行政院國家科學委員會專題研究計畫 成果報告
金屬-氧化鈦複合觸媒微結構之控制與改良(2/2)
計畫類別: 個別型計畫 計畫編號: NSC91-2214-E-002-007- 執行期間: 91 年 08 月 01 日至 92 年 07 月 31 日 執行單位: 國立臺灣大學化學工程學系暨研究所 計畫主持人: 吳乃立 計畫參與人員: 吳乃立 報告類型: 完整報告 處理方式: 本計畫可公開查詢中 華 民 國 92 年 10 月 20 日
hν, catal. hν, catal. hν, catal. hν, catal.
行政院國家科學委員會專題研究計畫成果報告
金屬-氧化鈦複合觸媒微結構之控制與改良(2/2)
Microstructural Engineering of metal-titania catalysts
計畫編號:NSC 91-2214-E-002-007
執行期間:91/08/01~92/07/31
主持人:吳乃立 台灣大學化工系教授
摘要(關鍵詞: 二氧化鈦,氫生成,光觸媒催化,煅燒氣氛,Ar,銅) 本研究主要探討二氧化鈦運用於光觸媒催化由甲醇水溶液中形成氫之相關議題。在此報告中將總 結兩項議題之研究結果,包括(1)觸煤煅燒氣氛(2)添加銅顆粒對觸媒活性之影響。二氧化鈦係經 由溶膠-凝膠法製成後於 Ar, 合成空氣(air),氮(N2), 氫(H2 3% in N2) and 真空(vacuum;~5 x 10-3torr) 。對於由甲醇水溶液中形成氫之活性高低次序為 Ar > air> N2 > vacuum ~ H2。光譜分析顯示
氫與真空導致之低活性源於低表面氫氧基與高晶格缺陷密度。Ar 導致之高活性源於高表面氫氧 基及較多之低能隙激發位置(site)。添加表面銅顆粒大幅提昇 TiO2活性。光譜分析顯示銅顆粒在
反應過程中氧化至具最佳活性之最佳氧化態,而此氧化態無法經由空氣中熱氧化達成。將銅離子 溶入氧化鈦晶格中降低活性。
Abstract (Keywords: Titania, hydrogen production, photocatalysis, calcination atmosphere, Ar, Cu)
TiO2 photocatalysts for hydrogen production from aqueous methanol solution is the
main subject of this work, Studies on two issues are reported here, including (1) the
effect of calcination atmosphere and (2) the effect of Cu metallization. TiO2 catalysts
were synthesized by a sol-gel process followed by calcination at 400 oC in Ar, air, N2,
H2 (3% in N2) and vacuum (~5 x 10-3 torr), respectively. Toward H2 production from a
water/methanol (vol. ratio= 1.4/1) solution, the catalysts exhibited activities in the
order, according to calcination atmosphere, of Ar > air> N2 > vacuum ~ H2. The low
activity resulting from either vacuum or H2 calcination was ascribed to a reduced
coverage of surface hydroxyl and high bulk defect density, based on the x-ray
photoelectron and UV-visible spectroscopic analyses, while the high activity from Ar calcination is to enhanced visible-light excitation. For studying the effect of Cu, Cu
particles have been deposited on TiO2 by incipient-wetness impregnation followed by
low-temperature (400 oC) calcination/reduction, and the metallization process leads to
significant, up to 10 folds, enhancement in photocatalytic activity of TiO2 for H2
production from aqueous methanol solution. Spectroscopic studies indicated that the metallic Cu particles were oxidized to have an optimum valence that can not be
achieved by thermal oxidation. Dissolution of Cu ion in TiO2 lattice, in contrast, led to
reduction in the activity. 1. Introduction
Photocatalytic production of hydrogen using sunlight as the energy input is a valuable sustainable- energy technology. In this case, solar energy is stored by driving
chemical reactions “up-hill” toward chemicals, such as H2, of higher chemical
potentials. Due to its high stability against photocorrosion and its favorite electronic
energy band structure, TiO2 has drawn tremendous attention for such applications. For
H2 production from water, many studies have concluded that direct
photo-decomposition of water into H2 and O2 has a very low efficiency due to rapid
reverse reaction. A much higher hydrogen production rate can be obtained by addition of the “so-called “sacrificial reagents,” such as alcohols and other organics which are
oxidized to products that are less reactive toward hydrogen. For H2 production from a
methanol (MeOH) and water solution, depending on reaction condition and whether metal catalyst used, the reaction could proceed either stepwise, involving stable intermediates, such as aldehyde and acid:
MeOH(l) ↔ HCHO(g) + H2(g) (1)
∆Go1= 64.1 KJ/mole
HCHO(g) + H2O(l) ↔ HCO2H(l) + H2 (g) (2) ∆Go2= 47.8 KJ/mole
HCO2H(l) ↔ CO2(g) + H2(g) (3) ∆Go3= -95.8 KJ/mole,
as suggested by Sakata et al., or in one-step on catalyst surface to give the overall reaction:
MeOH(l) + H2O(l) ↔ CO2(g) + 3 H2(g) (1) ∆Go= 16.1 KJ/mole,
The photocatalytic performance of TiO2 is well known to depend not only on its
bulk energy band structure but, to a great extent, on the surface property. The type and density of surface state are affected by, among others, the synthesis process and the use of additional metal particles. The interplay between processing condition and photocatalytic activity remains largely a state-of-art and beyond prediction at this
point. Crystalline TiO2 has typically been calcined and/or crystallized in oxidizing
atmospheres, such as air and oxygen. The effect of the so-called “inert” atmospheres,
such as N2, Ar, and vacuum, has mostly been overlooked. Deposition of Pt-group
metals, including Ni, Pd and Pt, on TiO2 photocatalyst has been shown to greatly
enhance the photocatalytic production of H2 from either pure water or
water/sacrificial reagent solutions. Sakata et al. [A4] attributed the enhancement to the
catalytic effects of the metal particles on H2 evolution. Millard and Bowker [B2], on
the other hand, suggested that the presence of Pd provides a reaction pathway which involves chemisorption and dehydrogenation of MeOH on Pd to produce chemisorbed
CO and subsequent oxidation of the chemisorbed CO to CO2.
Cu-containing TiO2 catalyst is well known for their photocatalytic activity toward
CO2 reduction [C1-C4] but much less known for its performance in H2 production
from water
In this report, we summarize our research results on two issues: (1) effect of
calcination atmospheres, including Ar, air, N2, H2 and vacuum (~5 x 10-3 torr); and (2)
the effect of Cu-metallization. 2. Experimental
TiO2 powder was synthesized by a conventional sol-gel process. TiCl4 was first
dissolved in an ethanol/water (volume ratio= 4:1) solution, and ammonia was then introduced into the solution to induce condensation until pH reached 7.5. The resulted
gelatinous precipitate was filtered and washed, primarily to remove Cl-, and then dried
at 65 oC in air. Calcination of xerogel was carried out in Ar, synthetic air (N2/O2 mol
ratio= 79:21), nitrogen, hydrogen (3mol % in N2), and vacuum (~5x10-3 torr),
respectively. Calcination procedure started with purging the reactor with the selected
gas (in the case of vacuum treatment, the reactor was evacuated to ~5x10-3 torr) for 1
hr, heated the powder at a rate of 100 oC/hr to 400 oC, and held the powder at 400 oC
for 1 hr before finally the powder being furnace-cooled.
Cu was loaded by an incipient-wetness method, in which an aqueous solution
containing CuCl2·2H2O was added to the oxide powder in an amount that was just
sufficient to wet completely the powder. The resulted powder was then dried and
calcined in synthetic air (N2/O2 mol ratio= 79:21) to make the catalyst. The powders
were calcined by following the standard calcination procedure. The catalyst powders
were stored as calcined, and they were further reduced at 400 oC by H2 (3 mol % in
N2) for 3 hrs prior to characterization works, including the spectroscopic and kinetic
analyses.
Kinetic experiments were carried out by using a vertical tubular batch reactor made of quartz. During a typical run, the entire reactor was enclosed inside a UV-light house (Rayonet photochemical reactor, RPR-100), and the reactor was half-filled with
a water/MeOH (H2O/MeOH vol. ratio= 1.4:1) solution, in which the oxide particles,
in an amount of 1.25 g/liter, were constantly dispersed by a magnetic stirrer. The light house was equipped with 16 UV-light (maximum intensity at 300 nm) tubes, each having a power of 12 W. The unfilled space above the solution was evacuated at the
beginning of reaction, and H2 concentration was determined intermittently by
extracting a small volume of the gas-phase product for gas chromatography (GC)
analysis. The accumulative H2 production data were then calculated from the
concentration data, assuming ideal-gas behavior. 3. Results and Discussion
3.1 Effect of calcination atmosphere
As shown in Fig. 1, upon UV irradiation, H2 was produced with steadily
increasing concentration with time until it saturated at a steady level. In a batch reactor, such as the one currently employed, a steady state represents equilibrium. The
H2 and vacuum calcined catalyst was found to exhibit the worst performance, while
the air- and N2-calcined catalysts showed another level of performance. The
Ar-calcined catalyst, on the other hand, gave overall the best performance in terms of both the maximum rate and equilibrium production level. It is also superior to the commercial P25 catalyst.
Figure 2 shows the diffuse reflectance UV-VIS spectra. The absorption edge
energy is determined by Tauc plot [(αhν)2 vs hν, where α is the absorbance] for
semiconductors with the direction transition. Except for the vacuum-calcined catalyst, which appears black, all the catalysts show a sharp UV absorption near 3.25 eV,
differences, however, exist in absorption within the visible-light range next to the
absorption edge. The Ar, air and N2 samples showed visible-light absorption
correlated consistently with their activities, showing increasing activity with
increasing absorption. On the other hand, the vacuum and H2 samples both showed
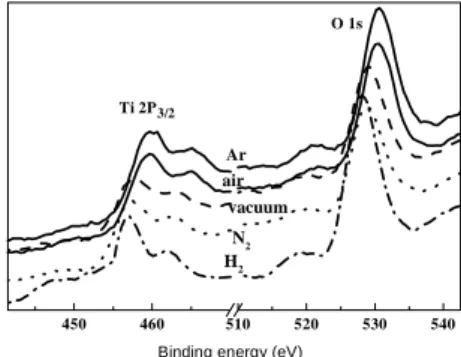
stronger visible-light absorptions but yet lower activities than the rest catalysts. XPS studies indicated mainly two types of surface properties. The Ar- and air-calcined catalysts showed higher Ti and O binding energies than the other catalysts
(Fig. 3). The binding energies of Ti (459.7 ± 0.2 eV for Ti 2P3/2) for the Ar and air
samples are typical of Ti+4. A red shift by ~2.2 eV observed on the other samples
indicates the presence of Ti+3 therein. For the O (1s) XPS peak, the differences
indicate the air- and Ar calcined catalysts have a higher surface coverage of hydroxyl group than the other catalysts.
For a semiconductor-photocatalyzed reaction, the reaction rate is basically determined by how fast the charged particles, including the valence-band hole and conduction-band electron, are generated and the how efficiently they are transferred to the reactants at surface before they recombine. Two opposite effects related to
photocatalysis could result from defects which provide energy states in the band gap. On the one hand, they could enable excitation with photon energy lower than the band gap energy and hence facilitates excitation to form the charged particles. On the other, they would also act as scattering centers to slow down the transport of charged
particles to surface where reaction takes place. As a result, an optimum density, as well as an optimum property, of the structure defect is expected for good
photocatalysis performance. This may explain the observation that there appears to be an optimum visible-light absorption level for maximum reaction rate (Fig. 2). For the
Ar, air and N2 samples, the photocatalytic activity was found to increase with
increasing the absorption. This may suggest that the defect density is low enough that the excitation effect is predominant. In contrast, the excitation effect might be
severely offset by the scattering effect in the cases of the vacuum and H2 samples,
which showed very strong visible-light absorptions but yet poor performance. Another strong clue to the cause of the reactivity variation is provided by the XPS data, which showed difference in the surface hydroxyl coverage. It has been shown that >TiOH, the so-called surface “titanol” group, is an important trapping center for the valence-band hole and hence the principal oxidizing site in the
photoactivated TiO2. Reduced hydroxyl coverage would thus impair the activity of the
catalyst. This might also contribute, in part, to the poor performance exhibited by the
vacuum- and H2-calcined samples. An intermediate level of visible-light absorption
level combined with high surface hydroxyl coverage render the Ar –calcined catalyst the highest photocatalytic activity.
Figure 1. H2 generation curves.
0 100 200 300 400 500 0 400 800 1200 1600 blank P25 H2 vacuum N2 air Ar H2 pr odu ct ion ( µ mo le )
Reaction time (min)
300 400 20 40 60 vacuum H2 Ar Air N2 R ef lec ta nce ( % ) Wavelength (nm)
Figure 2. Diffuse reflectance spectra of TiO2 .
Figure 3. XPS spectra of TiO2 catalysts.
3.2 The effect of Cu metallization
The blank TiO2 catalyst(Cu loading = 0 wt%) appeared yellowish. With
increasing Cu deposition up to 1.9 wt% of Cu, the catalysts turned increasing reddish,
but XRD did not detect reflection of Cu. The grain size of TiO2, being insensitive to
the Cu content, was slightly increased to ~ 10.0 nm, presumably caused by additional reduction heat treatment. The BET surface area is 110 m/g.
UV-VIS spectroscopic analysis (Fig. 4) of the blank TiO2 showed a major
UV-absorption near 350 nm wavelength and a minor one near 450 nm. The latter is consistent with its yellowish appearance. Determined by Tauc plot the major
absorption edge energy is close to the theoretical value (3.20 eV) of anatase TiO2.
Deposition of Cu caused a reduction in major edge energy to 3.09 eV and a dramatic increase in visible-light absorption. The absorption extends up to 700 nm in
wavelength, which is also consistent with their reddish appearance, and the
absorbance increased with increasing Cu content. XPS of the Cu-deposited catalysts detected only Cu, Ti, O, and C. The detected Cu (2p3/2) binding energy (930.5 eV; Fig. 5) is assigned to be metallic Cu. Quantitative analysis considering only the first
three elements gave almost the same ( ±2% variation) Cu content as the loaded value,
suggesting that Cu was indeed largely deposited on TiO2 surface, rather than
embedded within TiO2 matrix.
Figure 6 summarizes the H2-generation curves for catalysts of different Cu loading.
For comparison, data from a commercial TiO2 (P25, Degussa) was also shown (Fig.
6). The fitted dP/dtmax, Peq data are summarized in Fig. 7. They were found to follow
the same trend against Cu loading, showing a maximum at 1.2 wt% of Cu. The maximum production rate at the optimum Cu content is 10 times that of the blank
TiO2 and 4.5 times that of P25.
The occurrence of an optimum metal loading is indicative of interfacial active sites at and/or near the peripheries of the Cu particles. The total peripheral length increases with increasing Cu particle number and size until reactive domains begin to overlap. Further increase in Cu loading would only lead to reduction of the peripheral length, and hence the reactivity. It was also noticed that the catalysts, which appeared reddish at the beginning of the reaction, turned greenish after reaction, suggesting oxidation of Cu. This was confirmed by XPS (Fig. 5), which showed an increase by 1 eV in Cu (2p3/2) binding energy to ~931.5 eV. The result indicated that it is an
oxidized Cu species that is active. Two types of oxidized Cu-TiO2 catalysts (1.2 wt%
of Cu) were thus prepared for comparison. The first one was oxidized, after the thermal reduction procedure, by prolonged (5 days) exposure in air at room
temperature, while the second one was heated in air at 400 oC for 1 hr. The former
appeared dark brownish, while the latter dark green. Both catalysts did show significant photocatalytic activities (Fig. 8) but were inferior to the freshly reduced catalyst. XPS showed that Cu in these two catalysts exhibited higher binding energies, hence higher valences than that oxidized in situ during the reaction. In particular, the Cu (2p3/2) binding energy (932.3 eV) for the catalyst oxidized at room temperature
(Fig. 5) is consistent with Cu+1 in Cu2O, and therefore the Cu in the reaction-oxidized
catalyst has an average valence between 0 and +1.
Finally, it is pointed out that kinetic study on catalyst prepared by mixing the Cu
and Ti sources, namely CuCl2 and TiCl4, at the very beginning of the synthesis
process has also indicated that Cu-doping within the TiO2 lattice has a negative effect
on the overall catalytic activity.
450 460 510 520 530 540 H2 N2 vacuum air Ar 3/2 O 1s Ti 2P
6 Figur4. Diffuse reflectance spectra of Cu-TiO2 catalysts.
Figure 5. Cu XPS spectra of the Cu-TiO2 catalysts
Figure 6. H2 generation curves for the Cu-TiO2.
Figure 7. Effect of Cu loading on H2 production rate and equilibrium production.
300 400 500 600 700 1.9 1.2 0.3 Cu loading: 0 wt% R efl ec ta nc e ( a .u .) Wavelength (nm) 930 940 950 960 thermal oxidation reaction reduction Cu 2p3/2 Cu 2p1/2 In te ns it y (a .u.) Binding energy(eV) 0 100 200 300 400 0 1500 3000 4500 6000 7500 Cu loading: wt % P25 1.2 1.5 0.9 1.9 0.3 0.1 0 H2 p ro duc ti o n ( µ mo le )
Reaction time (min)
0.0 0.5 1.0 1.5 2.0 0 500 1000 1500 2000 2500 3000 3500 H2 pr o d u ction r a te ( µ m o le / h r) Cu loading (wt %) 1000 2000 3000 4000 5000 6000 7000 8000 Equi li bri u m pro ducti o n ( µ mo le ) 6000 8000 (2) (1) od uc ti on ( µ mo le )
Figure 8. Effect of Cu oxidation on H2 production. Key: (1) oxidation in situ by reaction; (2) thermal
oxidation at 400 oC in air for 1 hr; (3) thermal oxidation in air at room temperature for 120 hr. Cu loading is the same (1.2 wt %).
5. References
1. .T. Kawai and T. Sakata, J.C.S. Chem. Comm. (1980) 694.
2. T. Sakata, T. Kawai, and K. Hashimoto, Chem. Phys. Lett. 88, 50 (1982). B1. J. Chen, D. F. Ollis, W. H. Rulkens, and H. Bruning, Wat Res. 33, 669 (1999). B2. L.Millard and M. Bowker, J. Photochem. Photobiol. A 148, 91 (2002).