行政院國家科學委員會專題研究計畫 期中進度報告
利用氧化鯊烯環化酵素作為篩選胜胜適合體及其擬胜胜之
研究(1/3)
計畫類別: 個別型計畫
計畫編號: NSC94-2113-M-009-011-
執行期間: 94 年 08 月 01 日至 95 年 07 月 31 日
執行單位: 國立交通大學生物科技學系(所)
計畫主持人: 吳東昆
計畫參與人員: 張程翔,劉媛婷,張晉豪,王庭翊,陳令宗,林宏明、李文暄、
魏大景、溫皓宇
報告類型: 精簡報告
處理方式: 本計畫可公開查詢
中 華 民 國 95 年 5 月 18 日
行政院國家科學委員會專題研究計畫期中進度報告
計畫編號:NSC 94-2113-M-009-011-
利用氧化鯊烯環化酵素作為篩選胜肽適合體及其擬胜肽之研究
執行期限:94 年 08 月 01 日至 95 年 07 月 31 日
主持人:吳東昆 國立交通大學生物科技學系
計畫參與人員:張程翔,劉媛婷,張晉豪,王庭翊,陳令宗,林宏明、李文暄、魏大景、
溫皓宇
一、中文摘要 氧化鯊烯環化酵素為負責催化哺乳類中之膽 固醇與黴菌中之麥角脂醇生合成途徑中共同中間 產物,羊毛脂醇,之合成的重要酵素。由於此酵 素在生化及醫學中之學術重要性及其作為發展降 低膽固醇及抗黴菌藥物之重要標的之潛力,因而 成為學術研究與製藥廠中研發之重要題目。利用 隨機核酸片段與隨機胜肽序列庫作為藥物或其前 導藥物為目前非常熱門及極具潛力之藥物開發方 法。此類方法之共同特點為利用重複篩選、放大、 清洗之過程,可由隨機核酸片段庫或隨機隨機胜 肽序列庫中篩選出具親和力之核酸片段與胜肽序 列。本研究利用此類研究策略,已得到數段核酸 片段,經由 DNA 定序儀已測定其序列。而在胜肽 序列之篩選,亦已獲得數個對氧化鯊烯環化酵素 具有親和性的噬菌體胜肽序列,由西方墨點法測 定得到一個七胜肽序列和六個十二胜肽序列具有 反應。其中一個十二胜肽序列的樣品在西方點墨 法中可清楚看到其反應,現正進行親和力常數之 測量。 關鍵詞:氧化鯊烯環化酵素、隨機核酸片段庫、配 位體指數遞增演化、隨機胜肽序列庫、西方點墨 法、膠體遲滯電泳 AbstractOxidosqualene cyclase, a crucial enzyme in converting an acyclic polyene oxidosqualene to polycyclic lanosterol, plays important role in the biosynthesis of cholesterol and ergosterol. The enzyme fulfills a unique role in designing new antifungal, hypocholesteremic, and phytotoxic drugs, due to its great biological and medicinal significance and potential, and has attracted scientists for decades to study the structure-function-mechanism relationships and drug screening application. The application of random oligonucleotide and oligopeptide libraries holds great potential for drug screening. With the reiteration of selection, amplification, washing procedures, oligonucleotide and oligopeptide with high binding affinity and specificity could be obtained. We have applied the aforementioned strategies to identify several
oligonucleotides and oligopeptides and have determined the corresponding sequences. Following repeated biopanning process, one heptapeptide and six dodecapeptides, which showed OSC-binding affinity have been identified from Western blotting experiments. Among them, one oligopeptide which apparent binding affinity to OSC on dot blotting is now subjected to affinity binding constant determination.
Keywords: oxidosqualene-lanosterol cyclase, random oligonucleotide library, systematic evolution of ligands by exponential enrichment (SELEX), random oligopeptide library, western blotting, electrophoresis mobility shift assay (EMSA)
二、緣由與目的
Oxidosqualene-lanosterol cyclase enzymes have increasingly become targets for the development of antifungal, hypocholesterolemic, and phytotoxic drugs due to their unique roles in the biosyntheses of cholesterol, ergosterol, and phytosterols. The relationships between increase of coronary heart disease and elevated plasma LDL cholesterol are well documented and have attracted scientists to develop hypocholesterolemeric drugs. On the other hand, fungal infections have emerged as major causes of morbidity and mortality for individuals who become immunocompromised through infection with HIV, by treatment for organ transplanation, or during advanced chemotherapies. Both cholesterol and fungal sterol biosyntheses share a common biosynthetic pathway until oxidosqualene, the substrate which is catalyzed by oxidosqualene cyclase to form the sterol and related triterpenoid core structure.
Oxidosqualene cyclases catalyze the cationic cyclization/rearrangement of (3S)-2,3-oxidosqualene (OS) into a variety of sterols and triterpenes. The postulated cationic mechanism for the cyclization/rearrangement cascade is initiated by the epoxide protonation, ensued by a tetracyclic ring annulation, followed by a series of hydride and methyl groups rearrangement, and culminated by a highly specific deprotonation step. The multiple cyclization/rearrangement pathways, catalyzed by oxidosqualene cyclases, represent one of the most remarkable and fascinating biotransformations found
in nature and have fascinated both chemists and biochemists for more than half a century to study the structure-function-mechanism relationships of the enzyme as well as to design enzyme inhibitors for antifungal, hypocholesterolemic, and phytotoxic drugs applications.
Recent structure-reactivity studies of ERG7, via X-ray crystallographic analyses coupled with bioorganic and mutational data, have provided deeper insight into the catalytic cyclization/rearrangement reaction mechanism, diverse product profiles, and functional role of specific residues of the enzyme. Several critical amino acid residues (Tyr410Cys, Ala464Val, His477Tyr, Ile481Thr, and Tyr532His derived from oxidosqualene-cycloartenol cyclase (CAS) gene of A. thaliana) involved in determining product specificity, especially in the final termination of carbocation between lanosterol and cycloartenol formation, have been identified from a random mutagenesis coupled with plasmid shuffle strategies to change the product specificity. The result suggested that a single amino acid change is sufficient to alter the product specificity in the course of evolution and provided basis for further elucidation of other residues involved in determining substrate recognition, cyclization/rearrangement cascade, or changing product specificity for all cyclases. Three independent regions on ERG7 of S. cerevisiae and twenty-nine non-alanine residues located on the putative active site surface of CAS from A. thaliana have been mutated and assayed for their ability to complement the cyclase-deficient, ERG7 knockout, S.
cerevisiae strain. Critical residues, including
Phe-104-Thr, Trp-232-Arg, Trp-390-Ala, Trp443, Phe445, and Lys448, Phe-510-Arg, Trp-587-Ala, Phe-699-Ala, and Tyr-707-Ala that failed to complement the ERG7 disruption have been identified from the ERG7 counterselection process, indicating that these residues are also crucial for the catalytic function of the enzyme. Further site-saturated mutagenesis and product characterization of both Trp232 and His234 residues revealed their functional roles in the cyclization/rearrangement cascade and identified diverse product profiles. Several truncated cyclization or altered deprotonation products, including
monocyclic achilleol A, (13αH)-isomalabarica-14(26),17E,21-trien-3β-ol and
protosta-20,24-dien-3β-ol, truncated rearranged protosta-12,24-dien-3β-ol, lanosterol, and parkeol were isolated from the ERG7H234X site-saturated mutants. Among them, (13αH)-isomalabarica-14(26),17E,21- trien-3β-ol and protosta-20,24-dien-3β-ol were isolated, for the first time, from the ERG7 mutants. These products correspond to truncation of the cyclization/rearrangement cascade, precisely at two previously proposed and long-sought stages, the C-B 6-6-5 tricyclic cation and protosteryl cation, respectively. The results suggested that the His234 plays a key role in the following processes: stabilizing various carbocationic intermediates, and guiding
deprotonation reactions. In parallel, when Trp232 was subjected to site-saturated mutagenesis and product characterization, the results showed that all mutants, except Lys and Arg, produced protosta-12,24-dien-3β-ol, lanosterol, and parkeol. Overall, Trp232 plays a catalytic role in the influence of rearrangement process and determination of deprotonation position, but not involves intervention in the cyclization steps. The postulated mechanism for
oxidosqualene-lanosterol catalyzed cyclization/rearrangement cascade was shown in
Scheme 1.
Scheme 1. Proposed cyclization/rearrangement
pathways of oxidosqualene in S. cerevisiae TKW14 expressing ERG7H234X site-saturated mutations.
A new approach, SELEX, systematic evolution of ligands by exponential enrichment, which exploited nucleic acid’s shape- or sequence-specific recognition properties as well as the in vitro polymerase chain amplification has greatly contributed to the identification of novel nucleic acid moleculeas for specific bio-molecular interactions with a variety of binding partners, and is now widely applied to screen RNA or DNA oligonucleotides for binding against target small molecule organic dyes, nucleic acid binding proteins, or nucleic acids. With the application of the technologies such as synthesis of random oligonucleotide library, in vitro hybridization between target and library, enrichment of binding sequence via PCR amplification, iteration of selection cycle for binding affinity improvement, the SELEX has been proved to be a powerful technique for the identification of binding sequence.
A peptide based strategy, phage display, homologous to SELEX approach which specifically selects oligopeptides that can specifically recognize and strongly bind to a protein of interest, has become
another new tool for molecular medicine search. The selected peptide sequence was named peptide aptamer. The advantage of the peptide aptamers is that the selected sequences have the potential to dominantly interfere with specific activity of their target proteins and, therefore, could be used as in vivo inhibitors. In addition, peptide aptamers can also provide a basis for the development of novel diagnostic and therapeutic strategies, with implications for a broad variety of different disease entities, including metabolic disorders, infections, and cancer. For the diagnostic purposes, peptide aptamers could be employed as antibody-like molecules for the detection of the target, such as in western blot analyses. Following the immobilization of the target protein on a membrane, the peptide aptamer which specifically interact with the target protein could be visualized with secondary antibodies against the scaffold or against a tag sequence fused to the aptamer, which will become an alternative to the development of monoclonal antibodies. In the period of the research, we applied both the SELEX and phage display strategies to screen potential oligonucleotide and peptide aptamers that can selectively bind to the OSC.
三、結果與討論
Synthesis of Random Oligonucleotide Library
A pool of 72-mer oligonucleotides which contain a degenerate 36-nucleotide sequence flanked by two 18-nucleotide regions was synthesized using the solid-phase phosphoramidite chemistry. The nucleotide sequences for the flanking regions are as follow: 5’-CGTACGGTCGACGCTAGC-3’ and 5’-biotin-GGATCCGAGCTCCACGTG -3’. The synthetic DNA was purified by PAGE and amplified by PCR to generate DNA library. The resulting biotinylated double stranded DNA was applied to an avidin-agarose column pre-equilibrated with buffer A. After the binding of the biotinylated DNA to the avidin beads, the unbound DNA was washed with five times of buffer A. Matrix-bound double strand DNA denatured with NaOH and the free DNA was precipitated. Following centrifugation, the DNA library was re-dissolved in the selection buffer and stored at -20 ℃ for selection.
Proteins Immobilized and Screening of Oligonucleotide Aptamers on Microtiter Plate
To perform the aptamer selection, the ssDNA library was “panned” against protein immobilized on the surface of a microtiter plate. Different concentration of OSC was loaded onto the surface of the microtiter plate and incubated overnight at room temperature. After overnight incubation, linkers that are not derivatized with OSC were blocked with bovine serum albumin and incubated at 37 ℃ for 3 hr, followed by thrice washes with KPi/Tween 20. DNA library was first denatured at 95 ℃ for 5 min
and then placed on ice for 5 min. The ssDNA pool was added to a BSA-only well and then incubated at room temperature for 1 hr. Next, the solution was transferred to the “OSC target” well and incubated for 1 hr before washed thrice with selection buffer. The bound aptamer was eluted with guanidine thiocyanate after the plate was heated at 80 C for 15 min and precipitated. The eluted DNA was PCR amplified and subjected for next round of selection. After 3 to 5 rounds of selection the recovered PCR products were visualized by gel electrophoresis and compared to the ones obtained from the control wells.
Filter Binding Selection of Oligonucleotide Aptamers
To ensure the most accessible folding or stable structure of DNAs, samples were pre-thermally equilibrated and then passed through filters to remove species not bound to the filters. Bound DNAs were extracted from the filters by 7M urea and heat incubation. Following the incubation of DNA with OSC-bound filter, proteinase K was added to digest the protein portion. The digested protein was extracted with phenol/chloroform and the DNA portion was precipitated with NaOAc/Ethanol solution. The collected DNA was PCR amplified and subjected for next round of selection. Again, after 3 to 5 rounds of selection the recovered PCR products were visualized by gel electrophoresis and compared to the ones obtained from the control wells. Results from measurement of the amount of radioactively labeled DNA showed that only the DNA interact with the OSC was observed from filter binding assay. DNA did not interact with BSA as shown in Figure 1.
Figure 1. Filter binding assay of DNA with various concentrations of OSC ranging from 0.25, 0.5, 1.0, 2.0 to 4.0 µmol.
Electrophoretic Mobility Shift Assay (EMSA) of OSC-Binding Aptamers
Different concentrations of unlabeled aptamers were incubated with the OSC for 1 hour prior to the addition of the labeled probe. The reaction were incubated at 37 ℃ for 30 min and then fractioned using non-denatured polyacrylamide gel. The reactions either without OSC or without unlabeled aptamers, were performed as the positive or negative control experiment. The retardant radioisotope bands were clearly observed only from reaction without
addition of unlabeled probe. These retardant bands were blurred or even disappeared with addition of unlabeled probe. The results showed concentration dependence manner, indicating that the unlabeled probe can compete the binding site for the aptamer. Without OSC the retardant radioisotope bands were clearly appeared even in the presence of unlabeled probe, indicating the specificity of aptamer DNA. (Figure2).
Figure 2. Unlabeled probe competition
experiment: (1) Standard EMSA reaction addition of 20X unlabeled probe without OSC. (2) Standard EMSA reaction without OSC. (3) Standard EMSA reaction addition of 20X unlabeled probe. (4) Standard EMSA reaction addition of 10X unlabeled probe. (5) Standard EMSA reaction addition of 5X unlabeled probe. (6) Standard EMSA reaction addition of 1X unlabeled probe. (7, 8) Standard EMSA reaction.
DNA Sequencing
DNA sequencing of SELEX plasmids isolated through the OSC-bound targets was carried out by the dideoxy chain-termination method using ABI PRISM BigDye Terminator Cycle Sequencing Reaction kits on an Applied Biosystems 3100 DNA Sequencer. After several rounds of selection, 9 aptamers which showed binding affinity to OSC target were selected, cloned, and sequenced. Following analysis of the sequences using the NCBI Blast program, the results were shown in Figure 3.
Figure 3. Sequence alignment analysis of the
Oligonucleotide aptamer from cycle 6.
Biopanning Screening of OSC-Binding Peptide
Prepare a solution of 100 µg/mL of OSC in 5 mM
KPi (add 1 mM DTT and 0.2 %Trition X-100)(pH 7.4). Add 150 µL of OSC solution to each microtiter well. Store plates at 4°C overnight. At the time, inoculate 10 mL LB medium with ER2738 at 37 ℃. Next day pour off the coating solution form each well and washing each well rapidly 6X with TBST (TBS + 0.1% [v/v] Tween-20). Fill each well completely with blocking buffer. Incubate at least 1 hour at 4°C. Discaed the blocking solution and wash each well rapidly 6X with TBST buffer. Dilute 4 x 1010 phage (10 µL of original library) with 100 µL of TBST. Pipet onto coated plate and rock gently for 10–60 minutes at room temperature. Discard nonbinding phage by pouring off and slapping plate face-down onto a clean paper towel. Wash plates 20 times with TBST. Elute bound phage with 200 µL of an elution buffer and rock gently for 10 minutes at room temperature. Pipet eluate into a microcentrifuge tube of 15 mL 1 M Tris-HCl (pH 9.1). Titer a small amount (~1 µL) of the eluate.
ELISA Assay of OSC-Binding Peptide
For each clone to be characterized, inoculate 20 mL of LB medium with ER2738 and incubate at 37°C until slightly turbid. Alternatively, dilute an overnight culture of ER2738 1:100 in 20 mL LB. Add 5 µL of phage to each culture and incubate at 37°C with vigorous aeration for 4.5 hours. Transfer the culture to a centrifuge tube and spin 10 minutes at 10,000 rpm. Transfer supernatant to a fresh tube and re-spin. Pipet the upper 80% of the supernatant to a fresh tube and add 1/6 volume of PEG/NaCl. Allow phage to precipitate at 4°C for overnight. Spin PEG precipitation 15 minutes at 10,000 rpm at 4°C. Decant supernatant, re-spin briefly, and remove residual supernatant with a pipette. Suspend the pellet in 1 mL TBS. Transfer the suspension to a microcentrifuge tube and spin for 5 minutes at 4°C to pellet residual cells. Transfer the supernatant to a fresh microcentrifuge tube and re-precipitate with 1/6 volume of PEG/NaCl. Incubate on ice 60 minutes. Microcentrifuge for 10 minutes at 4°C. Discard supernatant, re-spin briefly, and remove residual supernatant with a micropipet. Suspend the pellet in 50 µL TBS. Titer the sample.
Coat one row of ELISA plate wells for each clone to be characterized with 100–200 µL of 100 µg/mL of OSC in 5 mM KPi buffer. Incubate at 4°C overnight. Shake out excess target solution and wash 6X with TBST ( TBS+0.1% tween-20 ). Fill each well completely with blocking buffer. Additionally, one row of uncoated wells per clone to be characterized should also be blocked in order to test for binding of each selected sequence to BSA-coated plastic. Incubate the blocked plates at 4°C, 1 hours. Shake out the blocking buffer and wash each plate 6 times with 1X TBST, slapping the plate face-down onto a clean section of paper towel each time. Carry out ten serial dilutions of the phage in 200 µL of TBS/Tween per well, starting with 1012 virions in the first well of a
0 0.5 1 1.5 2 2.5 posit ive 21 e7 a8 a2 a5 e6 e5 a6 sample A405
row and ending with 105 virions in the well. Incubate at room temperature for 1 hours with agitation. Wash plate 6 times with 1X TBST. Dilute HRP-conjugated anti-M13 antibody 1:5,000 in blocking buffer. Add 200 µL of diluted conjugate to each well and incubate at room temperature for 1 hour. Wash 6 times with 1X TBST. Add 1 µL 30% H2O2 to 10 mL of ABTS stock solution per plate to be analyzed. Add 200 µL substrate solution to each well, incubate at room temperature for 10–60 minutes. Read plates using a microplate reader set at 405 nm.
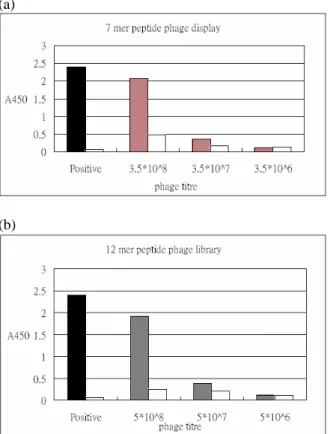
(a)
(b)
Figure 4. Biopanning results of (a) 7-mer peptide
phage library and (b) 12-mer peptide phage library
Figure 5. OSC-binding peptide sequences
Figure 6. Western blotting results of selected
peptide aptamers.
Figure 7. ELISA determination of OSC-binding
aptamer.ELISA 方法測定單一噬菌體對氧化鯊烯環 化酵素親和性結果。▓代表正向控制組,▧代表噬 菌體對氧化鯊烯環化酵素的反應,░ 代表負向控制 組。
四、計劃成果自評
Both the oligonucleotide and peptide aptamers that can bind to OSC have been selected from the corresponding libraries. Following the microtiter plate binding assay and electrophoretic mobility shift assay, we have successfully selected 15 oligonucleotide aptamers from SELEX cycle 6 for further binding constant determination. For the peptide aptamers selection, several aptamers were selected and showed interactions with OSC, as demonstrated by dot blotting and ELISA determination. The obtained results will be further evaluated by western blotting and BiaCore for association/disassociation constant determination. Further experiments on OSC affinity binding and specificity will be examined using the monoclonal antibody generated from the OSC enzyme. These experiments are now in progress. Therefore, the
research results open new avenue for future development of DNA or peptide-based antifungal and hypocholesteremic drug for therapeutical purposes. 五、參考文獻
1. Abe, I.; Rohmer, M.; Prestwich, G. D. Chem.
Rev. 1993, 93, 2189-2206.
2. Barth, M. M.; Binet, J. L.; Thomas, D. M.; de Fornel, D. C.; Samreth, S.; Schuber, F. J.; Renaut, P. P. J. Med. Chem. 1996, 39, 2302-2312.
3. Bacher, J. M., and Ellington, A. D. (1998) DDT
3, 265-273.
4. Bock, L. C., Griffin, L. C., Latham, J. A., Vermaas, E. H., and Toole, J. J. (1992) Nature
355, 564-566.
5. Brody, E. N., and Gold, L. (2000) Reviews Mol.
Biotechnol. 74, 5-13.
6. Buntel, C. J.; Griffin, J. H. J. Am. Chem. Soc. 1992, 114, 9711-9713.
7. Colin Cox, J., and Ellington, A. D. (2001)
Bioorg. & Med. Chem. 9, 2525-2531.
8. Corey, E. J.; Matsuda, S. P.; Bartel, B. Proc.
Natl. Acad. Sci.U.S.A. 1993, 90, 11628-11632.
9. Gold, L., Brown, D., He, Y.-Y., Shtatland, T., Singer, B. S., and Wu, Y. (1997) Proc. Natl.
Acad. Sci. USA 94, 59-64.
10. Golden, M. C., Collins, B. D., Willis, M. C., and Koch, T. H. (2000) J. Biotechnol. 81, 167-178.
11. Goldman, R. C.; Zakula, D.; Capobianco, J. O.; Sharpe, B. A.; Griffin, J. H. Antimicrob. Agents
Chemother. 1996, 40, 1044-1047.
12. Hart, E. A.; Hua, L.; Darr, L. B.; Wilson, W. K.; Pang, J.; Matsuda, S. P. T. J. Am. Chem. Soc. 1999, 121, 9887-9888.
13. Hermann, T., and Dinshaw, J. P. (2000)
Science 287, 820-825.
14. Hesselberth, J., Robertson, M. P., Jhaveri, S., and Ellington, A. D. (2000) Reviews Mol.
Biotechnol. 74, 15-25.
15. Hoppe-Seyler, F., Crnkovic-Mertens, I., Denk, C., Fitscher, B. A., Klevenz, B., Tomai, E., and Butz, K. (2001) J. Steroids Biochem. & Mol.
Biol. 78, 105-111.
16. Iqbal, S. S., Mayo, M. W., Bruno, J. G., Bronk, B. V., Batt, C. A., and Chambers, J. P. (2000)
Biosensors & Bioelectronics 15, 549-578.
17. James, W. (2001) Current Opinion in
Pharmacology 1, 540-546.
18. Jayasena, S. D. (1999) Clinical Chem. 45, 1628-1650.
19. Kushiro, T.; Shibuya, M.; Ebizuka, Y. J. Am.
Chem. Soc. 1999, 121, 1208-1216.
20. Kusser, W. (2000) Reviews Mol. Biotechnol. 74, 27-38.
21. Mark, M.; Muller, P.; Maier, R.; Eisele, B. J.
Lipid Res. 1996, 37, 148-158.
22. Meisenheimer, K. M., Meisenheimer, P. L., Willis, M. C., and Koch, T. H. (1996) Nucleic
Acids Res. 24, 981-982.
23. Meisenheimer, K. M., and Koch, T. H. (1997)
Critical Reviews Biochem. Mol. Biol. 32,
101-140.
24. Morris, K. N., Jensen, K. B., Julin, C. M., Weil, M., and Gold, L. (1998) Proc. Natl. Acad. Sci.
USA 95, 2902-2907.
25. Norris, C. L., Meisenheimer, P. L., and Koch, T. H. (1996) J. Am. Chem. Soc. 118, 5796-5803. 26. Willis, M. C., LeCuyer, K. A., Meisenheimer,
K. M., Uhlenbeck, O. C., and Koch, T. H. (1994) Nucleic Acids Res. 22, 4947-4952. 27. Wu, T. K., Liu, Y. –T., Chang, C. –H., Yu,
M. –T., Wang, H. –J. 2006, J. Am. Chem. Soc. 128, 6414-6419.
28. Wu, T. K., Yu, M. –T., Liu, Y. –T., Chang, C. –H., Wang, H. –J., and Diau, E. W. –G. 2006, Org. Lett. 8, 1319-1322.
29. Wu, T. K., Liu, Y. –T., and Chang, C. –H. 2005, ChemBioChem 6, 1177-1181.
30. Wu, T. K. and Chang, C. –H. 2004,
ChemBioChem 5, 1712-1715.
31. Wu, T. K., Huang, C.-Y., Ko, C.-Y., Chang, C.-H., Chen, Y.-J., and Liao, H.-K., 2004, Arch.
Biochem. Biophys. 421, 42-53.
32. Wu, T. K., and Griffin, J. H. 2002,