Organometallic ferrocenyl dendrimers: synthesis, characterization
and redox properties
Ching-Fong Shu* and Hsiu-Ming Shen
Department of Applied Chemistry, National Chiao T ung University, 1001 T a-Hsueh Road, Hsin-Chu, T aiwan, 30035, Republic of China
A series of dendritic poly(aryl ether)s containing 3, 6, 12 and 24 ferrocene functionalities located exclusively at the peripheries of the dendritic structures have been synthesized using the stepwise convergent approach. The structures of these dendrimers have
been characterized using1H and 13C NMR spectroscopy. Cyclic and normal pulse voltammetric studies indicate that the
ferrocenyl moieties located on the outer surfaces of these dendrimers are non-interacting redox centres, are electrochemically equivalent, and are oxidizable at the same potential. The results of controlled-potential coulometric oxidation show that nearly all the ferrocene residues in the dendrimers are accessible to electron transfers in electrode reactions.
Macromolecular materials with skeletons of transition-metal reported procedures. Other commercially available chemicals
were reagent grade and were used as purchased. atoms in close proximity are attracting increasing attention
because of their potentially interesting electrical, redox and
3-Bromopropylferrocene ( Fc-G0-Br)11
optical characteristics. Among the organotransition-metal complexes, ferrocene has been shown to have excellent thermal
To a mixture of 3-ferrocenylpropanol ( 5.43 g, 22.3 mmol ) and and photochemical stability, and to undergo a facile and
carbon tetrabromide (14.63 g, 44.6 mmol ) in 10 ml of THF reversible one-electron oxidation to ferrocenium cation; the
was added triphenylphosphine (11.53 g, 44 mmol ). The reac-reaction leads to a marked change in electrical and
spectro-tion mixture was stirred at room temperature under nitrogen scopic properties and may be effected chemically,
electrochemi-for 1 h. The resulting mixture was then poured into water and cally or photochemically. Ferrocenyl-based polymers have been
extracted with diethyl ether. The extract was dried over Na
2SO4
used in chemical modification of electrodes, in the construction
and evaporated to dryness. The crude product was purified by of amperometric biosensors, and in the area of non-linear
column chromatography (SiO
2), eluting with 1510 ethyl
acet-optical materials.1
ate-hexane, to give 4.67 g (69%) of Fc-G0-Br as an orange oil. Recently, dendritic macromolecules have received
consid-1H NMR (CDCl3)d 2.04 (m, 2H, CH2), 2.50 (t, 2H, J=7.8 Hz,
erable attention because of their unique hyperbranched
poly-CpCH2), 3.42 (t, 2H, J=6.3 Hz, CH2O), 4.07 (m, 4H, C5H4), meric structure and well defined three-dimensional
architec-4.10 (s, 5H, C5H5); 13C NMR (CDCl3) d 27.91 (CH2), 33.63 tures.2 These dendritic macromolecules are characterized by a
(CH2Br), 33.91 (CpCH2), 67.36, 6814 (C5H4), 68.57 (C5H5), central polyfunctional core, from which arise successive layers
88.25 (C5H4). of monomer units with branches occurring at each monomer
unit.3 This results in a nearly entanglement-free hyperbranched General procedure for the synthesis of Fc-Gx-OH (x=1, 2, 3)
structure that may adopt a spherical shape, the periphery of
A mixture of the appropriate Fc-Gx-Br (x=0, 1, 2) (2 equiv.),
which consists of a large number of chain ends or surface
3,5-dihydroxybenzyl alcohol (1 equiv.), potassium carbonate, functional groups.4 This unique macromolecular architecture
(3.5 equiv.) and 18-crown-6 (0.2 equiv.) in dry acetone was allows precise control of molecule size, as well as of the
heated at reflux and stirred vigorously under nitrogen for 48 h. disposition of the desired functionalities. The synthesis of well
The mixture was allowed to cool, added to water and extracted defined, highly branched macromolecules possessing functional
with CH2Cl2 (3×). The combined extracts were dried over components on their exterior surfaces, within their dendritic
MgSO4 and evaporated to dryness. The crude product was branches, and at their interior cores, giving rise to new
purified as outlined in the following text. materials with desirable properties has sparked much
inter-est.4–6 In this paper, we report full details on the synthesis,
General procedure for the synthesis of Fc-Gx-Br (x=1, 2, 3)12
characterizations and redox behaviour of a family of dendritic
poly(aryl ether) s that were synthesized using a stepwise conver- To a solution of Fc-Gx-OH (x= 1, 2, 3) (1 equiv.) in benzene
gent approach,7 and possess ferrocene functional groups
was added dropwise PBr3 (0.4 equiv.). The reaction mixture located exclusively at the peripheries of their dendritic
was stirred at room temperature under nitrogen for 4 h. For structures.
the latter generation (x=3), larger excesses of PBr
3 were
required to force the reaction to completion. PBr
3was added
in 0.4 equiv. amounts at hourly intervals until TLC showed
Experimental
no starting material. The mixture was then added to water,
Materials neutralized with saturated aqueous NaHCO
3, and extracted
with CH2Cl2 (3×). The combined extracts were dried over
Acetone was distilled from CaSO4. Benzene was distilled from MgSO4 and evaporated to dryness. The crude product was
CaH2. Tetrahydrofuran (THF) was distilled from Na/K alloy purified as outlined in the following text.
and benzophenone. n-Tetrabutylammonium
hexafluorophos-phate (TBAPF6) was prepared as described previously,8 and Fc-G1-OH
was purified by recrystallization three times from ethyl acetate
and dried in vacuo at 60°C. 3-Ferrocenylpropanol9 and 3,5- This was prepared from Fc-G0-Br and purified by column
chromatography (SiO2), eluting with 255 ethyl acetate–hexane, dihydroxybenzyl alcohol10 were synthesized according to the
to give Fc-G1-OH (79%) as an orange solid.1H NMR (CDCl3) to give Fc-G3-Br (24%) as an orange solid.1H NMR (CDCl3) d 1.98 (m, 16H, CH2), 2.51 (t, 16H, J=7.8 Hz, CpCH2), 3.96 d 1.98 (m, 4H, CH2), 2.51 (t, 4H, J=7.8 Hz, CpCH2), 3.96 (t, 4H, J=6.3 Hz, CH 2O), 4.06 (m, 4H, C5H4), 4.08 (m, 4H, C5H4), C(t, 16H, J=6.3 Hz, CH2O), 4.07 (m, 16H, C5H4), 4.09 (m, 16H, 5H4), 4.11 (s, 40H, C5H5), 4.40 (s, 2H, CH2Br), 4.95 (s, 12H, 4.11 (s, 10H, C 5H5), 4.62 (d, 2H, J=4.8 Hz, CH2OH), 6.40 (t,
1H, J=2.0 Hz, ArH), 6.52 (d, 2H, J=2.0 Hz, ArH); 13C NMR ArCH
2O), 6.42 (t, 4H, J=2.0 Hz, ArH), 6.54 (m, 3H, ArH),
6.57 (d, 8H, J=2.0 Hz, ArH), 6.60 (d, 2H, J=2.0 Hz, ArH), (CDCl 3)d 25.89 (CH2), 30.50 (CpCH2), 65.37, 67.27 (CH2O), 67.27, 68.14 (C 5H4), 68.55 (C5H5), 88.19 (C5H4), 100.57, 105.12, 6.68 (d, 4H, J(CH =2.0 Hz, ArH); 13C NMR (CDCl3) d 25.89 2), 30.47 (CpCH2), 33.59 (CH2Br), 67.27 (C5H4), 67.33, 143.26, 160.41 (ArC). 67.71, (CH2O), 67.74 (C5H4), 68.55 (C5H5), 70.10 (CH2O), Fc-G1-Br 88.25 (C5H4), 100.86, 101.62, 102.15, 105.75, 106.40, 108.12, 138.97, 159.92, 160.10, 160.36 (ArC). This was prepared from Fc-G1-OH and purified by column
chromatography (SiO2), eluting with 155 ethyl acetate–hexane General procedure for the synthesis of dendritic molecules
to give Fc-G1-Br ( 85%) as an orange solid.1H NMR (CDCl3)
d 1.98 (m, 4H, CH2), 2.51 (t, 4H, J=7.8 Hz, CpCH2), 3.95 (t, A mixture of Fc-Gx-Br (x=0, 1, 2, 3) (3 equiv.),
1,1,1-tris(4-4H, J=6.3 Hz, CH
2O), 4.06 (m, 4H, C5H4), 4.06 (m, 4H, C5H4), hydroxyphenyl)ethane (1 equiv.), potassium carbonate (4.5
4.11 (s, 10H, C
5H5), 4.41 (s, 2H, J=4.8 Hz, CH2Br), 6.39 (t, equiv.), and 18-crown-6 (0.3 equiv.) in acetone was heated at
1H, J=2.0 Hz, ArH), 6.52 (d, 2H, J=2.0 Hz, ArH); 13C NMR reflux and stirred vigorously under nitrogen for 72 h. The
(CDCl
3)d 25.89 (CH2), 30.44 (CpCH2), 33.76 (CH2Br), 67.27 mixture was allowed to cool, added to water and extracted
(CH
2O), 67.21, 68.09 (C5H4), 68.49 (C5H5), 99.10 (C5H4), with CH2Cl2 (3×). The combined extracts were dried over
101.42, 139.59, 160.27 (ArC). MgSO
4 and evaporated to dryness. The crude product was
purified as outlined in the following text. Fc-G2-OH
Dendrimer 1. This was prepared from Fc-G0-Br and purified This was prepared from Fc-G1-Br and purified by column
by column chromatography (SiO2), eluting with 154 ethyl
chromatography (SiO2), eluting with 153 hexane–chloroform acetate–hexane, to give dendrimer 1 (58%) as an orange solid.
to give Fc-G2-OH (85%) as an orange solid.1H NMR (CDCl3)
1H NMR (CDCl3) d 1.98 (m, 6H, CH2), 2.11 (s, 3H, CH3), d 1.98 (m, 8H, CH2), 2.51 (t, 8H, J=7.8 Hz, CpCH2), 3.96 (t, 2.51 (t, 6H, J=7.8 Hz, CpCH 2), 3.95 (t, 6H, J=6.3 Hz, CH2O), 8H, J=6.3 Hz, CH 2O), 4.05 (m, 8H, C5H5), 4.08 (m, 8H, C5H4), 4.05 (m, 6H, C5H4), 4.08 (m, 6H, C5H4), 6.79 (d, 6H, J=8.7 Hz, 4.11 (s, 20H, C
5H5), 4.62 (d, 2H, J=4.8 Hz, CH2OH), 4.96 (s, core Ar∞H), 6.99 (d, 6H, J=8.71 Hz, core Ar∞H); 13C NMR
4H, ArCH 2O), 6.42 (t, 2H, J=2.0 Hz, ArH), 6.54 (t, 1H, J= (CDCl 3) d 25.92 (CH2), 30.50 (CpCH2), 30.73 (CH3), 50.54 2 Hz, ArH), 6.57 (d, 4H, J=2.0 Hz, ArH), 6.61 (d, 2H, J= (CCH 3), 67.15 (CH2O), 67.17, 68.03, (C5H4), 68.46 (C5H5), 2.0 Hz, ArH); 13C NMR (CDCl3) d 25.89 (CH 2), 30.50 88.25 (C 5H5), 113.60, 129.56, 141.71, 156.92 (Ar∞C). (CpCH 2), 65.27 (CH2O), 67.23 (C5H4), 67.41 (CH2O), 68.41
(C5H4), 68.52 (C5H5), 70.03 (CH2O), 88.20 (C5H4), 100.80, Dendrimer 2. This was prepared from Fc-G1-Br and purified
101.27, 105.65, 105.73, 139.03, 143.37, 160.08, 160.36 (ArC).
by column chromatography (SiO
2), eluting with 151
chloro-Fc-G2-Br form–hexane to give dendrimer 2 ( 57%) as an orange solid.
1H NMR (CDCl3)d 1.98 (m, 12H, CH2), 2.11 (s, 3H, CH3),
This was prepared from Fc-G2-OH and purified by column
2.51 (t, 12H, J=7.8 Hz, CpCH
2), 3.95 (t, 12H, J=6.3 Hz,
chromatography (SiO
2), eluting with 253 hexane–chloroform CH2O), 4.05 (m, 12H, C5H4), 4.08 (m, 12H, C5H4), 4.95 (s, 6H,
to give Fc-G2-Br ( 44%) as an orange solid.1H NMR (CDCl3)
CH2O), 6.42 (t, 3H, J=2.0 Hz, ArH), 6.58 (d, 6H, J=2.0 Hz, d 1.98 (m, 8H, CH
2), 2.51 (t, 8H, J=7.8 Hz, CpCH2), 3.96 (t, ArH), 6.87 (d, 6H, J=8.7 Hz, core Ar∞H), 7.00 (d, 6H, J=
8H, J=6.3 Hz, CH 2O), 4.07 (m, 8H, C5H4), 4.09 (m, 8H, C5H4), 8.7 Hz, core Ar∞H); 13C NMR (CDCl 3)d 25.89 (CH2), 30.47 4.11 (s, 20H, C 5H5), 4.42 (s, 2H, CH2Br), 4.95 (s, 4H, ArCH2O), (CpCH 2), 30.76 (CH3), 50.60 (CCH3), 65.37 (CH2O), 67.21 6.43 (t, 2H, J=2.0 Hz, ArH), 6.55 (t, 1H, J=2.0 Hz, ArH), (C 5H4), 67.30 (CH2O), 68.08 (C5H4), 68.49 (C5H5), 70.01 6.57 (d, 2H, J=2.0 Hz, ArH), 6.64 (d, 2H, J=2.0 Hz, ArH); (CH 2O), 88.16 (C5H4), 100.77, 105.76, 113.95, 129.59, 139.35, 13C NMR (CDCl3) d 25.89 (CH2), 30.47 (CpCH2), 33.65 142.00, 156.80, 160.36 (Ar and Ar∞C). (CH 2Br), 67.27 (C5H4), 67.33 (CH2O), 68.14 (C5H4), 68.58 (C
5H5), 70.12 (CH2O), 88.25 (C5H4), 100.89, 102.17, 105.78, Dendrimer 3. This was prepared from Fc-G2-Br and purified
108.09, 138.80, 139.70, 159.98, 160.39 (ArC).
by column chromatography (SiO2), eluting with 152 chloro-form–hexane to give dendrimer 3 ( 59%) as an orange solid. Fc-G3-OH
1H NMR (CDCl3)d 1.97 (m, 24H, CH2), 2.11 (s, 3H, CH3),
This was prepared from Fc-G2-Br and purified by column 2.50 (t, 24H, J=8.1 Hz, CpCH
2), 3.95 (t, 24H, J=6.3 Hz,
chromatography (SiO
2), eluting with 153 hexane–chloroform, CH2O), 4.05 (m, 24H, C5H4), 4.08 (m, 24H, C5H4), 4.10 (s,
to give Fc-G3-OH (63%) as an orange solid.1H NMR (CDCl3)
60H, C5H5), 4.95 (s, 18H, CH2O), 6.42 (t, 6H, J=2.0 Hz, ArH),
d 1.97 (m, 16H, CH2), 2.51 (t, 16H, J=7.8 Hz, CpCH2), 3.95 6.57 (m, 15H, ArH), 6.68 (d, 6H, J=2.0 Hz, ArH), 6.87 (d, 6H,
(t, 16H, J=6.3 Hz, CH2O), 4.05 (m, 16H, C5H4), 4.08 (m, 16H, J=8.7 Hz, core Ar∞H), 7.02 (d, 6H, J=8.7 Hz, core Ar∞H);
C5H4), 4.10 (s, 40H, C5H5), 4.61 (d, 2H, J=4.8 Hz, CH2OH), 13C NMR (CDCl3) d 25.84 (CH
2), 30.44 (CpCH2), 50.72
4.96 (s, 8H, ArCH
2O), 4.97 (s, 4H, ArCH2O), 6.42 (t, 4H, J= (CCH
3), 67.18 (C5H4), 67.24 (CH2O), 68.06 (C5H4), 68.46
2.0 Hz, ArH), 6.54 (m, 3H, ArH), 6.57 (d, 8H, J=2.0 Hz, ArH), (C
5H5), 69.83, 70.01 (CH2O), 88.10 (C5H4), 100.81, 101.45,
6.60 (d, 2H, J=2.0 Hz, ArH), 6.68 (d, 4H, J=2.0 Hz, ArH); 105.73, 106.37, 113.89, 129.57, 139.38, 141.98, 156.69, 160.01,
13C NMR (CDCl3) d 25.89 (CH2), 30.47 (CpCH2), 65.23 160.30 (Ar and Ar∞C).
(CH2O), 67.27 (C5H4), 67.33 (CH2O), 67.74 (C5H4), 68.55
(C5H5), 69.95, 70.10 (CH2O), 88.25 (C5H5), 100.86, 101.21, Dendrimer 4. This was prepared from Fc-G3-Br and purified
101.56, 105.67, 105.79, 106.31, 138.97, 139.18, 143.37, 160.01,
by column chromatography (SiO2), eluting with 152 chloro-160.07, 160.36 (ArC).
form–hexane to give dendrimer 3 ( 41%) as an orange solid.
1H NMR (CDCl3)d 1.96 (m, 48H, CH2), 2.09 (s, 3H, CH3),
Fc-G3-Br
2.49 (m, 48H, CpCH2), 3.93 (t, 48H, CH2O), 4.05 (m, 96H, C5H4), 4.10 (s, 120H, C5H5), 4.94 (s, 36H, CH2O), 6.41 (m, This was prepared from Fc-G3-OH and purified by column
chromatography (SiO2), eluting with 253 hexane–chloroform 12H, ArH), 6.57 (m, 33H, ArH), 6.68 (d, 18H, J=2.0 Hz, ArH),
J. Mater. Chem., 1997, 7(1 ), 47–52
6.87 (d, 6H, J=8.7 Hz, core Ar∞H), 7.01 (d, 6H, J=8.7 Hz, rocene. The bromide Fc-G0-Br was obtained by bromination of 3-ferrocenylpropanol, which was prepared according to the
core Ar∞H); 13C NMR (CDCl
3)d 25.89 (CH2), 30.47 (CpCH2),
67.21 (C
5H4), 67.33 (CH2O), 67.71 (CH2O), 68.08 (C5H4), 68.28 literature procedures.restored the reactive bromomethyl functionality at the focal9 Bromination of Fc-G1-OH with PBr3
(CH
2O), 68.49 (C5H5), 70.10 (CH2O), 100.89, 105.79, 106.40,
113.92, 138.94, 160.10, 160.36, 160.48 (Ar and Ar∞C). point of the dendritic wedge to give the corresponding benzyl
bromide Fc-G1-Br. Reaction of Fc-G1-Br with 3,5-dihydroxy-benzyl alcohol resulted in the formation of the next-generation Electrochemical apparatus
benzyl alcohol G2-OH, and subsequent bromination of Fc-Electrochemical measurements were carried out with a BAS
G2-OH with PBr3 gave the second generation bromide Fc-G2-100B/W Electrochemical Workstation. The working electrode
Br. Repeated alkylation and bromination led to the third for cyclic voltammetry and normal pulse voltammetry was a
generation alcohol Fc-G3-OH and the bromide Fc-G3-Br. In platinum disk electrode ( 0.5 mm diameter, sealed in soft glass)
the convergent approach, the dendritic wedges obtained are that was polished prior to use with 1 mm diamond paste and
attached to a polyfunctional core. The polyfunctional core we rinsed thoroughly with water and acetone. For coulometry
chose was 1,1,1-tris (4-hydroxyphenyl) ethane. Coupling reac-a lreac-arge plreac-atinum greac-auze electrode wreac-as employed. All
poten-tions of the phenolic groups of the core molecule with each
tials are referenced to the Ag/Ag+ (0.01 mol dm−3 AgNO3,
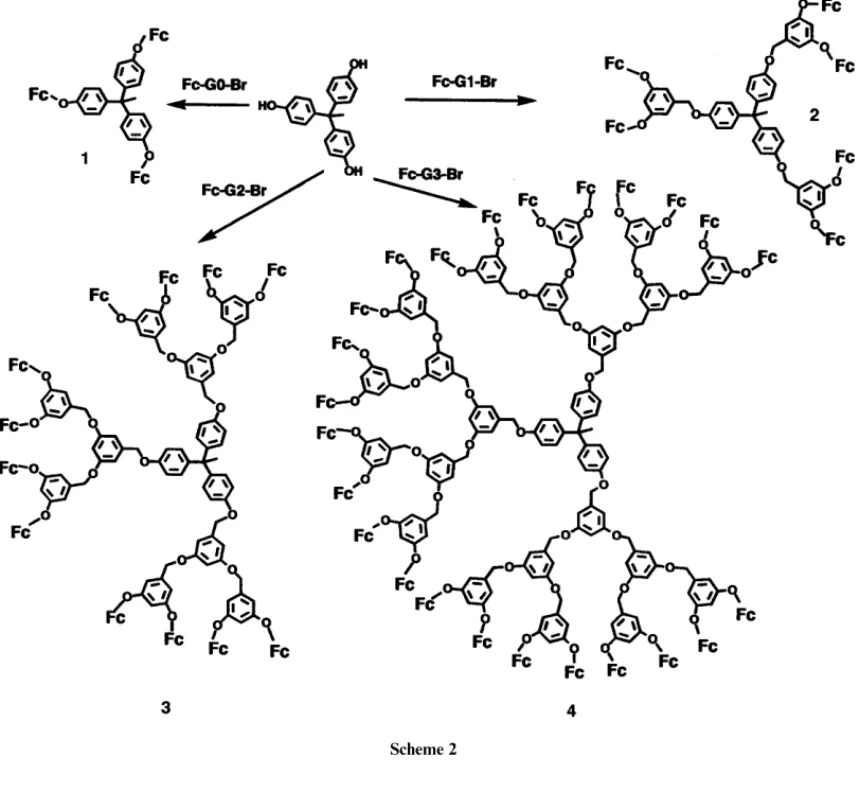
bromide generation, from Fc-G0-Br to Fc-G3-Br, as shown in
0.1 mol dm−3 TBAPF6–CH
3CN) reference electrode. A coiled Scheme 2, were carried out. Thus, a family of dendritic
poly-platinum wire was used as a counter electrode and the
electro-(arylether)s, dendrimers 1, 2, 3 and 4, containing 3, 6, 12 and chemical cells were of conventional design.
24 ferrocene functionalities, respectively, at their peripheries were obtained. The products were purified by column
chroma-tography, and their purities were evaluated by 1H and
Results and Discussion
13C NMR spectroscopy. No signal of the starting materials Synthesis of dendritic macromolecules
was detected in the NMR experiments. A series of poly(aryl ether) dendrimers containing ferrocene
functional groups located at the peripheries of their dendritic
Characterization structures were prepared using the convergent approach
devel-oped by Hawker and Fre´chet.7 The synthetic route to the The ferrocene-terminated dendrimers were characterized
struc-turally using 1H and 13C NMR spectroscopy. 1H NMR
ferrocene-terminated dendritic wedges is illustrated in
Scheme 1. This was made on the base of the Williamson ether spectroscopy is particularly crucial in confirming the structures
produced by our synthetic strategy.1H NMR spectra showed
synthesis for the formation of aryl ether from phenol and
benzylic bromide (or alkyl bromide). We used 3,5-dihydroxy- the exterior functional group gave five sets of resonances at
d 4.10 (C
5H5), 4.08, 4.05 (C5H4), 3.95 (CH2O), 2.50 (CpCH2)
benzyl alcohol as the monomer unit. The ferrocene groups
were introduced to the peripheries of the growing dendrimers and 1.98 (CH
2). This result is consistent with the expected
highly symmetrical structures of the ferrocene dendrimers. The by coupling two molecules of 3-bromopropyl ferrocene
Fc-G0-Br to the phenolic groups of the monomer unit in the resonances for the aromatic protons of the internal
3,5-dihydroxybenzyl group occurred in thed 6.50–6.70 region. All
presence of potassium carbonate and 18-crown-6 in refluxing
acetone, thereby obtaining the first generation alcohol Fc-G1- benzylic protons resonate atd 4.91–5.02, except those at the
focal point at which CH2OH appears at d 4.62 and CH2Br at
OH. We selected c-propylferrocene as the terminal group
because bromomethylferrocene, which is ana-functional ferro- d 4.42. Changes in resonance of benzylic protons occurred
upon conversion of the dendritic alcohol to the corresponding cene derivative, is hydrolytically unstable and readily converted
to ferrocenemethanol, and 2-bromoethylferrocene undergoes bromide, and on attachment of the bromide to the
polyfunc-tional core, were used to monitor product purities. When elimination during the alkylation reaction, producing
vinylfer-Scheme 1 Reagents and conditions: (a) 3,5-dihydroxybenzyl alcohol, K2CO3, 18-crown-6, reflux in acetone; (b) PBr3, C6H6, room temperature
Scheme 2
dendritic wedges were attached to the core molecule 1,1,1-tris(4-hydroxyphenyl )ethane, two doublets of the aromatic
rings of the core moiety were observed atd 6.87 and 7.02, and
were not obscured by the aromatic resonances of the dendritic
wedges; the methyl group of the core resonated at d 2.11.
Integration data for the exterior ferrocene groups, the internal aromatic groups, and the aromatic rings of the core moiety confirmed the structures and the generation numbers of the
dendrimers. The 13C NMR spectra provided further support
for the structural assignments. Each of the different carbon resonances was found to agree with the observed spectral resonances.
Electrochemistry
Electrochemistry studies of the ferrocene dendrimers in CH2Cl2
solutions containing 0.1 mol dm−3 TBAPF6as the supporting
electrolyte were performed. Cyclic voltammograms of 1 and 2 show the characteristics of a reversible one-electron oxidation, with production of soluble, stable cations. However, with 3 and 4, a stripping peak appeared on scan reversal. The shape of the reduction peak indicated that the oxidized products were insoluble and accumulated on the electrode’s surface. The precipitation problems led us to use the normal pulse voltam-metry (NPV) technique, which is less susceptible to problems of adsorption and precipitation as a means of determining the
wave parameters.13 The normal pulse voltammograms of 3
in CH2Cl2 are shown in Fig. 1A. Surprisingly, the slope of E vs. log(id−i)/i plot, where id is the limiting diffusion current, obtained from the normal pulse voltammogram is 42 mV,
Fig. 1 Normal pulse voltammograms for 50 mmol dm−3 of dendrimer
which is different from the slopes ( 60 mV) reported for most
3 in 0.1 mol dm−3 TBAPF6–CH2Cl2. A, Potential scanned in the
polymers (or dendrimers) containing multiple ferrocenyl moiet- forward direction, initial potential 0 mV vs. Ag/Ag+; B, potential
ies.13–15 We speculated that the reason why a value of 42 mV scanned in the reverse direction, initial potential 500 mV vs. Ag/Ag+.
Scan rate 20 mV s−1; sample width 17 ms; pulse width 50 ms.
was obtained instead of 60 mV may be due to severe adsorption
of the oxidized product. The effects of absorbed reactants and technique. Normal pulse voltammograms of the dendrimers are displayed in Fig. 3. The half-wave potentials, limiting products on the plateau currents and half-wave potentials in
normal pulse voltammograms have been reported by Anson diffusion currents (i
d), and slopes of E vs. log(id−i)/i plots,
obtained from the normal pulse voltammograms are given in et al.16 In the absence of an independent determination, it is
difficult to deduce the presence of product adsorption from Table 1. The E
1/2values, as determined by NPV, are similar
to those determined by cyclic voltammetry. Interestingly, the normal pulse voltammograms. However, the appearance of a
peak current in the voltammogram recorded in the reverse- E
1/2 values for the ferrocene redox couples remain essentially
constant upon successive generation buildup. This can be scan direction verifies the occurrence of adsorption. Indeed,
for dendrimer 3 there is a peak current recorded in the reverse- explained by noting that the ferrocene functional groups are
located on the outer surfaces of the dendrimers and their scan voltammogram, as shown in Fig. 1B.
In order to reduce perturbations in the shape of normal microenvironments are independent of dendrimer generation.
This result is in contrast to the recent observation by Diederich pulse voltammograms induced by adsorption, we used a mixed
solvent, 355 CH3CN–CH2Cl2 in the electrochemical studies, et al.17 that for zinc porphyrin dendrimers the redox potential
shifts as generations build up. Because the redox group is where the adsorption of the dendrimers and their oxidized
products was minimized. Fig. 2 shows the cyclic voltammo- located in the interior of the porphyrin macromolecules, the
electrophore environment, as well as the redox potential of the gram of the ferrocene-terminated dendrimers in the mixed
solvent containing 0.1 mol dm−3 TBAPF
6 as the supporting zinc porphyrin, changes markedly with increasing generationalcascading. It was observed that oxidation of the terminated
electrolyte. The cyclic voltammogram of each dendrimer
exhib-its a single reversible oxidation wave corresponding to oxi- ferrocenyl units in the dendrimers occurred at a potential ca.
40 mV more negative than the corresponding process in ferro-dation of the exterior ferrocene groups. The peak current is
linearly proportional to the square root of the scan rate, the cene. This was due to the electron-donating effect of the alkyl
group on the ferrocenyl moiety, making the chain-end ferro-peak shape is symmetrical, with i
pc/ipa#1, and Epis
indepen-dent of the scan rate. The peak potential separation DEp is ca. cenyl groups more easily oxidized. For the dendrimers, the
values of DEp determined from CV and the values of slopes 59 mV for dendrimers 1 and 2, 52 mV for 3 and 50 mV for 4.
The smaller DEp values for dendrimers 3 and 4 may be obtained from NPV are close to the theoretical value of 59 mV
for a reversible one-electron transfer reaction, indicating that attributable to minor adsorption of the dendrimers (or
oxi-dation products) on the electrode surface. The half-wave poten- all the ferrocenyl redox centres located at the peripheries of
tials (E1/2) and DEp obtained from the cyclic voltammograms are given in Table 1. Note that the sharp anodic or cathodic peak current due to the adsorption of reactant or product is not observed in the mixed solvent system. The electrochemical properties of the dendrimers were also studied using the NPV
Fig. 3 Normal pulse voltammograms for the oxidation of A, Fig. 2 Cyclic voltammograms for A, 200 mmol dm−3 of 1; B,
100 mmol dm−3 of 2; C, 50 mmol dm−3 of 3; and D, 25 mmol dm−3 200 mmol dm−3 of 1; B, 100 mmol dm−3 of 2; C, 50 mmol dm−3 of 3; and D, 25 mmol dm−3 of 4 in 0.1 mol dm−3 TBAPF
6–355 CH3CN–CH2Cl2. of 4 in 0.1 mol dm−3 TBAPF
6–355 CH3CN–CH2Cl2. Scan rate
60 mV s−1. Scan rate 20 mV s−1; sample width 17 ms; pulse width 50 ms.
Table 1 Results of cyclic voltammetry, normal pulse voltammetry and controlled-potential electrolysis for the oxidation of ferrocene-terminated dendrimers 1–4a compound E 1/2b/mV E1/2c/mV DEp/mV sloped /mV id/10−1 mA Qce/10−1 C npf 1 211 208 59 60 8.16 2.90 ( 0.03) 3.0 ( 0.1 ) 2 211 208 59 58 7.21 2.95 ( 0.03) 6.1 ( 0.1 ) 3 213 212 52 56 5.90 2.74 ( 0.06) 11.4 ( 0.2 ) 4 214 212 50 58 5.14 2.70 ( 0.05) 22.4 ( 0.4 )
aExperimental conditions are given in the captions to Fig. 2 and 3. bBy cyclic voltammetry. cBy normal pulse voltammetry. dSlope of plot of E vs. log (id−i)/i. eThe electrolysis solutions were 5 ml, and the concentration of each solution was as indicated in the captions to Fig. 2 and 3. Values are an average of three independent determinations, and standard deviations are listed in parentheses. fNumber of ferrocenes per molecule oxidized.
and M. S. Sigman, Macromolecules, 1992, 25, 6055; A. Togni and
these dendrimers are equivalent, non-interacting and exchange
G. Rihs, Organometallics, 1993, 12, 3368.
electrons with the electrode in waves characteristic of
one-2 D. A. Tomalia, A. M. Naylor and W. A. Goddard, Angew. Chem.,
electron transfer processes.
Int. Ed. Engl., 1990, 29, 138; H. Mekelburger, W. I. Jaworke and
To evaluate the total number of electrons transferred during F. Vo¨gtle, Angew. Chem., Int. Ed. Engl., 1992, 31, 1571;
oxidation without knowing the relative diffusion coefficients, J. M. J. Fre´chet, Science, 1994, 263, 1710.
3 D. A. Tomalia, H. Baker, J. R. Dewald, M. Hall, G. Kallos,
controlled-potential electrolyses were carried out. Coulometric
S. Martin, J. Roeck, J. Ryder and P. Smith, Macromolecules, 1986,
oxidation of the dendrimers was performed at a large-area
19, 2466; G. R. Newkome, Z. Yao, G. R. Baker, V. K. Gupta,
platinum gauze electrode in a CH
2Cl2–CH3CN mixture at P. S. Russo and M. J. Saunders, J. Am. Chem. Soc., 1986, 108, 849;
Eapp 150 mV more positive than the respective anodic potential. G. R. Newkome, A. Nayak, R. K. Behara, C. N. Moorefield and
The results are given in Table 1. It is shown that values of np, G. R. Baker, J. Org. Chem., 1992, 57, 358; H. Uchida, Y. Kabe,
K. Yoshino, A. Kawamata, T. Tsumuraya and S. Masamune,
the number of electrons per molecule consumed in the
oxi-J. Am. Chem. Soc., 1990, 112, 7077.
dation, calculated from the total number of coulombs, come
4 J. L. Fillaut and D. Astruc, J. Chem. Soc., Chem. Commun., 1993,
close to matching the number of ferrocenyl groups anticipated
1320; F. Moulines, L. Djakovitch, R. Boese, B. Gloaguen, W. Thiel,
in the synthetic strategy bound to the chain-ends of these J. L. Fillaut, M. H. Delville and D. Astruc, Angew. Chem., Int. Ed.
dendrimers. Note that the values of np obtained for 3 and 4 Engl., 1993, 32, 1075; Y-H. Liao and J. R. Moss, J. Chem. Soc.,
Chem. Commun., 1993, 1774; B. Alonso, I. Cuadrado, M. Moran
are somewhat smaller than the number of ferrocene groups
and J. Losada, J. Chem. Soc., Chem. Commun., 1994, 2575;
Y-attached to these dendrimers. This may have been because the
H. Liao and J. R. Moss, Organometallics, 1995, 14, 2130; K. Lorenz,
rate of electrolysis of dendrimers 3 and 4 slowed drastically
R. Muhaupt, H. Frey, U. Rapp and F. J. Mayer-Posner,
near the end of coulometric oxidation, and at the time the Macromolecules, 1995, 28, 6657.
controlled-potential electrolysis was stopped, the ferrocene 5 T. Nagasaki, M. Ukon, S. Arimori and S. Shinkai, J. Chem. Soc.,
Chem. Commun., 1992, 608; G. R. Newkome, F. Cardullo,
redox groups that were bound to the dendrimers in the solution
E. C. Constable, C. N. Moorefield and A. M. W. C. Thompson,
were not totally oxidized.
J. Chem. Soc., Chem. Commun., 1993, 925; S. Achar and R. J. Puddephatt, Angew. Chem., Int. Ed. Engl., 1994, 33, 847; G. R. Newkome, C. N. Moorefield, J. M. Keith, G. R. Baker and
Conclusion
G. H. Escamilla, Angew. Chem., Int. Ed. Engl., 1994, 106, 701; S. Serroni, G. Denti, S. Campagna, A. Juris, M. Ciano andUsing the stepwise convergent approach we have synthesized
V. Balzani, Angew. Chem., Int. Ed. Engl., 1992, 104, 1540.
a series of dendritic poly(aryl ether) s that contain 3, 6, 12 and 6 R-H. Jin, T. Aida and S. Inoue, J. Chem. Soc., Chem. Commun.,
24 ferrocene functionalities located exclusively at the peripher- 1993, 1260; C. J. Hawker, K. L. Wooley and J. M. J. Fre´chet, J. Am.
Chem. Soc., 1993, 115, 4375; K. L. Wooley, C. J. Hawker,
ies of their dendritic structures. The structures of these
dendri-J. M. dendri-J. Frechet, F. Wudl, G. Srdanov, S. Shi, C. Li and M. Kao,
mers were characterized using1H and 13C NMR spectroscopy.
J. Am. Chem. Soc., 1993, 115, 9836; P. J. Dandliker, F. Diederich,
The results of electrochemical studies show that electron
J-P. Gissebrecht, A. Louati and M. Gross, Angew. Chem., Int. Ed.
transfers from the dendritic macromolecules yielded voltam- Engl., 1995, 34, 2725.
metric waves with shapes matching those of corresponding 7 C. Hawker and J. M. J. Fre´chet, J. Am. Chem. Soc., 1990, 112, 7638;
K. L. Wooley, C. J. Hawker and J. M. J. Fre´chet, J. Chem. Soc.,
molecules with electroactive centres, but with magnitudes
Perkin T rans. 1, 1991, 1059; C. J. Hawker and J. M. J. Fre´chet,
determined by the total number of redox centres present. On
J. Am. Chem. Soc., 1992, 114, 8405.
the basis of this observation, we conclude that the ferrocenyl
8 D. T. Sawyer and J. L. Roberts Jr., Experimental Electrochemistry
moieties located on the outer surfaces of these dendrimers are for Chemists, Wiley, New York, 1979.
non-interacting redox centres, are electrochemically equivalent 9 K. Schlogel, Monatsh. Chem., 1957, 88, 601.
and oxidizable at the same potential. This fully reversible 10 C. G. Pitt, H. H. Seltzman, Y. Sayed, C. E. Twine Jr. and
D. L. Willians, J. Org. Chem., 1979, 44, 677.
multi-electron redox system may be useful for multi-electron
11 J. Hooz and S. S. H. Gilani, Can. J. Chem., 1968, 46, 86.
redox catalysis.
12 E. Reimann, Chem. Ber., 1968, 102, 2887.
13 J. B. Flanagan, S. Margel, A. J. Bard and F. C. Anson, J. Am. Chem. Soc., 1978, 100, 4248.
Dr. Fred C. Anson (California Institute of Technology) is
14 T. W. Smith, J. E. Kuder and D. Wychich, J. Polym. Sci., 1976,
gratefully acknowledged for helpful discussions. We thank the
15, 2433.
National Science Council (ROC) (NSC 84-2113-M-009-003 )
15 C. D’Silva, S. Afeworki, O. L. Parri, P. K. Baker and
for its financial support of this research. A. E. Underhill, J. Mater. Chem., 1992, 2, 225.
16 J. B. Flangan, K. Takahashi and F. C. Anson, J. Electroanal. Chem., 1977, 85, 257.
17 P. J. Dandliker, F. Diederich, M. Gross, C. B. Knobler, A. Louati
References
and E. M. Sanford, Angew. Chem., Int. Ed. Engl., 1994, 33, 1739. 1 P. D. Hale, J. Inagaki, H. I. Karan, Y. Okamoto and
T. A. Stotheim, J. Am. Chem. Soc., 1989, 111, 3482; M. E. Wright Paper 6/04225B; Received 17th June, 1996