3+ Si2H5reaction giving
rise to various products are shown in Figure 5b−e. The rate constants for production of Si3H8 (k6) and various products
(k7−10) at 500 K covering the wide pressure range are shown in
Figure 6a. The branching ratios of thesefive product channels are shown in Figure 6b. The product channel giving SiH4 + SiH3SiH is predominant up to 1800 K. The competitive nature
of reactions 6−10 shown in these figures is qualitatively similar to that presented above for reactions 1−5, as one would expect. The only difference between the two bimolecular reactions lies in the amount of internal energies carried by the chemically activated Si3H8*, as shown in the PES.
We have done the rate constant calculations for the forward reactions of SiH2+ Si2H6and SiH3+ Si2H5, which form various
products with and without multiple reflection corrections. At 300 K (the lowest temperature) and 760 Torr and the high-pressure limit, in the SiH2 + Si2H6 → Si3H8 reaction, the
forward reaction rate constant with the multiple reflection correction is around 2 orders of magnitude lower than that without the correction. There is no effect of multiple reflection corrections above 700 K (see Supporting Information Figure S1A
and B). At 300 K and 760 Torr, the multiple reflection
correction for SiH2+ Si2H6→ SiH4+ SiH3SiH (k2) is around 1
order of magnitude lower than that without the correction, and no effect of correction is seen above 500 K (see Supporting Information Figure S1C). For other product channels of H2 and SiH3(k3, k4, and k5) in the above SiH2+ Si2H6reaction, the effect of the multiple reflection correction is negligible. For the forward reactions of SiH3+ Si2H5forming various low-energy
product rate constants, we compared the results with and without multiple reflection corrections at 300 K and 760 Torr of pressure, as shown in Figure S2 (Supporting Information). In this case, no multiple reflection correction effect was observed. Thermal Decomposition of Si3H8. The thermal
decom-position of Si3H8under similar conditions as those given above
for the association process produces predominantly SiH4 +
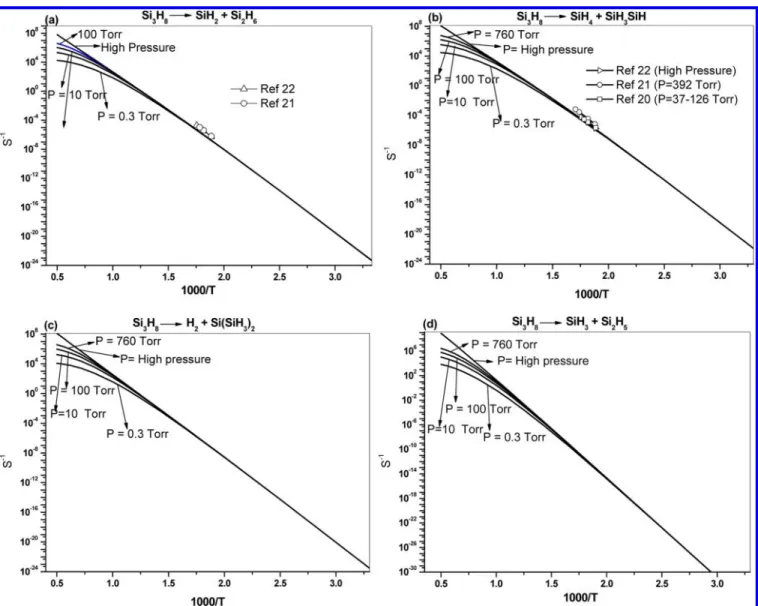
SiH3SiH because of its lower-energy barrier compared with those for SiH2+ Si2H6, H, and H2elimination from the primary and secondary positions. As shown in Figure 7 and the rate constant expressions summarized in Table 6 obtained by least-squares fitting to the predicted values, both reactions have Table 6. Arrhenius Parametersafor the Unimolecular Decomposition of Si3H8Giving Different Products at Various Pressures
and Temperatures Including High-Pressure (k∞) and Low-Pressure (k0) Limits
reaction P (Torr) A n Ea/R (K) kP(500 K) Si3H8→ SiH2+ Si2H6(k) k∞ 7.64× 1033 −6.23 2887 9.89× 10−9 k0 3.70× 1033 −11.2 31263 1.60× 10−24 0.3 1.07× 1052 −12.2 31263 9.28× 10−9 1 5.70× 1047 −10.8 30736 9.66× 10−9 10 2.64× 1040 −8.36 29774 9.86× 10−9 760 2.43× 1034 −6.39 28942 9.89× 10−9
Si3H8→ SiH4+ SiH3SiH (k) k∞ 8.59× 1038 −7.68 29126 8.24× 10−8
k0 5.87× 1039 −13.1 31567 1.18× 10−23 0.3 1.70× 1058 −14.1 31567 6.81× 10−8 1 4.52× 1054 −12.8 31182 7.63× 10−8 10 1.66× 1047 −10.4 30255 8.16× 10−8 760 4.87× 1039 −7.93 29231 8.24× 10−8 Si3H8→ H2+ Si(SiH3)2(k) k∞ 9.46× 1022 −2.83 27534 2.72× 10−8 k0 4.18× 1026 −9.1 30465 4.47× 10−25 0.3 1.21× 1045 −10.1 30465 2.59× 10−9 1 1.30× 1040 −8.4 29843 2.67× 10−9 10 4.82× 1031 −5.66 28735 2.71× 10−9 760 7.24× 1023 −3.12 27659 2.72× 10−9
Si3H8→ H2+ SiH3SiH2SiH k∞ 8.86× 1012 0.09 26513 1.45× 10−10
k0 3.66× 1011 −4.48 28824 2.47× 10−26 0.3 1.06× 1030 −5.48 28824 1.43× 10−10 1 2.15× 1025 −3.94 28209 1.45× 10−10 10 1.91× 1018 −1.64 27255 1.45× 10−10 760 2.07× 1013 −0.03 26565 1.45× 10−10 Si3H8→ SiH3+ Si2H5 k∞ 2.02× 1023 −1.83 38031 2.14× 10−15 k0 8.68× 1037 −11.8 42161 2.73× 10−31 0.3 2.51× 1056 −12.80 42161 1.58× 10−15 1 1.79× 1052 −11.37 41734 1.81× 10−15 10 1.90× 1043 −8.36 40683 2.06× 10−15 760 5.86× 1028 −3.60 38787 2.13× 10−15 ak(T) = ATnexp(−E
positive-pressure dependence, reflecting the nature of colli-sional activation. Experimentally, Vanderwielen et al.20 kineti-cally measured the unimolecular decomposition of Si3H8 to
SiH4+ SiH3SiH and SiH2+ Si2H6in the 530−561 K
tempera-ture range and 37−126 Torr of Si3H8 pressure range. Later,
Martin et al.21measured the rate constant for the decomposi-tion pathways of Si3H8→ SiH4+ SiH3SiH by pyrolysis at 532−
586 K and 392 Torr of H2 pressure. In 1992, Moffat el al.
22
studied the rate constant for the decomposition channels of trisilane to form SiH4 and SiH2products by RRKM numerial
analysis in the temperature range of 530−570 K. The
experimental results are in good agreement with our predicted values, as shown in Figure 7a and b.20−22The rate constants for other production channels by H2elimination from primary and
secondary Si−H bonds are also shown in Figure 7c and d. Figure 8 shows the branching ratios of Si3H8 dissociation
products; the low-energy channel producing SiH4+ SiH3SiH is
dominant up to 1500 K, beyond which SiH3+ Si2H5becomes
dominant. It should be pointed out that although the SiH2 +
Si2H6product pair energetically lies below SiH3+ Si2H5by as
much as 19 kcal/mol, its branching ratio was predicted to be less than that of the latter above 1300 K, attributable to the tighter transition state TS1 than the variational TS for the latter production.
■
CONCLUSIONThe mechanisms, rate constants, and product branching ratios for the SiH2+ Si2H6and SiH3+ Si2H5reactions and the thermal
unimolecular decomposition of Si3H8have been investigated at the QCISD(T)/CBS level of theory based on QCISD/6-311+ +G(d,p) optimized geometries in conjunction with VTST and RRKM calculations. The formation of the most favorable low-energy products for the SiH2+ Si2H6reaction occurs readily by Si−H insertion, yielding the excited intermediate Si3H8via the
van der Waals complex with 8.3 kcal/mol of binding energy. The excited intermediate carrying as much as 55.0 kcal/mol of internal energy can readily dissociate into SiH4 + SiH3SiH// Si2H4 following the 2,1-H migration involving one of the H
atoms in the secondary SiH2group. The excited intermediate can also decompose by H2 elimination from the secondary
position with 2.8 kcal/mol lower energy than that from the primary position, which requires 51.6 kcal/mol. The dissociation
energies for breaking of the Si−Si and the primary and
secondary Si−H bonds in Si3H8were predicted to be 74.0, 85.0,
and 86.9 kcal/mol, respectively. The values agree well with the known heats of formation of the decomposition products.
The computed heats of formation ΔfH0° at 0 K for Si3H8, SiH3, SiH3SiH, Si2H4, i-Si3H7, n-Si3H7, Si(SiH3)2, and SiH3SiH2SiH
are 33.5, 48.3, 77.3, 68.0, 66.9 ± 1.0, 68.7 ± 1.0, 87.1, and 87.3 kcal/mol, respectively, with an estimated error of ±1.2 kcal/mol. The results are in good agreement with available experimental values. Furthermore, the rate constants for the bimolecular association/decomposition reactions (SiH2+ Si2H6 and SiH3 + Si2H5) and the thermal unimolecular
decom-position of Si3H8 for all of the product channels have been calculated using the VTST method and/or the RRKM theory by solving the master equation involved over a wide range of P,T conditions covering those typically employed in the a-Si/H CVD process by PECVD and/or Cat-CVD. The predicted results have been tabulated for modeling and optimizing homo-geneous large-area growth of a-Si/H thinfilms by these methods.
■
ASSOCIATED CONTENT*
S Supporting InformationCalculated moments of inertia and vibrational frequencies of the species involved in the SiH2reactions with Si2H6computed
at the QCISD/6-311++G(d,p) level are given in Table S1, and Arrhenius plots of rate constants for the SiH2+ Si2H6and SiH3+
Si2H5reactions forming various products are given in Figures S1 and S2, respectively. This material is available free of charge via the Internet at http://pubs.acs.org.
■
AUTHOR INFORMATIONCorresponding Author
*E-mail: [email protected].
Notes
The authors declare no competingfinancial interest.
■
ACKNOWLEDGMENTSThe authors deeply appreciate the support by Taiwan’s National Science Council (NSC) under Contract No. NSC100-2113-M-009-013 and by the Ministry of Education’s ATU program. M.C.L. also acknowledges the support from the NSC for the distinguished visiting professorship at National Chiao Tung University in Hsinchu, Taiwan. We are also grateful to the National Center for High-Performance Computing for computer time and the use of its facilities.
■
REFERENCES(1) Gaspar, P. P. In Silylenes, Reactive Intermediates; Jones, M., Moss, Eds.; Wiley: New York, 1985; Vol. 3.
(2) Becerra, R.; Walsh, R. In Research in Chemical Kinetics; Compton, R.G., Hancock, G., Eds.; Elsevier: New York, 1995; Vol. 3, p 263.
(3) Jasinski, J. M.; Becerra,; Walsh, R. Direct Kinetic Studies of Silicon Hydride Radicals in the Gas Phase. Chem. Rev. 1995, 95, 1203−1228.
(4) Dietrich, T. R.; Chiussi, S.; Marek, M.; Roth, A.; Comes, F. J. Role of Sllylene in the Deposition of Hydrogenated Amorphous Silicon. J. Phys. Chem. 1991, 95, 9302−9310.
(5) Jasinski, J. M.; Whittaker, E. A.; Bjorklund, G. C.; Dreyfus, R. W.; Estes, R. D.; Walkup, R. E. Detection of SiH2in Silane and Disilane Glow Discharges by Frequency Modulation Absorption Spectroscopy. Appl. Phys. Lett. 1984, 44, 1155−1157.
(6) Jasinski, J. M.; Gates, S. M. Silicon Chemical Vapor Deposition One Step at a Time: Fundamental Studies of Silicon Hydride Chemistry. Acc. Chem. Res. 1991, 24, 9−15.
Figure 8.Branching ratios of Si3H8 decomposition products at high pressure.
(7) Lecomber, P. G.; Spear, W. E; Ghaith, A. Amorphous-Silicon Field-Effect Device and Possible Application. Electron. Lett. 1979, 15, 179−181.
(8) Carlson, D. E.; Wronski, C. R. Amorphous Silicon Solar Cell. Appl. Phys. Lett. 1976, 28, 671−673.
(9) Kushner, M. J. A Model for the Discharge Kinetics and plasma Chemistry During Plasma Enhanced Chemical Vapor Deposition of Amorphorous Silicon. J. Chem. Phys. 1988, 63, 2532−2551.
(10) Matsuda, A. Thin-Film Silicon Growth Process and Solar Cell Application. Jpn. J. Appl. Phys. 2004, 43, 7909−7920.
(11) Matsumura, H. Formation of Silicon-Based Thin Films Prepared by Catalytic Chemical Vapor Deposition (Cat-CVD) Method. Jpn. J. Appl. Phys. 1998, 37, 3175−3187.
(12) Matsumura, H.; Umemoto, H.; Masuda, A. Cat-CVD (Hot-Wire CVD): How Different from PECVD in Preparing Amorphous Silicon. J. Non-Cryst. Solids 2004, 338−340, 19−26.
(13) Roth, A.; Chiussi, S.; Dietrich, T. R.; Comes, F. J. Hydrogenated Amorphous Silicon by Infrared Multiphoton Absorption with a Pulsed CO2-Laser. Ber. Bunsen-Ges. Phys. Chem 1990, 94, 1105−1110.
(14) John, P.; Purnell, J. H. Arrhenius Parameters for Silene Insertion Reactions. J. Chem. Soc., Faraday Trans. 1 1973, 69, 1455−1461.
(15) White, R. T.; Espino-Rios, R. L.; Rogers, D. S.; Ring, M. A.; O’Neal, H. E. Mechanism of the Silane Decomposition. I. Silane Loss Kinetics and Rate Inhibition by Hydrogen. II. Modeling of the Silane Decomposition (All Stages of Reaction). Int. J. Chem. Kinet. 1985, 17, 1029−1065.
(16) Inoue, G.; Suzuki, M. Reactions of SiH2(X−1A1) with H2, CH4, C2H4, SiH4and Si2H6at 298 K. Chem. Phys. Lett. 1985, 122, 361−364. (17) Jasinski, J. M.; Chu, J. O. Absolute Rate Constants for the Reaction of Silylene With Hydrogen, Silane, and Disilane. J. Chem. Phys. 1988, 88, 1678−1687.
(18) Baggott, J. E.; Frey, H. M.; Lightfoot, P. D.; Walsh, R.; Watts, I. M. Absolute Rate Constants for the Gas-Phase Reactions of Silylene with Silane, Disilane and the Methylsilanes. J. Chem. Soc., Faraday Trans. 1 1990, 86, 27−33.
(19) Becerra, R.; Frey, H. M.; Manson, B. P.; Walsh, R. Time-Resolved Gas-Phase Kinetic Studies of the Reactions of Silylene with Disilane and Trisilane. J. Organomet. Chem. 1996, 521, 343−349.
(20) Vanderwielen, A. J.; Ring, M. A.; O’Neal, H. E. Kinetics of the Thermal Decomposition of Methyldisilane and Trisilane. J. Am. Chem. Soc. 1975, 97, 993.
(21) Martin, J. G.; O’Neal, H. E; Ring, M. A. Thermal Decomposition Kinetics of Polysilanes: Disilane, Trisilane, and Tetrasilane. Int. J. Chem. Kinet. 1990, 22, 613−632.
(22) Moffat, H. K.; Jensen, K. F.; Carr, R. W. J. Estimation of Arrhenius Parameters for the 1,l Elimination of H, from Si2H6and the Role of Chemically Activated Disilane in Silane Pyrolysis. J. Phys. Chem. 1992, 96, 7695−7703.
(23) Kumata, K.; Itah, U.; Toyoshima, Y.; Tanaka, N.; Anzai, H.; Matsuda, A. Photochemical Vapor Deposition of Hydrogenated Amorphous Silicon Films from Disilane and Trisilane Using a Low Pressure Mercury Lamp. Appl. Phys. Lett. 1986, 48, 1380−1382.
(24) Kanoh, H.; Sugiura, O.; Matsumura, M. Chemical Vapor Deposition of Amorphous Silicon Using Tetrasilane. Jpn. J. Appl. Phys. 1993, 32, 2613−2619.
(25) Wu, S. Y.; Raghunath, P.; Wu, S. J.; Lin, M. C. Ab Initio Chemical Kinetic Study for Reactions of H Atoms with SiH4 and Si2H6: Comparison of Theory and Experiment. J. Phys. Chem. A 2010, 114, 633−639.
(26) Varma, D. H.; Raghunath, P.; Lin, M. C. Ab Initio Chemical Kinetics for the Reaction of an H Atom with Si3H8. J. Phys. Chem. A 2010, 114, 3642−3648.
(27) Raghunath, P.; Lin, M. C. Ab Initio Chemical Kinetics for SiH3 Reactions with SixH2x+2 (x = 1−4). J. Phys. Chem. A 2010, 114, 13353−13361.
(28) Raghunath, P.; Lee, Y. M.; Wu, S. Y.; Wu, S. J.; Lin, M. C. Ab Initio Chemical Kinetics for Reactions of H Atoms with SiHx(x = 1− 3) Radicals and Related Unimolecular Decomposition Processes. Int. J. Quantum. Chem. 2013, 113, 1735−1746.
(29) Pople, J. A.; Head-Gordon, M.; Raghavachari, K. Quadratic Configuration Interaction. A General Technique for Determining Electron Correlation Energies. J. Chem. Phys. 1987, 87, 5968−5975.
(30) Matsumoto, K.; Klippenstein, S. J.; Tonokura, K.; Koshi, M. Channel Specific Rate Constants Relevant to the Thermal Decomposition of Disilane. J. Phys. Chem. A 2005, 109, 4911−4920.
(31) Pei, K.; Li, H. Ab Initio and Kinetic Calculations for the Reactions of Cl with SiHnCl4−n(n=1−4). J. Chem. Phys. 2004, 121, 6738−6742.
(32) Peterson, K. A.; Woon, D. E.; Dunning, T. H., Jr. Benchmark Calculations with Correlated Molecular Wave Functions. IV. The Classical Barrier Height of the H + H2→ H2+ H Reaction. J. Chem. Phys. 1994, 100, 7410−7415.
(33) Woon, D. E., Jr.; Dunning, T. H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. V. Core-Valence Basis Sets for Boron through Neon. J. Chem. Phys. 1995, 103, 4572−4585.
(34) Frisch., M. J.; Trucks, G. W.; Schlgel, H. B.; Scuseria, G. E.; Robb, M. A. Cheeseman, J. R.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; et al. Gaussian 03, revision C.02; Gaussian, Inc.: Wallingford, CT, 2004.
(35) Klippenstein, S. J.; Wagner, A. F.; Dunbar, R. C.; Wardlaw, D. M.; Robertson, S. H. VARIFLEX, version 1.00; Argonne National Laboratory: Argonne, IL, USA, 1999.
(36) Klippenstein, S. J. An Efficient Procedure for Evaluating the Number of Available States within a Variably Defined Reaction Coordinate Framework. J. Phys. Chem. 1994, 98, 11459−11464.
(37) Klippenstein, S. J. A Bond length Reaction Coordinate for Unimolecular Reactions. II. Microcanonical and Canonical Imple-mentations with Application to the Dissociation of NCNO. J. Chem. Phys. 1991, 94, 6469−6482.
(38) Werner, H.-J.; Knowles, P. J., with contributions from Almlof, J.; Amos, R. D.; Berning, A.; et al. MOLPRO, version 2009.1, a Package of ab initio programs; University College Cardiff Consultants Limited: Cardiff, U.K., 2009.
(39) Callomnon, J. H.; Hirota, E.; Kuchitsu, K.; Lafferty, W. J.; Maki, A. G. ; Pote, C. S. Structure Data on Free Polyatomic Molecules (Landolt-Bornstein, New Series, Group 11); Springer-Verlag: Berlin, Germany, 1976, Vol. 7.
(40) Haaland, A.; Rypdal, K.; Stuger, H.; Volden, H. V. Molecular-Structures and Conformational Composition of Trisilane by Gas-Phase Electron-Diffraction. Acta Chem. Scand. 1994, 48, 46−51.
(41) Katzer, G.; Ernst, M. C.; Sax, A. F.; Kalcher, J. Computational Thermochemistry of Medium-Sized Silicon Hydrides. J. Phys. Chem. A 1997, 101, 3942−3958.
(42) Chase, M. W. Jr. NIST-JANAF Thermochemical Tables, 4th ed., J. Phys. Chem. Ref. Data 1998; Monograph No. 9 (Parts I and II).
(43) Rustic, B.; Berkowitz, J. Photoionization Mass Spectrometric Studies of the Transient Species Si2Hn(n=2−5). J. Chem. Phys. 1991, 95, 2416−2432.
(44) Miller, W. H. Unified Statistical Model for “Complex” and “Direct” Reaction Mechanisms. J. Chem. Phys. 1976, 65, 2216−2223. (45) Chakraborthy, D.; Hsu, C.-C.; Lin, M. C. Theoretical Studies of Nitroamino Radical Reactions: Rate Constants for the Unimolecular Decomposition of HNNO2 and Related Bimolecular Processes. J. Chem. Phys. 1998, 109, 8887−8896.
(46) Xu, Z. F.; Hsu, C. -H.; Lin, M. C. Ab Initio Kinetics of the Reaction of HCO with NO: Abstraction versus Association/ Elimination Mechanism. J. Chem. Phys. 2005, 122, 234308/1− 234308/11.
(47) Coltrin, M. E.; Kee, R. J.; Miller, J. A. A Mathematical Model of Silicon Chemical Vapor Deposition. J. Electrochem. Soc. 1986, 133, 1206−1213.