ELSEVIER
22 August 1997
Chemical Physics Letters 275 (1997) 19-27
CHEMICAL
PHYSICS
LETTERS
Ab initio molecular orbital study of excited electronic states of
the vinyl radical
Alexander M. Mebel a, Yit-Tsong Chen a,b, Sheng-Hsien Lin a,b
~t Institute of Atomic and MolecularSciences, Academia Sinica, P.O. Box 23-166, Taipei 10764, Taiwan, ROCb Department of Chemist~, National Taiwan University Taipei 106, Taiwan, ROC
Received 15 January 1997; in final form 25 April 1997
Abstract
Eight excited doublet electronic states of C2H 3 have been studied using multireference configuration interaction calculations. Close agreement of the excitation energies with experiment is found for the ~2N,, 5 2~ (~-n), and 3 2N, ( v - R ) states where the experimental data are available. The undiscovered B 2 2~, (n-~r * ) state is predicted to have the adiabatic and vertical excitation energies of 33530 and 38546 c m - t, respectively. The ~r--rr * (~ 2 2A' state with the vertical energy of 45001 c m - ~ is predicted to be difficult to observe because of the small oscillator strength. © 1997 Elsevier Science B.V.
I. Introduction
The vinyl radical, C 2 H 3 , plays an important role in hydrocarbon fuel combustion chemistry [1-3]. Thermal decomposition o f C2H 3 and its reactions with hydrogen [4] and oxygen [5] are of fundamental importance in combustion processes. C 2 H 3 is also a significant intermediate in such chemical reactions as the addition to and the polymerization of an acetylenic bond, and the decomposition of ethenoid compounds. Identification of electronic spectra of vinyl is relevant to the measurements of its gas phase reactivity.
Geometry and energetics of the ground (X 2~;) and the first exited (~2~,) states of C 2 H 3 are well established. A visible absorption spectrum of the vinyl radical due to the A ~-J~ electronic transition has been observed by Hunziker et al. [6], using modulation spectroscopy. They detected a band ori- gin at 20020 cm ~ with a vibrational progression
toward the blue with an observed maximum extinc- tion coefficient of 30 1 mol ~ cm-~ at 23629 cm and an estimated F r a n c k - C o n d o n maximum at 24815 cm -1. Hunziker et al. [6] optimized the geometry of
Jk2*Z~ ' C2H 3 at the HF level, and calculated the vertical and adiabatic excitation energy to be 26100 and 18100 cm - j , respectively, using the multirefer- ence configuration interaction (MRCI) approach. Paddon-Row and Pople [7,8] reported the adiabatic energy to be 18607 cm -1 at the U M P 4 / / U H F / 6 - 31G * * level. A recent MRCI study by Wang et al. [9] with a better basis set gives the .~ ~ X vertical excitation energy of 25529 c m - ~.
Little is known about the electronic exited states of C 2 H 3 higher than ,~2~/,. Fahr and Laufer [10] detected two absorption features at 59407 and 60713 cm -1 from the vacuum ultraviolet flash photolysis of Sn(C2H3) 2 and Hg(C2H3) 2 and assigned them as due to the transitions to the Rydberg states of vinyl radical. Fahr and Laufer used these bands to measure 0009-2614/97/$17.00 © 1997 Elsevier Science B.V. All rights reserved.
20 A.M. Mebel et al. / Chemical Physics Letters 275 (1997) 19-27 the rate constant of the C e l l 3 + 02 reaction [10]. Of
theoretical calculations, Paddon-Row and Pople [7,8] considered a n-Tr * state within C2v symmetry and found that adiabatically it lies 31512 c m - l above the ground state. Wang et al. [9] obtained the vertical excitation energy for a ~r-~r * (2 2?¢) transition of 43910 cm -~ at the MRCI level.
We present here an ab initio MRCI study of the electronic spectra for C2H 3 corresponding to the lowest lying eight excited doublet electronic states. For the first three excited states, we also discuss the equilibrium geometries and the adiabatic excitation energies. Using a recently developed method to theo- retically investigate the vibronic spectra of poly- atomic molecules and radicals [11], we calculated the vibronic spectrum of the C e l l 3 ~k2~ ' ~'- X 2/~ transi- tion and compared it to experiment. In the computa- tions of vibrational overlap integrals and Franck- Condon factors, we took into account the distortions, displacements and normal mode mixings in the ex- cited state.
[17]. Vertical and adiabatic excitation energies have been computed using internally contracted multiref- erence configuration interaction (MRCI) method [18]
with (5,6) and (7,7) active spaces. The
C A S S C F ( I I , l l ) wavefunction was used as a refer- ence for the MRCI(5,6) and MRCI(7,7) calculations. Besides ANO(2 + ), the MRCI calculations have been carried out with the ANO(2 + ) * * basis set which additionally includes polarization [ 3 f ] / ( l f ) functions on C and [3d]/(ld) functions on H [16]. For the A ~ X transition, adiabatic excitation energy has also been calculated by the restricted open shell coupled cluster (RCCSD(T)) method [19] with the ANO(2 + )** and Dunning's correlation consistent pVTZ and aug-pVTZ basis sets [20]. Oscillator strength for each excited state has been calculated using MRCI(7,7)/ANO(2 + )* * transition moments and energies. For the calculations, we used GAUSS- IAN 94 [21], MOLCAS-3 [22], and MOLPRO-96 [23] programs.
2. Theoretical methods
Geometries of the ground and excited states of C2H 3 have been optimized using the CASSCF method [12]. A small active space, including 3 elec- trons ( C - C xr bond and an unpaired electron) dis- tributed at 11 orbitals (7a' + 4a"), was used for the calculations of the X 2A and fik2/5~ states. For these states, we additionally carried out optimization em- ploying the hybrid density functional B3LYP ap- proach [13]. Vibrational frequencies for X and ,g, have been calculated at the CASSCF(3,11) and B3LYP levels with the 6-311G(2 + ) G * basis set which is the standard 6-311G* basis set [14] with two additional sp diffuse functions on carbon with the exponents of 0.0438 and 0.013928 [15]. Geome- tries of ~ t 2 ~ ' and other excited states have also been optimized at the CASSCF level with a larger (11,11) active space and ANO(2 + ) basis set. The (11,11) active space includes all valence electrons distributed at 11 orbitals, 9a' + 2a". ANO(2 + ) is the ANO basis set (4s3p2d for C, 3s2p for H) [16] augmented with several diffuse functions for the carbon atom (s exponents: 0.012138 and 0.00422482; p exponents: 0.0080150 and 0.0028052; d exponent: 0.028512)
3. Results and discussion
3.1. V e r t i c a l e x c i t a t i o n e n e r g i e s
Vertical excitation energies for the lowest lying eight doublet excited states of C 2 H 3 are collected in Table 1. The ,g27¢, state corresponds to a "rr-n transition. The A ~ X vertical energy is found to be in the 26000-27000 c m - 1 range from various MRCI calculations. The most reliable value is 26102 c m - obtained at the MRCI(7,7)/ANO(2 + )* * level with Davidson correction for quadruple excitations, desig- nated as MRCI + D(7,7). This value agrees well with previous theoretical results, 26100 cm -~ by Hunziker et al. [6] and 25529 cm i by Wang et al. [9] at the M R C I ( 5 , 6 ) / A N O level. The calculated vertical excitation energy is higher than the esti- mated Franck-Condon maximum at 24815 cm -~ in the experiment [6]. The calculated oscillator strength f o r , g , ~ X is 1 . 2 2 × 1 0 3.
The second excited state, B 2 2~,, corresponds to a transition of the unpaired electron to a ~r * antibond- ing orbital. At our best level, M R C I + D(7,7)/ANO(2 + )* *, the vertical excitation energy is 38546 cm -1, and the oscillator strength for the ,-- X transition, 1.36 × l0 -3, is similar to that of
A.M. Mebel et al. / Chemical Physics Letter7 275 (1997) 19-27
Table 1
Vertical and adiabatic excitation energies (cm -j ) for various excited states of C 2 H 3
21 12N' 7r-n 2 2•, n - ' l T * 2 2N "rr-'rr * 3 2N n - R 4 2A' n - R 52A ' 3 2N' ~r-n 7r-R 4 2N' I r - R vertical energies a C A S S C F ( I 1,1 I ) / A N O ( 2 + ) 28302 44889 M R C I ( 5 , 6 ) / A N O ( 2 + ) 26645 40374 M R C I + D ( 5 , 6 ) / A N O ( 2 + ) 26393 38281 M R C I ( 7 , 7 ) / A N O ( 2 + ) 26936 40434 M R C I + D ( 7 , 7 ) / A N O ( 2 + ) 26416 38784 M R C I ( 7 , 7 ) / A N O ( 2 + ) ** 26851 40446 M R C I + D ( 7 . 7 ) / A N O ( 2 + ) * * 26102 38546 experiment 24815 b oscillatorstrength d 1.22 × 10 - 3 adiabatic energies ~ 48536 45941 45412 45768 49771 5 4 1 6 l 65691 59303 64247 45290 49901 54460 58093 59949 64881 45787 50257 54540 66654 59878 64755 45001 50422 54862 58991 60327 65202 59407 ~ 60713 c 1.36 × 10 - 3 3.05 X 10 4 7.04 × 10 3 0.0172 0.0190 0.0199 3.37 × 10 3 12P~'-pl. 12A'-tw. ~T--n 7r-n 2 2P~'-pl. 2 2A'-tw. 2 2A'-pl. 2 2,N'-tw. n-~ n-Tr 7r-~r ~r-~r CASSCF(11,11 ) / A N O ( 2 + ) 20003 M R C I ( 5 , 6 ) / A N O ( 2 + ) 19536 M R C I + D ( 5 , 6 ) / A N O ( 2 + ) 19252 M R C I ( 7 , 7 ) / A N O ( 2 + ) * * 18426 MRC1 + D ( 7 , 7 ) / A N O ( 2 + ) * * 19122 (19175) J R C C S D ( T ) / A N O ( 2 + ) * * (19404) f R C C S D ( T ) / p V T Z (19812) f R C C S D ( T ) / a u g - p V T Z (19652) f experiment 20020 b 39221 39043 40527 37543 35412 35769 39164 36582 33609 34007 38353 35736 20894 33697 34542 37063 35078 20866 33530 34068 37700 35212
The total energies of the ground state C2H 3 (in hartree) at various levels are the following: CASSCF(I 1,11)/ANO(2 + ): - 7 7 . 5 4 0 2 4 ; M R C I ( 5 , 6 ) / A N O ( 2 + ): - 7 7 . 7 0 5 2 8 ; M R C I + D ( 5 , 6 ) / A N O ( 2 + ): - 7 7 . 7 2 9 5 7 : M R C I ( 7 , 7 ) / A N O ( 2 + ): - 7 7 . 7 1 1 3 9 ; M R C I + D ( 7 , 7 ) / A N O ( 2 + ): - 7 7 . 7 3 9 6 8 ; M R C I ( 7 , 7 ) / A N O ( 2 + ) * *: - 7 7 . 7 2 3 3 5 ; MRC1 + D ( 7 , 7 ) / A N O ( 2 + ) * ": - 7 7 . 7 5 2 5 0 .
b From ref [6].
From ref [9].
,I At the M R C I + D ( 7 , 7 ) / A N O ( 2 + ) * * level. Without zero-point energy correction. With zero-point energy correction.
,-- X. The energy is not very sensitive to the basis set and active space used in the MRCI calculations. On the other hand, the Davidson correction is essen- tial. The vertical energy decreases by about 2000 cm ~ with the correction. The third doublet excited state has 2N symmetry and a ,rr-'rr * character, and can be denoted as (~ 2 2~. Previous calculations at the M R C I ( 5 , 6 ) / A N O level gave this state (repre- sented by B
2A'
in that paper) a vertical energy of 43910 cm J [9]. The present calculations with the larger active space and basis set give 45001 c m - ~ at the MRCI + D ( 7 , 7 ) / A N O ( 2 + )* * level. The transi- tion of C ,-- )( is weak and the oscillator strength is only 3.05 × 10 -4.Of the higher states shown in Table 1, 5 2~ and 3 2A:' are worth mentioning. The 5 2p~ state is due to the cr-n transition of an electron moving from the C - C ~r bond to the lone pair of the carbon atom.
1
Our best vertical excitation energy is 58991 cm which agrees fairly well with the experimental band observed by Fahr and Laufer at 59407 cm -1 [10]. The second band, at 60713 cm -~, reported by these authors is apparently due to the 'rr-R transition to the 3 2A' electronic state, although one cannot exclude the possibility of vibrational spacing mentioned in [10]. The best calculated vertical energy of 3 2N is 60327 cm -l . 5 2A; (~r-n) and 3 2A' ( w - R ) have large oscillator strength of 0.019 and 0.020, respectively.
22 A.M. Mebel et al. / Chemical Physics Letters 275 (1997) 19-27
The M R C I + D ( 7 , 7 ) / A N O ( 2 + )* * calculations un-
derestimate the energies of these two states by --~ 400
cm -1, but the energy difference between the two is reproduced within the accuracy of 30 c m - 1 .
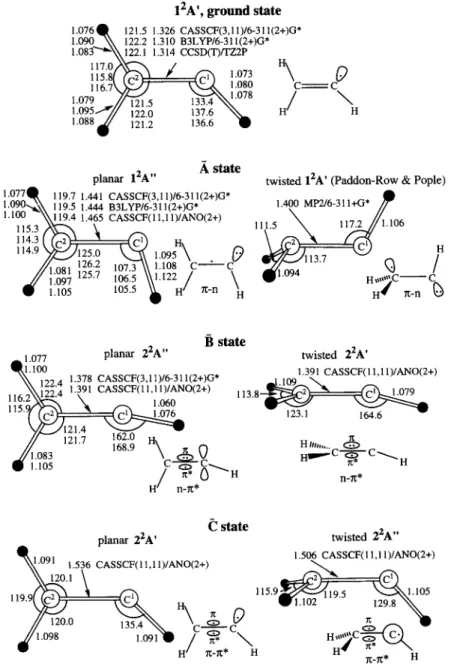
3.2. Geometries and adiabatic excitation energies Optimized geometries of the A, B, and C excited states are shown in Fig. 1. For
~k2~ ',
the bondlengths and angles, calculated at three different lev- els of theory agree well with each other and with the earlier results of UHF calculations [7,8]. The major geometric changes in ~2g, as compared to the ground state are the elongation of the CC bond by 0.11-0.13
and the decrease of the C2CIH angle by = 30 °. The elongation of the CC bond is because the C - C w-bonding population is halved in the ~2~, state. 12A ', ground state
1.076Q 121.5 1.326 CASSCF(3,11)/6-311(2+)G* 1.090 \ \ 122.2 1.310 B3LYP/6-311(2+)G* 1.083"'~\\122.1 1.314 CCSD(T)/TZ2P I17.0 ,~'-"N . / Ilk 115.8/ ~ 1.073 \ 0 1 i g S \ ~ 1.080 --C C 1.079 ~" 1/~91 ~ 133.~N,~1.078 / 1"095""~// i2210 137.6 N ~ - H / H 1.088 , ~ 121.2 136.6 "O state
planar 12A '' twisted lZA ' (Paddon-Row & Pople) 1.077Q 119.7 1.441 CASSCF(3,11)/6-31 l(2+)G* 1.400 MP2/6-311+G* / ~ 1.090,,~\ 119.5 1.444 B3LYP/6-31 l(2+)G* 1.100 ~\\119.4 1.465 CASSCF(II,ll)/ANO(2+) ~ _ 1 1 1 . 5 ~ ~ 1 1 7 . 2 1.106
115.3 /'x~--~
114.3 { (c,2~ | "q k Q 114.9 ~___j]125.0 x,~A\ t.097~ ~ H 1.081 " 107.3 \\ 1.108 C'--:--" C.
1.105 II122/ zc-n ~H
H,,,~C--C
H x-n [3 state1 077 planar 22A '' twisted 22A '
11100 199 a 1.378 CASSCF(3,11)/6-311(2+)G* _| 1091"3"~ CASSCF(II'll)/ANO(2+) ,~ ~i2214 1.391 CASSCF(11,11)/ANO(2+) 113 8 .... 2 c,l 1.079
11116:2
1.060
/ / ~ " 168.9 n \ 1 ~ Hit, ... .//1083 . . . . \__~__V r t , , . - - c - ~ - - - c ~ ." 1.105
?'--~, 8 ~
~
rl
n-~lt'* H n lt* H / s t a t eplanar 22A ' twisted 22A ''
.o91 1.536 CASSCF(II,ll)/ANO(2+) 1.506x~SSCF(II,I1)/ANO(2+)
1 " ~ ' 1 ~
/,~
1 1 5 . 9 ~ . 1 0 5'g-/t* H
Fig. 1. Optimized geometries (bond lengths in ,~, bond angles in degrees) and schematic presentation of the electronic structure for the ground and excited states of the vinyl radical. The MP2/6-311 + G * optimized geometry of the twisted 12A' structure is taken from [8].
A.M. Mebel et al. / Chemical Physics Letters 275 (1997) 19-27 23 The drastically bent
C2CIH
is caused by the largerepulsion from the two or-type lone-pair electrons of the C ~ atom. The adiabatic excitation energy at the MRCI + D(7,7)/ANO(2 + )* * level is 19175 c m - 1 if the zero-point energy correction (ZPE), calculated at the B3LYP level, is taken into account. The ZPE is small, about 50 cm ~. Our value for the adiabatic ~23;, ,__ ~ 23; excitation energy is 500-1000 cm -1 higher than the earlier results of Hunziker et al. [6] and of Paddon-Row and Pople [7,8]. According to Paddon-Row and Pople [7,8], another minimum ex- ists on the first excited doublet state PES. As shown in Fig. 1, it has non-planar C~ geometry with a twisted CH 2 group and the electronic term is 123; (w-n). The electronic structure has been described [7,8] in terms of three nonbonding electrons, one of them occupying the tricoordinate carbon nonbonding AO and the remaining two occupying the bicoordi- nate carbon nonbonding AO. Both nonbonding A O ' s lie in the same plane. As a consequence, both carbon atoms are highly bent, i.e., C 2 is considerably pyra- midalized and the C2C~H angle is 117.2 °. At this geometry, :3;' is the ground state. The adiabatic excitation energy for non-planar 12p~ is calculated to be 20866 c m - J at the MRCI + D(7,7)/ANO(2 + )** level. Thus, this structure lies = 1700 cm -1 higher in energy than planar 123;,. At M P 2 / 6 - 3 1 G *, Paddon-Row's 123; structure has no imaginary fre- quencies [8].
Two geometries have been optimized for the (n-~r *) state. The planar structure has the 2 23;, electronic term. The non-planar C~ symmetric struc- ture with a twisted CH 2 group is optimized for the 2 23; state. Both planar 2 23;, and twisted 2 23; have similar electronic structures and, consequently, simi- lar bond lengths and bond angles. The unpaired electron is moved from the nonbonding AO of C ~ to the ~r * antibonding orbital. This results in the elon- gation of the CC bonds to 1.39 A, and the C2C~H angles increase to 165-169 ° . In the twisted 223; structure, the tricoordinated C 2 atom is not pyrami- dalized. The geometry of 2 23; is quite different from that suggested by Paddon-Row and Pople for twisted 123;. This is due to the fact that the two geometries correspond to the stationary points on PES's of two different electronic states, the first and the second 23;, and their electronic structures are different, r r - n versus n-Tr *. The MRCI + D(7,7)/ANO(2 + )* *
energy of the 2 23; state calculated at Paddon-Row's geometry is by 1.8 eV higher than the energy at the twisted geometry optimized for 2 23;.
The adiabatic excitation energies for planar 2 23;, and twisted 2 23; are close, 33530 and 34068 cm J, respectively. Paddon-Row and Pople [7,8] calculated the n - r r * state within C2v symmetry with a linear C 2 C I H fragment and obtained the adiabatic energy of 31512 cm -j at the U H F / 6 - 3 1 G * level. At this level, the C2v structure has two imaginary frequen- cies and collapses to the twisted 12p~ structure upon optimization allowing an out-of-plane distortion.
Two stationary points have been found on the PES for the 7r-Tr" (~ excited state. For both planar 223; and twisted 2 23;, states, the optimized geometry is characterized by the long CC distance (1.51-1.54 A), while the C 2 C I H angle (130-135 °) does not change much as compared to that in the ground state. The adiabatic excitation energies are calculated to be 37700 cm ~ for the planar 2 23; structure and 35212 c m - J for the twisted 2 23;,.
For the four optimized geometries of B and (~ states we carried out the Hessian matrix calculations at the C I S / 6 - 3 1 1 ( 2 + ) G * level [24]. The results show that all of them, planar 2 23;' and 2 23; as well as twisted 2 23; and 2 23;,, have no imaginary fre- quencies and correspond to minima on the PES's. Higher level calculations may change this conclu- sion. However, one can at least expect that the structures with lower energy, planar 2 23;, for the state and twisted 2 23;, for (~, are real minima. 3.3. V i b r o n i c s p e c t r a
Vibrational frequencies of the ground and excited ,~ state, calculated at various levels of theory are presented in Table 2. For the X state, Wang et al. [9] reported the C C S D ( T ) / T Z 2 P frequencies. The fre- quencies, calculated at the B3LYP/6-311(2 + ) G * level, agree well with the CCSD(T) ones. Therefore, we used the B3LYP frequencies of the ground and excited states for the calculations of the vibronic spectrum. In the A state, vibrational frequencies change substantially. For instance, frequency u 2 (a") corresponding to a HCCH torsion increases from 820 to 1142 cm - I . Two a' normal modes, Q n and Q4, responsible for the coupling between the C2C1H bending the C2H rocking, increase their frequencies
24 A.M. Mebel et al. / Chemical Physics Letters 275 (1997) 1 9 - 2 7 Table 2
Vibrational frequencies (cm-1 ) of the X 2A' and g2~, electronic states of CzH 3, calculated at various levels of theory
2 X ~2~,
U H F / CASSCF/ B3LYP/ CCSD(T)/ U H F / CASSCF/ B3LYP/
6-31G* a 6-311(2+)G* 6-311(2+)G* TZ2P b 6-31G* 6-311(2+)G* a 6-311(2+)G* v I (~) 827 762 713 764 v] (~) 1017 998 949 v 2 ( 4 ) 884 559 820 830 v~ (4) 1002 1513 1142 v 3 (~') 959 979 920 944 v~ ( 4 ) 779 904 850 (d) 1194 1092 1046 1098 v~ (d) 1386 1363 1290 v 5 (d) 1407 1451 1391 1411 v~ (~) 1257 1216 1191 v r ( ~ ) 1635 1640 1651 1609 v~ (~) 1656 1632 1531 u7 (~) 3279 3256 3038 3049 v~ (if) 3268 3243 3020 v 8 (d) 3372 3332 3136 3156 v~ (~) 3369 3349 3141 v 9 (if) 3431 3359 3236 3215 u~ (d) 3157 3129 2942 a From Ref. [7,8]. b From Ref. [9].
from 713 and 1046 cm - l in -~2~ to 949 and 1290 cm - l in ,~2p~,. The frequency change in v 1 and v 4 is caused by the decrease of the C 2 C I H bond angle in the excited state. The Q5 and Q6 normal modes, corresponding to the CC stretching coupled with the C 2H bending, exhibit a decrease of their frequencies from 1391 and 1651 cm 1 to 1191 and 1531 cm -1, respectively. This is due to the elongation of the CC bond in the excited state. The /)9 frequency of the C IH stretch vibration also decreases from 3236 to 2943 c m - 1.
The greatest displacement was calculated due to the QI and a n normal coordinates; as seen in Table 3, AQ 1 and A Q 4 are 0.3327 and 0.3587 A a m u 1/2, respectively. Qs, Q r , and Q9 are also substantially displaced in the ~2E, state, with A Q ' s of 0.09-0.14 A amu 1/2. Besides the distortion and displacement described above, normal coordinates are rotated, i.e., mixed in the excited state. The corresponding Duschinsky matrices are shown in Table 3. The mixing between the a" modes, Q2 and Q3, is rela- tively small. Q1, Q4, Q8, and Q9 are rotated to a larger extent where the mixing is especially signifi- cant between QI, Q4, and Q9- The third group of the rotated normal modes consists of Qs, Q6, and Q7. The largest mixing among the three is found between Q5 and Q6.
The Duschinsky matrices, AQ's, and B3LYP vi- brational frequencies for the X and A states were used to calculate the vibronic overlap integrals and Franck-Condon factors, employing the algorithm
described earlier [11]. The calculated vibronic spec- trum for the ~2~, ~ ~ 2 A transition is shown in Fig. 2, and positions of the most intense peaks are com- pared to the experimental bands [6] in Table 4. In the experiment, the first band was observed at 20020 c m - t. However, Hunziker et al. [6] did not rule out a possibility that the origin of the spectrum lies at a lower energy. M R C I / A N O ( 2 + )* * calculations Table 3
Duschinsky matrices and displacement of the normal modes AQ (,~amu 1/2) in the excited g2p~, state of C2H 3
(a) QI Q4 Q8 Q9 Q] 0.8928 0.3178 0.0943 0.3068 Q~ -0.2332 0.8499 0.1539 -0.5088 Q~ 0.0160 -0.1342 -0.9923 -0.0600 Q~ 0.4139 -0.1959 0.0642 -0.8098 AQ 0.3327 0.3587 0.0326 0.1023 (b) Q2 Q3 Q~ 0.9061 -0.2965 Q~ 0.4999 0.9224 AQ 0.0 0.0 (c) Q5 Q6 Q7 Q~ 0.7325 0.1292 -0.2212 Q~ -0.9805 0.5823 -0.0916 Q~ -0.0414 -0.3670 0.9651 ~ Q 0.0925 0.1445 0.0381
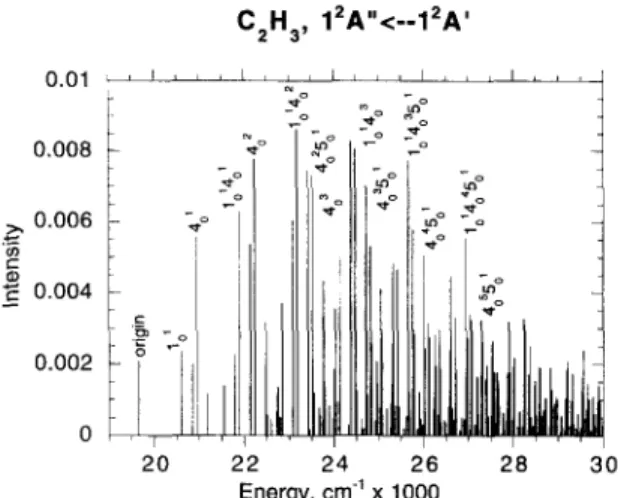
A.M. Mebel et al. / Chemical Physics Letters 275 (1997) 19-27 25 C 2 H a , 1 2 A " < - - 1 2 A ' 0 . 0 1 , i , , ~ t , , I . . . I , , 0 . 0 0 8 :~ 0 . 0 0 6 0 . 0 0 4 0 . 0 0 2 ~o 2 0 2 2 .,_o ,n.o L o o
lt il
' I ' ,I 2 4JJll] lLb,IJ,l k
2 6 ' 2 1 8 ' ' E n e r g y , c m ~ x 1 0 0 0 30Fig. 2. Calculated vibronic spectrum for the A 2 A " ~ X 2 A ' transi-
tion in C~_H 3.
maximum Franck-Condon factor occurs 3529 c m - above the origin (1~42o), while the observed absorp- tion maximum appears 4795 cm-~ above the origin and was assigned to 44 [6]. Franck-Condon factors nearly as large as those for the former are found for the transitions predicted to lie in the energy range between V0o + 4 0 0 0 and v00 + 5000 cm ~; it is likely that the observed maximum corresponds to one of these, particularly to 1~43, which is predicted to lie at v00 + 4 8 1 9 cm -~. It is worth mentioning that the largest Franck-Condon factors are predicted for the transitions between 22232 and 25662 cm -~, while the calculated vertical excitation energy is 26102 cm -~. Thus, the simple assumption that "vertical excitation e n e r g y " = "absorption maxi- m u m " , often used in the theoretical analysis of electronic spectra, is violated here.
give the spectral origin at 19175 cm -~. In order to confirm this result, we additionally carried out RCCSD(T) calculations with the ANO(2 + )* *, pVTZ, and aug-pVTZ basis sets. The adiabatic exci- tation energy calculated by RCCSD(T) with ZPE is in the 19404-19812 cm -] range. The highest level result, RCCSD(T)/aug-pVTZ, where the basis set (5s3p3d2f for C and 4s3p2d for H) contains 161 contracted basis functions, is 19652 cm ]. Since the calculated Franck-Condon factor for the origin is not much lower than those for the other intense peaks, we believe that the feature observed at 20020 c m - J is truly the band origin. The best theoretical value underestimates the experimental result by 368 cm -~ The accuracy may be increased using the RCCSD(T) method with pVQZ and pV5Z basis sets, which is beyond our computational facilities.
The calculated spectrum reproduces the main fea- tures of the observed one, as can be seen in Table 4. The spectrum contains two vibrational progressions with the spacings of 1290 and 949 cm l, compared to 1200 and 920 cm-~ in the observation, owing to the Qj and Q4 normal modes. Additionally, Q5 is active in the spectrum, with the frequency of 1191 c m - J in the excited state. The v 4 and u~ frequen- cies in C2H 3 ~2~, differ by about 100 cm - l in our calculations, therefore, the peaks due to excitation of
1 1 9 2 1
Q4 and Qs, such as 4050 and 4 °, 4~, 4050 and 43 etc., are quite close to each other and might merge if the resolution is not high enough. The calculated
3.4. Correlation diagram between C~. H 3 and C: H 2 + H
A diagram describing the correlation between the ground and valence excited states of C2H 3 and its
Table 4
Experimental and calculated positions of the peaks (cm - t ), Franck-Condon factors, and assignment of the vibronic spectrum for the A, zx' ~ X 2k; transition in the vinyl radical
Experimental Theoretical Franck-Condon Assignment
band a peak factor
20020 19652 0.0021 origin 21222 20942 0.0056 4~ I I 22148 21891 0.0063 1 o4o 22427 22133 0.0054 41~51~ 22232 0.0077 40 23348 23082 0.0060 1]o4~5~ I 2 23629 23181 0.0086 I o4o 23423 0.0074 4~511 23522 0.0073 4~ I 2 I 24522 24372 0.0083 104o50 I 3 24815 24471 0.0081 104o 24713 0.0070 4i~ 50 24812 0.0053 4~ 25727 25662 0.0077 111,4~ 5~1 I 4 25981 25761 0.0058 104o 4 I 26003 0.0051 4 o 5 o 26918 26952 0.0056 1 ~445~]~ 27137 27293 0.0030 4~ 511) a From ref. [6].
26 A.M. Mebel et al. / Chemical Physics Letters 275 (1997) 19-27 Energy, eV
I
C2H2(2tAu,21A2)+H8
..:...'.2";
C2H2(1Bu,IB2)+H ..-.-;'" . CEH2(t A2)+H 7" . . . . -'.'; -"" ...."".. C2H2 ( 1Aa)+i.i _ _.1 - - - . . . - " . . - .." . - " ~ ~ 1 - 1 2 L ~ ¢ 2 ( A2)+I'I . . . . ... 6" ~, . . . . . ' " ..'" . . . . -" ;'..::::." . . . . C2H2( ' ~ ) + t i .'" . . . ..-." . - ' . . ~ . ~ " . . . . .: . _~C2H2(°Bu)+H 5" /~ . . - " - ' . ~.~.~%'*.'-""" ,~,'~'.'" ~ H2Cc( B2)+H A 2A" ,,,,,~*, "" ..- H2CC(IAI)+H 3 T °°;'" "'" / C2H3Fig. 3. Correlation diagram between the ground and valence excited states of C2H 3 and its dissociation products C2H 2 + H and
H 2 C C + H.
dissociation products C 2 H 2 - I - H and H 2 C C + H is shown in Fig. 3. The correlation diagram is based on a simple symmetry consideration and the adiabatic excitation energies for acetylene and vinylidene were
taken from the most accurate theoretical
calculations 1, while the bond energies of C 2 H 3 are taken as 3.47 eV (H2CC + H) and 1.45 eV (HCCH + H) [30]. Both singlet and triplet states of C 2 H 2 and HECC together with a hydrogen atom in the doublet state can be formed from the doublet states of C E H 3. T h u s , adiabatically, the ground X 2a/ state of the vinyl radical can dissociate to C E H 2 ( l E g ) +
H, H E C C (1A l ) + H, C E H 2 (cis-3B2 or trans-3Bu) + H, and H E C C (3B 2) + H. Dissociation of the first excited state, ,~2N, can give six different products:
i The adiabatic excitation energies relative to the ground state acetylene are the following: 3.82 eV for 3B2, 4.16 eV for 3B u, 4.44 eV for 3Au, and 4.76 eV for 3A 2 states of CEH 2 [25,26];
I I
5.43 and 5.78 eV for the A u and A 2 states of C2H 2, respec- tively, [27]; 6.70 eV for the IB u and IB 2 states and 6.94 eV for
I I
the 2 A u and 2 A 2 states of C2H 2 [28]; 4.07 eV for 3B2, 4.75
3 1
eV for A2, and 5.12 eV for A 2 states of HECC [29].
trans-C2H 2 (1A u a n d 3Au) , c i s - C 2 H 2 (IA 2 a n d 3A 2) as well as H 2 C C (3A 2 a n d IA2). C 2 H 3 (B2 2N) is correlated to the 2 1A u and 2 1A 2 singlet states and a triplet state of C 2 H 2 originated from 3E u. The C 2 2K state of the vinyl radical is connected to C E n 2 (1B u and ~B z) and a triplet state correlated to 3A u .
The difference in the strengths of the C~H and C2H bonds is large for the ground state but becomes small for the ,4 state of C2H 3. According to the recent results of Stanton and coworkers [27,29], vinylidene lies lower in energy than acetylene on the S l surface. Therefore, the C 1H bond in C 2 H 3 -~ 2N is weaker than the C2H bonds. Dissociation mecha- nism of the B and C states of the vinyl radical would apparently involve internal conversion to the ,~ or state. However, one cannot exclude that the dissocia- tion takes place via intersystem crossing into the quartet manifold which we do not consider here. Experimental measurements of Lee and coworkers [31] have shown that upon the absorption of a 193 nm (51813 cm -1) photon CEH 3 fragments to H + H E C C (1A 1 o r 3BE) , which are adiabatically corre- lated to the X state.
A.M. Mebel et al. / Chemical Physics Letters 275 (1997) 19-27 27
Acknowledgements
AMM is grateful to Academia Sinica for the fellowship at IAMS. This work supported in part by the National Science Council of ROC under grant No. NSC-86-2113-M-001-043-CT. We thank Profes- sor Paddon-Row and other reviewers for their help- ful comments.
References
[1] H. Okabe, Photochemistry of Small Molecules, Wiley, New York, 1978.
[2] W.C. Gardiner Jr. (Ed.), Combustion Chemistry, Springer, New York, 1984.
[3] I.K. Puri (Ed.), Environmental Implications of Combustion Processes, CRC Press, Boca Raton, FL, 1993.
[4] A. Fahr, A. Laufer, R. Klein, W. Braun, J. Phys. Chem. 95 (1991) 3218.
[5] D.J. Donaldson, I.V. Okuda, J. Sloan, J. Chem. Phys. 193 (1995) 37.
[6] H.E. Hunziker, H. Kneppe, A.D. McLean, P. Siegbahn, H.R. Wendt, Can. J. Chem. 61 (1983) 993.
[7] M.N. Paddon-Row and J.A. Pople, J. Phys. Chem. 89 (1985) 2768.
[8] M.N. Paddon-Row, reviewer's comments for this Letter. [9] J.-H. Wang, H.-C. Chang, Y.-T. Chen, Chem. Phys. 206
(1996) 43.
[10] A. Fahr, A.H. Laufer, J. Phys. Chem. 92 (1988) 7229. [11] (a) A.M. Mebel, Y.-T. Chen, S.H. Lin, Chem. Phys. Lett.
258 (1996) 53; (b) A.M. Mebel, Y.-T. Chen, S.H. Lin, J. Chem. Phys. 105 (1996) 9007.
[12] (a) H.B. Schlegel, M.A. Robb, Chem. Phys. Lett. 93 (1982) 43; (b) F. Bernardi, A. Bottoni, J.J.W. McDougall, M.A. Robb, H.B. Schlegel, Faraday Symp. Chem. Soc. 19 (1984)
137.
[13] (a) A.D. Becke, J. Chem. Phys. 98 (1993) 5648; (b) C. Lee, W. Yang, R.G. Parr, Phys. Rev. B 37 (1988) 785.
[14] R. Krishnan, M. Frisch, J.A. Pople, J. Chem. Phys. 72 (1988) 4244.
[15] K.B. Wiberg, C.M. Hadad, J.B. Foresman, W.A. Chupka, J. Phys. Chem. 96 (1992) 10756.
[16] P.-O. Widmark, P.-,~. Malmqvist, B.O. Roos, Theor. Chim. Acta. 77 (1990) 291.
[17] L. Serrano-Andres, M. Merchan, I. Nebot-Gil, R. Lindh, B.O. Roos, J. Chem. Phys. 98 (1993) 3151.
[18] (a) H.-J. Wemer, P.J. Knowles, J. Chem. Phys. 89 (1988) 5803; (b) P.J. Knowles, H.-L Wemer, Chem. Phys. Lett. 145 (1988) 514.
[19] P.J. Knowles, C. Hampel, H.-J. Werner, J. Chem. Phys. 99 (1994) 5219.
[20] T.H. Dunning, J. Chem. Phys. 90 (1989) 1007.
[21] M.J. Frisch, G.W. Trucks, H.B. Schlegel, P.M.W. Gill, B.G. Johnson, M.A. Robb, J.R. Cheeseman, T. Keith, G.A. Peters- son, J.A. Montgomery, K. Raghavachari, M.A. AI-Laham, V.G. Zakrzewski, J.V. Ortiz, J.B. Foresman, J. Cioslowski, B.B. Stefanov, A. Nanayakkara, M. Challacombe, C.Y. Peng, P.Y. Ayala, W. Chen, M.W. Wong, J.L. Andres, E.S. Re- plogle, R. Gomperts, R.L. Martin, D.J. Fox, J.S. Binkley, D.J. DeFrees, J. Baker, J.P. Stewart, M. Head-Gordon, C. Gonzalez, J.A. Pople, Gaussian 94, Revision B.2, Gaussian, Pittsburgh, PA, 1995.
[22] MOLCAS-3, K. Andersson, M.R.A. Blomberg, M.P. Fulscher, G. Kadstrom, V. Kello, R. Lindh, P.-,~. Malmqvist, J. Noga, J. Olsen, B.O. Roos, A.J. Sadlej, P.E.M. Siegbahn, M. Urban, P.-O. Widmark, University of Lund, Sweden. [23] MOLPRO is a package of ab initio programs written by H.-J.
Werner and P.J. Knowles, with contributions from J. Alml6f, R.D. Amos, M.J.O. Deegan, S.T. Elbert, C. Hampel, W. Meyer, K. Peterson, R. Pitzer, A.J. Stone, P.R. Taylor, R. Lindh.
[24] J.B. Foresman, M. Head-Gordon, J.A. Pople, M.J. Frisch, J. Phys. Chem. 96 (1992) 135.
[25] J.K. Lundberg, R.W. Field, C.D. Sherrill, E.T. Seidl, Y. Xie, H.F. Schaefer III, J. Chem. Phys. 98 (1993) 8384. [26] Y. Yamaguchi, G. Vacek, H.F. Schaefer III, Theor. Chim.
Acta 86 (1993) 97.
[27] J.F. Stanton, C.-M. Huang, P.G. Szalay, J. Chem. Phys. 101 (1994) 356.
[28] M. Peric, S.D. Peyerimhoff, R.J. Buenker, Mol. Phys. 62 (1987) 1339.
[29] J.F. Stanton, J. Gauss, J. Chem. Phys. 101 (1994) 3001. [30] K.M. Ervin, J. Ho, W.C. Lineberger, J. Chem. Phys. 91
(1989) 5974.
[31] B.A. Balko, J. Zhang, Y.T. Lee, J. Chem. Phys. 97 (1992) 935.