RESEARCH ARTICLE
Comparison of Six DNA Extraction Procedures and the Application of
Plastid DNA Enrichment Methods in Selected Non-photosynthetic Plants
Shin-Yi Shyu(1) and Jer-Ming Hu(1*)
1. Institute of Ecology and Evolutionary Biology, National Taiwan University, Taipei, Taiwan 106. * Corresponding author. Tel: +886-2-33662472; Fax: +886-2-23686750; Email: [email protected] (Manuscript received 27 May 2013; accepted 06 September 2013)
ABSTRACT: Genomic DNA was isolated using three DNA extraction commercial kits and three CTAB-based methods for two non-photosynthetic plants, Balanophora japonica and Mitrastemon kanehirai. The quality of the isolated DNA was evaluated and subjected to following restriction enzyme digestions. All six procedures yielded DNA of sufficient quality for PCR, and the method described by Barnwell et al. (1998) performed well in isolating DNA from both species for restriction enzyme digestion. In addition, we succeeded to enrich plastid DNA content by using the methods depending on a high salt buffer to deplete nuclear material. The ‘high salt’ methods based on protocol presented by Milligan (1989) were able to increase plastid DNA effectively and significantly reduce nuclear DNA from M.kanehirai. The plastid DNA enrichment protocols are inexpensive and not time-consuming, and may be applicable to other non-photosynthetic plants.
KEY WORDS: CTAB, DNA isolation, heterotrophic plants, plastid DNA, polysaccharide.
INTRODUCTION
Many heterotrophic plants contain polysaccharides and other secondary metabolites that interfere with DNA isolations (Hayata, 1913; Do and Adams, 1991; Scott and Playford, 1996; Nickrent et al., 2000; Tsai et al., 2008; Yu et al., 2010; Wang et al., 2012). These compounds sometimes prevent enzymes to access DNA, and therefore inhibiting follow-up experiments such as polymerase chain reaction (PCR) or restriction enzyme digestions. Many DNA extraction methods, including most commercial kits, are generally designed for cultivated species, which contain much less interfering compounds for isolating DNAs, and therefore might be inapplicable for heterotrophic plants (Do and Adams, 1991; Scott and Playford, 1996). Previous studies (Nickrent et al., 1997a; Nickrent et al., 1997b) showed that the CTAB-based method described by Nickrent (1994) can successfully extract DNA from non-photosynthetic plants with quality good enough for PCR, but is insufficient for enzyme digestions.
We have applied the common DNA extraction methods (Doyle and Doyle, 1987) to the non-photosynthetic parasitic Balanophora species, and it is generally fine to obtain good quality DNA for PCR amplification (Su and Hu, 2012; Su et al., 2012). However, the Balanophora DNA from such methods sometimes failed to perform well in enzyme digestions in our preliminary surveys. Furthermore, ordinary DNA extraction methods cannot guarantee to have enough plastid DNA since very few plastids are present in the cells of the non-photosynthetic plants (dePamphilis and
Palmer, 1990; Nickrent et al., 1997b; Nickrent et al., 2000). All available methods for plastid DNA isolation are designed for isolating ordinary non-reduced chloroplasts in green plants, for example, the gradient-based methods for plastid isolation (Kolodner and Tewari, 1975; Palmer, 1986), and others to enrich organelles (e.g. Herrmann, 1982; Palmer, 1982; Bookjans et al., 1984; Milligan, 1989; Triboush et al., 1998; Kausch et al., 1999). The isolation methods with DNAase-I treatment postulated by Herrmann (1982) require a large amount of plant samples, which is also unpractical in our study, since the plant materials are usually limited.
However, some methods (e.g. Milligan, 1989; Triboush et al., 1998) have been used without the requirement on the color of materials and just need a small amount of tissue. The Milligan’s procedure (1989) depends on a high salt buffer to solubilize nuclear material in order to obtain a chloroplast fraction, and Triboush et al. (1989) isolated organelles by differential centrifugation. These methods are thus have potentials for plastid isolation in non-photosynthetic plants.
In our preliminary survey on 12 DNA extraction methods for two non-photosynthetic plants,
Balanophora japonica Makino and Mitrastemon kanehirai Yamamoto, six of them showed promising
results, while the others performed badly, with low or no yield of DNA (see Table S1). In this report, we compared the performance of the six procedures that successfully isolated DNA from these two non-photosynthetic plants, and evaluated with the
following PCR and restriction enzyme digestion. The procedures include three DNA extraction commercial kits, the two methods mentioned above and another CTAB-based method that Barnwell et al. (1998) developed for the highly mucilaginous succulent plants. At the same time, we attempted to enrich plastid DNA content during extraction of M. kanehirai DNA for studying its plastid genome by using and modifying the Milligan and Triboush methods. The results were compared with the proportion of plastid vs. nuclear DNA content among the three different plastid enrichment methods.
MATERIALS AND METHODS
Plant Materials
Balanophora japonica and M. kanehirai are both
non-photosynthetic plants native to Taiwan. The materials (B. japonica: Mt. Datong, Taipei County, Sep. 29, 2005, Hu1567; M. kanehirai: Lienhuachih, Nantou County, Oct. 12, 2010, Hu1810) were freshly collected and stored in -20°C. Frozen tissues were used for each DNA extraction method.
DNA extraction methods
The methods used for comparison are listed in Table 1, including three commercial kits (Method 1‒3) and three CTAB-based methods (Method 4‒6).
Total genomic DNAs were extracted by Method 1‒3 according to the corresponding manufacturer’s protocols. Method 1, the DNeasy Plant Mini Kit (QIAGEN), uses the QIAshredder spin column to remove initial precipitates and cell debris, and a DNeasy column to capture DNA. Method 2, the Tri-Plant Genomic DNA Reagent Kit (Geneaid), and
Method 3, the TRI Reagent (Molecular Research
Center), both use their own particular reagents to lyse plant samples and then follow by an isopropanol or ethanol precipitation.
Method 4 denoted for the standard CTAB method
described by Doyle and Doyle (1987). Plant materials were ground in liquid nitrogen and then incubated in 10 volumes of preheated 2× CTAB buffer (100 mM Tris-HCl, 1.4 M NaCl, 20 mM EDTA, 2% w/v CTAB, 2% w/v PVP40 and added 0.2% β-mercaptoethanol just before use.) at 65°C for 1 h with occasional swirling. The solution was mixed with 10 mL of Chloroform:isoamyl alcohol (24:1, v/v) and was blended thoroughly. This was followed by a centrifugation at 9,000 g for 10 min, and the aqueous phase was transferred to a new centrifuge tube. The DNA was precipitated by adding 0.7 volume of isopropanol and incubated at -20°C for 30 min. The DNA pellet was collected by a centrifugation for 10 min at 10,000 g and washed with cold 75% ethanol. The
pellet was resuspended in 2 mL of TE buffer, and then RNase digestion was performed.
Method 5 denoted for the “delayed hot CTAB”
method described by Nickrent (1994). The sample was cut into small pieces and homogenized with hot 95°C CTAB buffer (about 25 mL for every 2‒3 g of plant tissue). The modified 2× CTAB buffer is composed of 100 mM Tris-HCl, 1.4 M NaCl, 30 mM EDTA, 2% w/v CTAB, 5 mM ascorbic acid, 4 mM diethyldithiocar-bamic acid and 2% w/v PVP40, the latter two ingredients were added just before use. The extract was strained through cheesecloth into 50-mL centrifuge tube and then incubated at 70‒80°C for 30 min with occasional swirling. The sample was briefly centrifuged without pausing, and the supernatant was transferred to a new tube. Chloroform:isoamyl alcohol (24:1) (0.7 volume) was added and the solution was mixed for 5 min. This was then centrifuged at 9,000 g for 15 min, and the aqueous phase was transferred to a new centrifuge tube. The DNA was precipitated by adding 0.7 volume of ice-cold isopropanol and incubated at -20°C for at least 1 h. The DNA pellet was collected by a centrifugation for 20 min at 10,000 g. Then the pellet was resuspended in 3 mL of TE and 2 mL of 4 M NH4OAc. This was followed by an extraction with an
equal volume of phenol:chloroform (1:1), and 2 volumes of ethanol were added to the aqueous phase. The content was incubated at -20°C for at least 30 min, and the DNA pellet was collected by a centrifugation, then proceeded an RNase treatment.
Method 6 denoted for the extraction procedure
developed by Barnwell et al. (1998) for the highly mucilaginous succulent plants. The frozen plant tissue was ground to powder, 5 volumes of extraction buffer (100 mM Tris-HCl, 1.4 M NaCl, 20 mM Na2 EDTA,
2% w/v CTAB, 1% w/v PVP40) were added and mixed. The homogenate was then incubated at 65°C for 30 min with occasional shaking followed by a centrifugation at 3,000 g for 5 min. The supernatant was mixed with 1.25 volumes of 10% CTAB (w/v, in 0.7 M NaCl), and the mixture was vortexed for 10 s and centrifuged at 3,000 g for 5 min. The supernatant was thoroughly mixed with 3 volumes of precipitation buffer (50 mM Tris-HCl, 10 mM Na2 EDTA, 1% w/v CTAB). The
mixture was incubated at room temperature for 30 min and then centrifuged at 5,000 g for 15 min. The pellet was dissolved in high salt TE buffer (10 mM Tris-HCl, 1.0 M NaCl, 1 mM Na2 EDTA), and 2 volumes of
ice-cold ethanol were added followed by incubation at -20°C for 1 h. The DNA was pelleted by a centrifugation and washed twice with 70% ethanol.
Plastid DNA enrichment methods
Three plastid enrichment methods were analyzed in this study. Method PE1 denoted for the procedure
Table 1. The methods used in this study.
Method Reference
DNA isolation
Method 1 Column-based commercial kit DNeasy Plant Mini Kit, QIAGEN, Manchester, UK
Method 2 Particular reagent- based commercial kit Tri-Plant Genomic DNA Reagent Kit, Geneaid, New Taipei City,
Taiwan
Method 3 Particular reagent- based commercial kit TRI Reagent – RNA, DNA, protein isolation reagent, Molecular
Research Center, Cincinnati, OH, USA
Method 4 The standard CTAB method Doyle and Doyle, 1987
Method 5 The delayed hot CTAB method Nickrent, 1994
Method 6 The increased CTAB method Barnwell et al., 1998
Plastid DNA enrichment
PE1 Depleted nuclear material by using a high salt buffer Milligan, 1989
PE2 Combined PE1 with Method 2 Milligan, 1989
PE3 Isolated organelles by differential centrifugation Milligan, 1989; Triboush et al., 1998
developed by Milligan (1989) that incorporates several other methods of extracting chloroplast DNA. The procedure is summarized below. The tissue was ground with 6 volumes of ice-cold isolation buffer (50 mM Tris-HCl, 1.25 M NaCl, 5 mM EDTA, 0.1% w/v BSA, 0.1% β-mercaptoethanol), and the homogenate was filtered through 4 layers of cheesecloth. The plastids were pelleted by a centrifugation at 3,000 g for 10 min and then resuspended in 10 mL of cold isolation buffer. The centrifugation and resuspension were repeated once, and 0.1 volume of 10% CTAB was added to lyse the plastids. The extract was then incubated at 60°C for 1 h, and followed by extraction with Chloroform:iso-amyl alcohol (24:1). The DNA was precipitated by adding 0.7 volume of cold isopropanol to the aqueous phase and incubated at -20°C for at least 30 min. During the isolation of plastids, the materials should be kept at 4°C.
Method PE2 denoted for a method combined with
Milligan’s protocol (1989) and the Tri-Plant Genomic DNA Reagent Kit. Plastid pellet was isolated by centrifugation at 6,000 g for 20 min instead of 3,000 g for 10 min following Milligan’s protocol. The DNA was then extracted from pellet by using Tri-Plant Genomic DNA Reagent Kit.
Method PE3 denoted for the method mainly based
on Triboush’s DNA extraction method (Triboush et al., 1998), combined with Milligan’s protocol (1989), as described below. All the operations of isolating plastids were conducted in ice. The sample was homogenized with 6 volumes of STE buffer (50 mM Tris-HCl, 400 mM sucrose, 20 mM Na2EDTA, 0.2% w/v BSA, 0.2%
β-mercaptoethanol). The homogenate was filtered through 4 layers of cheesecloth, and centrifuged at 200 g for 20 min. The supernatant was centrifuged at 3,700 g for 20 min, and the pellet was resuspened in 20 mL of isolation buffer (based on Milligan’s protocol). It was then repeated the centrifugation and resuspension once,
and then the DNA was obtained by following Milligan’s protocol.
Real-time PCR
Real-time PCR and data analysis were performed in the CFX96TM Real-Time PCR Detection Systems
(Bio-Rad Laboratories). Sequences of nuclear and plastid SSU fragments were amplified from M.
kanehirai DNA extracted by different methods. The
primers SSU1594F: 5’-CTACGTCCCTGCCCTTTGT A-3’ and SSU1703R: 5’-GGACTTCTCGCGGCATCA CGAG-3’ were used to amplify a nuclear 18S rDNA fragment; the primers 16S298F: 5’-GGAAACAGCCC AGATCATCA-3’ and 16S436R: 5’-GCCGACATTCT CACTTCTGC-3’ were used to amplify the plastid 16S rDNA. The primers were designed based on
Mitrastemon kanehirai sequences in our preliminary
survey. The PCR mixture (20 μL) contained 10 μL KAPA SYBR FAST qPCR Master Mix (Kapa Biosystems), 50 nM (nr18S rDNA) or 100 nM (pt16S rDNA) of each primer and 20 ng of extracted DNA was used as template. The amplification program initiated at 95°C for 3 min, followed by 40 cycles at 95°C for 10 s and 64°C for 30 s, and finally 95°C for 10 s. Melting curve analysis was carried out after amplification. All experiments were performed in triplicate.
RESULTS AND DISCUSSIONS
Performance of different isolation procedures
Balanophora japonica and M. kanehirai both lost
their photosynthetic ability completely and plants are very rich in polysaccharides and other secondary metabolites (Hayata, 1913; Wang et al., 2012). Many of the commercial kits and methods failed to extract high quality DNA from heterotrophic plants (listed in supplementary data Table S1). Here we show the six methods that can successfully extract the DNAs from
these two non-photosynthetic plants.
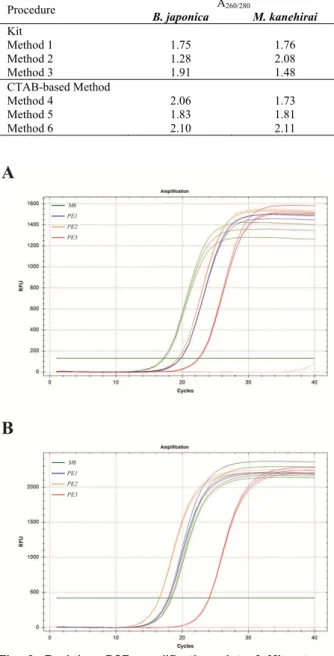
The ratios of the absorbance at 260 and 280 nm (A260/280) of DNA isolated from two species with
different procedures are in the range of 1.28‒2.11 (Table 2). Two of the three CATB-based methods (Method 4 and 6) yielded better results than all the others, including the commercial kits, for B. japonica. In comparison, Method 2 and 6 worked best for M.
kanehirai.
Only Method 6 (Barnwell et al., 1998) performed well on both B. japonica and M. kanehirai, with acceptable A260/280 ratio. However, Methods 2 and 4
showed inconsistent results between Balanophora and
Mitrastemon. Nonetheless, the quality of all DNA
isolated by different procedures were good enough for the following PCR (data not shown).
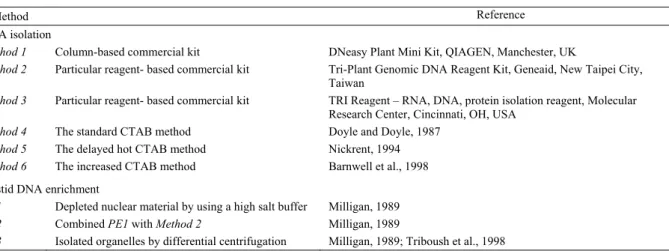
To further examine the DNA quality, we proceeded with restriction enzyme digestion on the obtained DNA extracts. The result shows that the Method 6 (Barnwell protocol) performed better than the other procedures in extracting DNA from B. japonica (Fig. 1A), and
Method 2 (the Tri-Plant Genomic DNA Reagent Kit)
performed best in M. kanehirai (Fig. 1B). In general, all procedures performed better in M. kanehirai which is likely because it contains less polysaccharides than B.
japonica. However, Method 3 (the TRI Reagent) and Method 4 (the Doyle and Doyle protocol) could not
produce DNA bands for M. kanehirai (M3 and M4 in Fig. 1), compared to B. japonica. From all of the commercial kits tested, we found that most kits extracted DNA by using columns did not perform well. It is probably because these kits were unable to eradicate polysaccharides that prevent the elution of DNA from columns and result in the low yields (Fleischmann and Heubl, 2009). Among the three CTAB-based DNA isolation methods, it seems that shortening the initial incubation time, and increasing the incubation temperature could improve DNA purity, since the Method 5 (Nickrent protocol) performed better than Method 4. However, Method 6 that Barnwell et al. (1998) developed increases CTAB concentration in a step-wise manner in order to avoid co-precipitation of polysaccharides with DNA, which was the most effective procedure to obtain DNA with high quality. As for yields, all the commercial kits produced more amount of DNA than the CTAB-based methods. The
Method 6 produced least but the purest DNA among all
procedures, which might result from the protocol’s additional purification steps and less efficient precipitation buffer.
Plastid DNA enrichment
The Milligan’s ‘high salt buffer’ method (1989) and the Triboush’s differential centrifugation method (1998) were used and modified to enrich plastid DNA content
Fig. 1. The results of restriction enzyme digestion on the isolated DNAs. A: Balanophora japonica. B: Mitrastemon
kanehirai. 4 μg DNA was digested with 4 U EcoRI/μg DNA
for 1 h at 37°C, and then were separated on a 0.8% TAE agarose gel. U, uncut DNA; C, cut DNA; λ, lambda DNA/HindIII marker; M1, QIAGEN DNeasy Plant Mini Kit; M2, Geneaid Tri-Plant Genomic DNA Reagent Kit; M3, Molecular Research Center TRI Reagent; M4, the Doyle and Doyle protocol; M5, the Nickrent protocol; M6, the Barnwell protocol.
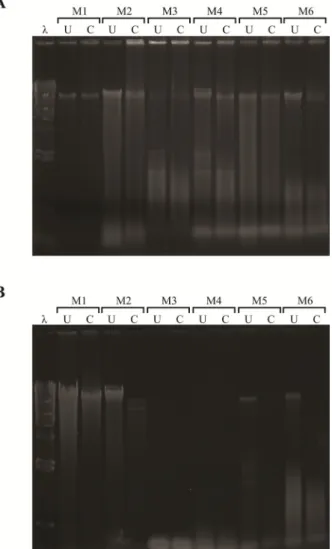
for extracting M. kanehirai DNA. The performance of these procedures was evaluated by relative quantification of nr18S and pt16S rDNA in the DNAs extracted by different methods with Barnwell protocol as the reference. DNA isolated by all methods (included the Barnwell protocol) could amplify nr18S and pt16S rDNA fragments by real-time PCR (Fig. 2). However,
Method PE3 (Triboush-based method) not only failed to
enrich plastid DNA in our tests but also yielded the lowest quality DNA that the Cq (quantification cycle) values of PE3 were the largest among all methods in
Table 2. A260/280 ratios of DNA extracted by commercial kits
and CTAB-based methods.
Procedure A260/280 B. japonica M. kanehirai Kit Method 1 1.75 1.76 Method 2 1.28 2.08 Method 3 1.91 1.48 CTAB-based Method Method 4 2.06 1.73 Method 5 1.83 1.81 Method 6 2.10 2.11
Fig. 2. Real-time PCR amplification plot of Mitrastemon
kanehirai DNA. A: nr18S rDNA. B: pt16S rDNA. M6, the
Barnwell protocol; PE1, the Milligan protocol; PE2, the Milligan protocol combined with Geneaid Tri-Plant Genomic DNA Reagent Kit; PE3, the Triboush method combined with Milligan protocol.
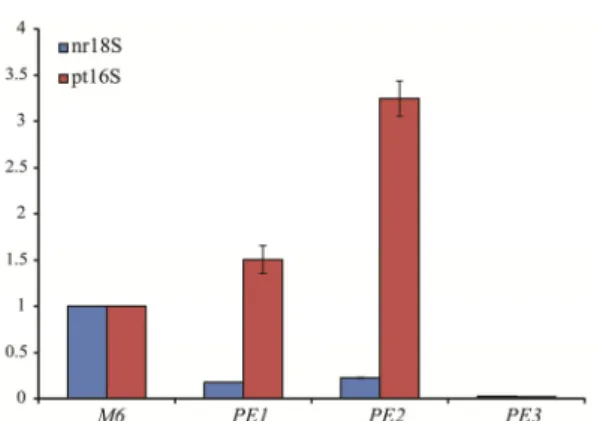
both nr18S and pt16S rDNA. The two Milligan-based protocols (Method PE1 and PE2) were capable of increasing plastid DNA content more than 1.5 times and meanwhile reduced the proportion of nuclear DNA effectively (Fig. 3). The pt16S/nr18S rDNA ratios of extracted DNA by using these two procedures are significantly higher than Method 6 (Fig. 4). The best performed method was Method PE2, the Milligan
protocol combined with the Tri-Plant Genomic DNA Reagent Kit, which yielded pt16S/nr18S rDNA ratio almost 15 times higher than Method 6 with good quality of DNA from M. kanehirai. It suggests that increasing centrifugal speed in the beginning step of collecting plastids could enrich plastid DNA further since Method
PE3 performed better than Method PE2 (Fig. 3 and Fig.
4).
The result shows that the procedure based on differential centrifugation failed to apply to our studying materials. Although the high salt buffer-based protocols could not eliminate nuclear DNA completely, they still could enrich plastid DNA content significantly (Method
PE2 and PE3). These protocols are inexpensive and not
time-consuming, and may be applicable to other non-photosynthetic plants, which will be useful in studying the plastid genome of heterotrophic plants.
Overall, the Barnwell protocol performed best among all examined methods, but it is inapplicable for small amount of plant samples. However, our results suggest that there is no DNA isolation protocol can be applied to all plants because of the presence of various compounds in the plant tissue and it cannot have DNA with both the highest quality and quantity from the same protocols. Additional effort to find out or modify isolation procedures is necessary in order to obtain high quality DNA for non-photosynthetic plants.
LITERATURE CITED
Barnwell, P., A. N. Blanchard, J. A. Bryant, N. Smirnoff and A. F. Weir. 1998. Isolation of DNA from the highly mucilagenous succulent plant Sedum telephium. Plant Mol. Biol. Rep. 16: 133‒138. doi: 10.1023/A:1007473302551
Bookjans, G., B. M. Stummann and K. W. Henningsen. 1984. Preparation of chloroplast DNA from pea plastids isolated in a medium of high ionic strength. Anal. Biochem. 141: 244‒247. doi: 10.1016/0003-2697(84)9045 2-4
Croy, E. J., T. Ikemura, A. Shirsat and R. R. D. Croy. 1993. Plant nucleic acids. In R.R.D. Croy, (ed.). Plant molecular biology labfax. Oxford, pp. 21‒48.
dePamphilis, C. W. and J. D. Palmer. 1990. Loss of photosynthetic and chlororespiratory genes from the plastid genome of a parasitic flowering plant. Nature. 348: 337‒339. doi: 10.1038/348337a0
Do, N. and R. P. Adams. 1991. A simple technique for removing plant polysaccharide contaminants from DNA. Biotechniques. 10: 162, 164, 166.
Doyle, J. J. and J. L. Doyle. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19: 11‒15.
Fleischmann, A. and G. Heubl. 2009. Overcoming DNA extraction problems from carnivorous plants. Anal. Jardín Bot. Mad. 66: 209‒215. doi: 10.3989/ajbm.2198
Hayata, B. 1913. On the systematic position of Mitrastemon as a genus representing a special tribus of the Rafflesia-ceae. Icones Plantarum Formosanarum. pp. 199‒213.
Fig. 3. Relative quantification of nr18S and pt16S rDNA.
Mitrastemon kanehirai DNAs extracted by three methods
were compared with Barnwell protocol as the reference. M6, the Barnwell protocol; PE1, the Milligan protocol; PE2, the Milligan protocol combined with Geneaid Tri-Plant Genomic DNA Reagent Kit; PE3, the Triboush method combined with Milligan protocol.
Fig 4. The ratio of pt16S/nr18S rDNA in extracted
Mitrastemon kanehirai DNAs. The pt16S rDNA content was
compared with nr18S rDNA in the same DNA samples extracted by four different procedures. M6, the Barnwell protocol; PE1, the Milligan protocol; PE2, the Milligan protocol combined with Geneaid Tri-Plant Genomic DNA Reagent Kit; PE3, the Triboush method combined with Milligan protocol.
Herrmann, R. G. 1982. The preparation of circular DNA from plastids. In M. Edelman, R.B. Hallick, and N.H. Chua, (eds.). Methods in chloroplast molecular biology. Elsevier Biomedical Press, Amsterdam, pp. 259‒280. Kausch, A. P., T. P. Owen, Jr., S. Narayanswami and B. D.
Bruce. 1999. Organelle isolation by magnetic immunoabsorption. Biotechniques. 26: 336‒343.
Kolodner, R. and K. K. Tewari. 1975. The molecular size and conformation of the chloroplast DNA from higher plants. BBA-Nucleic Acids and Protein Synthesis. 402: 372‒390. doi: 10.1016/0005-2787(75)90273-7
Milligan, B. 1989. Purification of chloroplast DNA using hexadecyltrimethylammonium bromide. Plant Mol. Biol. Rep. 7: 144‒149. doi: 10.1007/BF02669630
Nickrent, D. L., R. J. Duff and D. A. M. Konings. 1997a. Structural analyses of plastid-derived 16S rRNAs in holo parasitic angiosperms. Plant Mol. Biol. 34: 731‒743. doi:
10.1023/A:1005808615763
Nickrent, D. L. 1994. From field to film - rapid sequencing methods for field-collected plant species. Biotechniques. 16: 470‒475.
Nickrent, D. L., R. J. Duff, A. E. Colwell, A. D. Wolfe, N. D. Young, K. E. Steiner and C. W. dePamphilis. 2000. Molecular phylogenetic and evolutionary studies of parasitic plants. In D.E. Soltis, P.S. Soltis, and J.J. Doyle, (eds.). Molecular Systematics of Plants II: DNA sequencing. Kluwer Academic Publishers, Norwell, Massachusetts, USA, pp. 211‒241. doi: 10.1007/978-1-4615-5419-6_8
Nickrent, D. L., O. Y. Yan, R. J. Duff and C. W. dePamphilis. 1997b. Do nonasterid holoparasitic flowering plants have plastid genomes? Plant Mol. Biol. 34: 717‒729.
Palmer, J. D. 1982. Physical and gene mapping of chloroplast DNA from Atriplex triangularis and Cucumis sativa. Nucleic Acids Res. 10: 1593‒1605. doi: 10.1093/nar/10.5.1593 Palmer, J. D. 1986. Isolation and structural analysis of
chloroplast DNA. In H.W. Arthur Weissbach, (ed.). Methods in Enzymology. Academic Press, pp. 167‒186. doi: 10.1016/0076-6879(86)18072-4
Scott, K. D. and J. Playford. 1996. DNA extraction technique for PCR in rain forest plant species. Biotechniques. 20: 974, 977, 979.
Su, H.-J. and J.-M. Hu. 2012. Rate heterogeneity in six protein-coding genes from the holoparasite Balanophora (Balanophoraceae) and other taxa of Santalales. Ann. Bot. 110: 1137‒1147. doi: 10.1093/aob/mcs197
Su, H.-J., J. Murata and J.-M. Hu. 2012. Morphology and phylogenetics of two holoparasitic plants, Balanophora japonica and Balanophora yakushimensis (Balanophora- c eae), and their hosts in Taiwan and Japan. J. Plant Res. 125: 317‒326. doi: 10.1007/s10265-011-0447-5
Sytsma, K. J. 1994. DNA extraction from recalcitrant plants: long, pure, and simple? In R.P. Adams, J.S. Miller, E.M. Golenberg, and J.E. Adams, (eds.). Conservation of plant genes II. Missouri Botanical Garden, Missouri, pp. 69‒81. Triboush, S. O., N. G. Danilenko and O. G. Davydenko.
1998. A method for isolation of chloroplast DNA and mitochondrial DNA from sunflower. Plant Mol. Biol. Rep. 16: 183‒189.
Tsai, T.-H., G.-J. Wang and L.-C. Lin. 2008. Vasorelaxing alkaloids and flavonoids from Cassytha filiformis. J. Nat. Prod. 71: 289‒291. doi: 10.1021/np070564h
Wang, X., Z. Liu, W. Qiao, R. Cheng, B. Liu and G. She. 2012. Phytochemicals and biological studies of plants from the genus Balanophora. Chem. Cent. J. 6: 79. doi: 10.1186/1752-153X-6-79
Yu, F.-R., Y. Liu, Y.-Z. Cui, E.-Q. Chan, M.-R. Xie, P. P. McGuire and F.-H. Yu. 2010. Effects of a flavonoid extract from Cynomorium songaricum on the swimming endurance of rats. Am. J. Chin. Med. 38: 65‒73. doi: 10.1142/S0192415X10007774
Supplementary data
Table S1. Other methods have been tested in this study.
Method Reference
DNA isolation
Column-based commercial kit Fast ID Genomic DNA Extraction Kit, Genetic ID, Fairfield, IA, USAa Column-based commercial kit Plant Genomic DNA Mini Kit, BIOMAN, New Taipei City, Taiwana Column-based commercial kit Plant Genomic DNA Purification Kit, GeneMark, Taichung City, Taiwana Particular reagent- based commercial kit TRIzol® Reagent, Invitrogen, Carlsbad, CA, USAb
The rainforest method Scott and Playford, 1996c
Modified CTAB methods Doyle and Doyle, 1987; Croy et al., 1993; Sytsma, 1994c Plastid DNA enrichment
The sunflower method Triboush et al., 1998b
a The yields of DNA from these methods were very low, and A
260/280 ratios of DNAs were below 1.2.
b The methods failed to extract DNA from B. japonica and M. kanehirai. c The methods failed to improve DNA quality compared with Method 4.
DNA 萃取方式以及分離葉綠體 DNA 方法在非光合作用植物中的比較與應用
徐馨怡(1)、胡哲明(1*)
1. 國立臺灣大學生態學與演化生物學研究所,臺北,臺灣。
* 通信作者。Tel: +886-2-33662472; Fax: +886-2-23686750; Email: [email protected]
(收稿日期:2013年5月21日;接受日期:2013年9月6日)