ContentslistsavailableatScienceDirect
Journal
of
Power
Sources
j ou rn a l h o m e pa g e :w w w . e l s e v i e r . c o m / l o c a t e / j p o w s o u r
Electrochemical
degradation
of
Nafion
ionomer
to
functionalize
carbon
support
for
methanol
electro-oxidation
Yu-Chi
Hsieh, Jing-Yu
Chen,
Pu-Wei
Wu
∗DepartmentofMaterialsScienceandEngineering,NationalChiaoTungUniversity,Hsin-chu300,Taiwan
a
r
t
i
c
l
e
i
n
f
o
Articlehistory:
Received8April2011
Receivedinrevisedform24May2011
Accepted25May2011
Available online 24 June 2011 Keywords: Nafionionomer Carbonfunctionalization Cyclicvoltammetry Methanolelectro-oxidation Pt
a
b
s
t
r
a
c
t
Aneffectiveelectrochemicalroutetoproducefunctionalgroupsoncarbonsurfaceisdemonstrated. Cyclicvoltammetric(CV)sweepsareperformedin0.5MH2SO4 electrolyteonelectrodescontaining carboncloth,VulcanXC72R,andNafionionomer.Withsupplyofambientoxygen,thegenerationof hydroxylradicalsfromtheoxygenreductionreactionduringCVcyclesinitiatesthedecompositionof Nafionionomerthatleadstoformationofoxygenatedfunctionalgroupsonthecarbonsurface.Ion chro-matographyconfirmsthedissolutionofsulfateanionsuponCVscans.Ramananalysissuggestsaminor alterationforthecarbonstructure.However,X-rayphotoelectronspectroscopyindicatesasignificant increaseofoxygenatedfunctionalgroupsinconjunctionwithnotablereductioninthefluorinecontent. TheamountoftheoxygenatedfunctionalgroupsisdeterminedbycurvefittingofC1sspectrawith knownconstituents.Thesefunctionalgroupscanalsobefoundbyimmersingtheas-preparedelectrode inasolutioncontainingconcentratedresiduesfromNafionionomerdecomposition.Thefunctionalized electrodeallowsa170%incrementofPtionadsorptionascomparedtothereferencesample.After elec-trochemicalreductions,thefunctionalizedelectroderevealssignificantimprovementsinelectrocatalytic abilitiesformethanoloxidation,whichisattributedtotheoxygenatedfunctionalgroupsthatfacilitates theoxidationofCOonPt.
© 2011 Elsevier B.V. All rights reserved.
1. Introduction
Carbonaceousmaterialshavebeenwidelyusedasthesubstrates
for catalystimpregnations inroom tempeaturare fuelcells like
polymermembranefuelcellsanddirectmethanolfuelcells[1–10].
Itisbecausewiththeselectionofcarbonsassupports,
nanopartic-ulatecatalystssuchasPtandPtRuareabletodistributeuniformly,
leadingtoreducedloadingandbettercatalystutilization.Todate,
carbonsinarichvarietyofformsincludingcarbonblacks,carbon
nanotubes(CNTs),mesoporouscarbons,carbonnanocapsules,
acti-vatedcarbons,andcarbonxerogelshavebeeninvestigatedasthe
catalystsupportswithimpressiveresults[11–16].Theinteractions
betweenthecarbonandcatalystarecriticalbecausealackof
suf-ficientbondingbetweenthem causespossibledetachmentsand
colaescence,which resultsin undesirableperformance
degrada-tion[17].Unfortunately,untreatedcarbonsareoftenhydrophobic
innaturethat allowpooradsorptionofcatalyst precursorsand
catalysts.Therefore,itisnecessarytocarryoutadditional
function-alizationtreatmentsonthecarbonstorenderahydrophilicsurface
instead.Afterpropersurfacefunctionalizations,thecarbonsare
∗ Correspondingauthor.Tel.:+88635131227;fax:+88635724727.
E-mailaddress:[email protected](P.-W.Wu).
expectedtoadsorbmorecatalystprecursorsforalargeramount
ofcatalystdeposition.
Earlierstudiesonthecarbonfunctionalizationsareconcerned
withcarboncorrosionsbecauseundertheoperationconditionsof
phosphoricacidfuelcells,thecarbonis pronetooxidation loss
bytheformationofsurfaceoxidizedgroups[18–22].Ingeneral,
thefunctionalizationofcarboninvolvesanodizationtreatmentsin
concentratedacidsatmoderatetemperature[23,24].Forexample,
Kangasniemietal.imposedpotentiostatictreatmentsonthe
Vul-canXC72(XC72) in1MH2SO4 solution,anddeterminedthatat
roomtermperature,asignficantoxidationwasoccurringforthe
anodizingvoltageof1.2Vfor16hbut0.8Vwassufficentat65◦C
toproducethesameeffect[25].Asimilaranodizationtreatment
of2VwasemployedbyStevanovicet al.tointroduceselective
functionalgroupsontheglassycarbons[26].Thedegreeof
sur-facefunctionalizationalsodependsonthetypeofcarbonmaterials
becausetheirsurfaceareaandmicrostructuredifferconsiderably.
For instance, theCNTsreveal notable oxidation resistanceover
theXC72 while theBP2000, witha larger specificsurface area
(m2g−1),experiencesmoreoxidationlossasopposedtotheXC72
withasmallerspecificsurfacearea[27].Sofar,after
functionaliza-tion,surfaceoxidizedgroupssuchasphenols,carbonyls,carboxylic
acids,ethers,quinones,andlactoneshavebeenidentified.Theexact
mechanismresponsiblefortheformationofselectivefunctional
groupsiscontingentontheprocessingstepsinvolvedandthetype
0378-7753/$–seefrontmatter © 2011 Elsevier B.V. All rights reserved.
ofcarbonmaterials.Sincethecarbonisrelativelyinertin
corro-siveelectrolytes,typicalsurfacefunctionalizationstepsarerather
time-consuming.Therefore,itisofparticularinteresttodevelopa
simpleandefficientprocessforfunctionalizationpurpose.
Analternativeapproachtofunctionalizethecarbonmaterials
isbychemicalalterationofpolymericbinders.Inelectrode
fabri-cations,Nafionionomerisoftenaddedinmixturewithcarbons,
servingsimultaneouslyasabinderandconductivepathforproton
transport.Therefore,itispossiblethattheNafionionomerwould
sufferfromstructuraldamageandlossofsulfonicacidsidechains
ifdeliberateelectrochemicaltreatmentsareapplied.Previously,
extensiveeffortshavebeendevotedtounderstandtheresponsible
mechanismforNafionmembranedegradationindifferent
environ-mentsandfactorsincludinghumidity,temperature,andoxygen
concentrationarefoundtoberelevant[28,29].Accordingto
litera-ture,hydroxyl(•OH)andperoxy(•OOH)radicalsformedduringfuel
celloperationsareabletoreactwithpolymerendgroupsthatstill
containresidualterminalH-groups[30–32].Furtherstudies
indi-catethatthesulfonicacidsidechainsarealsosusceptibletoradicals
[29,33,34].Inaddition,thedegradedspeciesofNafioncontainfree
radicalsthathavebeenreportedtoattackcarbonsandchemically
bondtotheirsurface[35–37].Moreover,itissuggestedthatthe
presenceoffunctionalizedgroupsonthecarbonsurfaceis
possi-bletoengenderadditionaloxidizedgroups[38–40].Inlightofthis
information,werealizethattheintentionaldegradationofNafion
ionomermightprovideaneffectiverouteforcarbon
functionaliza-tion.
In this work, we conducted multiple cyclic voltammetric
scans (CV) to introduce functional groups on the electrode
structure,followed byPtcationadsorptionand electrochemical
reduction to produce nanoparticulate Ptimpregrenated onthe
functionalizedsupport.Electrocatalyticanalysisonthemethnaol
electro-oxidationwasperformedtoelucidatetheeffectof
func-tionalizedsupportforcatalyticactions.
2. Experimental
2.1. Carbonsurfacefunctionalization
Theelectrodefor surfacefunctionalizationwasfabricatedby
depositingacarbon/Nafionmixtureontoacommerciallyavailable
carboncloth(E-TEK).First,10mgNafionionomersolution(5wt%)
and 8mg carbonpowders (VulcanXC72R) were mixedin 5mL
99.5wt%ethanolfor60minundersonicationtoformanink
dis-persion.Thedispersionwasdepositedrepeatedlyona4cm2carbon
clothwhichwaskeptat80◦Catopahotplatetoevaporateresidual
solvent.TheloadingsfortheXC72RandNafionionomeronthe
car-bonclothwere2and0.125mgcm−2,respectively.Subsequently,
thesurfacefunctionalizationwasperformedbyimposingCVscans
ontheelectrode(activeareaof0.785cm2)for20cyclesbetween
−0.2and1.1V(vs.Ag/AgCl)at50mVs−1inanaqueouselectrolyte
of0.5MH2SO4.APtfoilof10cm2wasusedasthecounterelectrode.
ThedurationfortheCVscanswas17min.Inordertointroduce
oxygenduringtheCVscans,thebacksidefortheelectrodewas
pressedagainstastainlesssteelfoilthatwaspartiallyexposedto
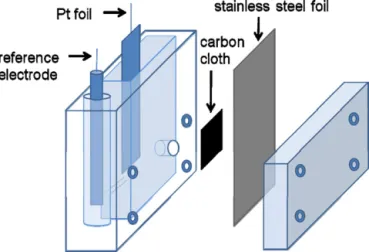
theambientoxygen.Aschematicforthecelldesignisillustratedin
Fig.1.Forcomparisonpurpose,wealsoimmersedtheas-prepared
electrodesin0.5MH2SO4or0.1MHClaqueoussolutionwith
con-centratedresiduesfromNafionionomerdecompositiontoanalyze
theirsurfacefunctionalgroups.
2.2. Electrochemicalanalysis
Thefunctionalizedelectrode wasimmersedin5mMH2PtCl6
aqueoussolution(pHadjustedto8)at40◦C.Theimmersionlasted
Fig.1.AschematicoftheelectrochemicalcellforCVscansin0.5MH2SO4aqueous
solution.Thecarbonclothispartiallyexposedtoambientoxygen.
for48htoensuresufficientadsorptionofPtCl62−.Toreducethe
adsorbedPtions,CVscanswerecarriedoutbetween−0.2and0.2V
in0.5MH2SO4aqueoussolutionat50mVs−1.Toevaluatethe
elec-trochemicalsurfacearea(ECSA)forthedepositedPt,weconducted
CVscansbetween−0.2and0.9Vin0.5MH2SO4at50mVs−1.The
ECSAwasestimatedbytheintegratedchargeinthehydrogen
des-orptionregion.Formethanolelectro-oxidation,multipleCVscans
wereperformedbetween−0.2and 0.9Vat50mVs−1 in500mL
aqueoussolution of0.5MH2SO4 and 1M CH3OH.The areafor
theworkingelectrodewas0.785cm2.Forlifetimedetermination,
chronoamperogramswererecordedat0.5Vfor30minin500mL
of0.5MH2SO4and1MCH3OH.TheAg/AgClandPtfoil(10cm2)
wereusedasthereferenceand counterelectrodes,respectively.
Surfacefunctionalization,PtCl62−reduction,ESCAdetermination,
andmethanolelectro-oxidationwerecarriedoutat26◦Cina
three-electrodearrangementusingaEG&G263Apotentiostat.
2.3. Materialscharacterizations
X-rayPhotoelectronSpectroscopy(XPS;ThermoMicrolab350)
wasadoptedtoevaluatetheoxygenatedfunctionalgroupsonthe
functionalizedelectrodes.RamanSpectroscopy(LabRAMHR800)
wasconductedtodetectthemicrostructurevariationontheXC72R
afterCVscans.Ionchromatography(DionexDX120)wasutilized
toanalyzetheconcentrationofdissolvedSO42−andfluorocarbons
fromthedecompositionofNafionionomer.TransmissionElectron
Microscope(TEM;PhilipsTecnai-20)wasusedtoobservethe
mor-phologiesanddistributionsforthePtnanoparticles.TheaveragePt
sizewasobtainedbyTEMimageanalysis(Image-ProPlus6.0).The
amountofPtloadingswasdeterminedbyanInductivelyCoupled
PlasmaMassSpectrometry(ICP-MS;SCIEXELAN5000)wherethe
samplesweredissolvedinasolutioncontainingHCl,HNO3,andHF
ata2:2:1volumeratio.
3. Resultsanddiscussion
3.1. ElectrochemicaldegradationofNafionionomer
Fig.2 providestheCVprofilesatvariouscyclesfor the
elec-trodescontainingcarboncloth,Nafionionomer,andXC72Rwith
thesupplyofambientoxygen.Asshown,theCVprofilesexhibited
acharacteristicbehaviorforcapacitorswithsymmetricresponses
inwhichconsiderableanodicandcathodiccurrentswereobserved
above0.9Vandbelow−0.1V,respectively.Notably,thecurrent
fromtheanodicscanforthefirstcyclewasnegligibleuntil0.9V
1.2 1.0 0.8 0.6 0.4 0.2 0.0 -0.2 -6 -4 -2 0 2 4 6 20th 10th I (mAcm -2 ) E (V) vs. Ag/AgCl 5th 1st
Fig.2.ProfilesfrommultipleCVscanswithambientoxygenforelectrodes
contain-ingcarboncloth,XC72R,andNafionionomer.
currentsforthecathodicscan,suggestingsurfaceactivationatan
oxidativepotentialabove0.9Vinthefirstcycle.Interestingly,both
theanodicandcathodiccurrentsdemonstratedsteadyincrements
withincreasingCVcycles.Weunderstoodthattherecorded
cur-rentsweremostlyfromtheXC72Rasthecarbonclothcontributed
aninsignificantamountwithitsrelativelyreducedsurfacearea.
However,inourobservation,samplesofXC72Rdepositedonthe
carbonclothrevealedCVcurvesthatwereinsensitivetoincreasing
cycles,agenericbehaviorforelectrochemicaldouble-layer
capac-itors.Therefore,werealizedthattherewaschemicaldegradation
ofNafionionomerthatledtotheincreasingcurrents.
InordertoobservetheeffectofNafionionomerdegradation
more clearly, we need to remove the capacitive currentsfrom
theXC72R.Therefore,wecarriedoutadditionalexperimentswith
theelectrodescontainingcarbonclothandNafionionomeronly.
Fig.3(a)exhibitstheCVprofilesforthesampleswiththesupply
ofambientoxygen.Asshown,thereappearedobviousoxidation
and reduction peaks centering around 0.55 and 0.34V,
respec-tively.Inaddition,thesesignalsincreasedsteadilywithincreasing
cycles. According to literature, these peaks were attributed to
hydroquinone-quinone redox couple on the carbon substrates,
suggestingtheformationofoxygenatedfunctionalgroupsonthe
surface[25–27].Alsoshownisthecarbonclothwithoutthe
addi-tionofNafionionomerbutwiththeoxygensuppliedfromambient.
Obviously,therewasnegligiblecurrentintheCVscans,indicating
thatwithoutNafionionomer,oxygenatedfunctionalgroupsonthe
carbonsurfacewerenotformedatnoticeableamount.
EarlierstudiesontheNafionmembranedegradationhave
iden-tifiedthehydroxyl (•OH)and peroxy(•OOH)radicalstobethe
activespeciestoattackthechemicalstructureofNafion.Itwas
sug-gestedthatthedissolvedoxygendiffusestotheanodesidereacting
withthehydrogentoformhydrogenperoxide[29,30].Inourcase,
withsufficientsupplyofambientoxygen,theCVscansinanacidic
environmentonthecarbonelectrodeswerelikelytoinitiatethe
oxygenreductionreactionbyatwo-electronroutewhichledto
theformationofhydrogenperoxide[41,42].Thishydrogen
perox-idesubsequentlyengenderedthedecompositionofNafionionomer
thatfurtheracceleratedtheoxidationofcarbon.Toverifythe
sig-nificanceofoxygeninthisprocess,werepeatedtheexperiments
withtheelectrodescontainingcarbonclothandNafionionomerbut
withoutthesupplyofambientoxygen.Theeliminationofoxygen
wasachievedbyimmersingtheworkingelectrodetotheelectrolyte
completelyinasealedthree-electrodecellinconjunctionwith
suf-ficientargonpurgingtoremoveanydissolvedoxygen.Theresulting
CVprofilesaredisplayedinFig.3(b).Interestingly,therewas
neg-1.2 1.0 0.8 0.6 0.4 0.2 0.0 -0.2 -3 -2 -1 0 1 2 3 I (mAcm -2 ) E (V) vs. Ag/AgCl
a
20th 10th 5th 1st carbon cloth only 1.2 1.0 0.8 0.6 0.4 0.2 0.0 -0.2 -3 -2 -1 0 1 2 3 E (V) vs. Ag/AgCl I (mAcm -2 ) 20th 1stb
Fig.3.ProfilesfrommultipleCVscans(a)withambientoxygenand(b)without
ambientoxygenforelectrodescontainingcarbonclothandNafionionomer.Also
shownin(a)istheelectrodewithcarbonclothonly.
ligibledifferencefortheCVresponsesbetweenthefirstand20th
cycle,andthehydroquinone–quinoneredoxcouplewasrather
sub-dued.Thisminuteamountofredoxcouplewaspossiblypresentin
thesamplebeforetheCVscanswereimposed.AccordingtoFig.3,
withoutthesimultaneouspresenceofoxygenandNafionionomer,
theamountofnewlyformedoxygenatedfunctionalgroupsonthe
carbonsurfacewasinsignificant.Analternativeapproachto
pro-ducethehydroxylradical(•OH)isthedirectoxidationofwater
[30].ThisisascenariothatispossibleintheCVscanswithoutthe
supplyofambientoxygen.However,fromFig.3(b)weconcluded
thatthedirectoxidationofwaterwasunabletoproducesufficient
hydroxylradicals(•OH)forNafionionomerdegradation.Therefore,
theprincipalcausefortheformationofoxygenatedfunctionalized
groupsonthecarbonsurfacewastheoxygenreductionroutethat
engenderedthedecompositionofNafionionomer.
TofurthervalidatethecontributoryroleofNafionionomerand
oxygenforcarbonfunctionalizations,additionalexperimentson
thecarbonclothandNafionionomerwerecarriedout.We
per-formed the CVscans with and withoutthe supply of ambient
oxygen,andrecordedtheiranodiccurrentsatthe20thcycle.Fig.4
exhibitsthecomparisonfortheanodiccurrentat0.5Vforboth
sam-ples,aswellasdatafromFigs.2and3(a),respectively.Apparently,
withoutthesupplyofambientoxygen,theanodiccurrentbecame
relativelysubduedforeverysample.Ingeneral,thepresenceof
1E-5 1E-4 1E-3 0.01 0.1 1 10 without O2 with O2 CC/ XC72R Nafion ionomer CC/ Nafion ionomer CC I (mAcm -2 ) Nafion ionomer
Fig.4.Comparisoninthecurrentvalueobtainedat0.5Vfromthe20thCVcycle
forelectrodescontainingcarboncloth(CC),Nafionionomer,CC/Nafionionomer,
andCC/XC72R/Nafionionomer.TheseCVexperimentsareperformedwithambient
oxygenandwithoutambientoxygen,respectively.
oxidationcurrent.However,withtheadditionofNafionionomer,
theeffectofoxygenbecamemorepronounced.Itistherefore
con-cludedthatthedegradationofNafionionomer,promotedbythe
presenceofoxygen,ledtoacceleratedcarbonfunctionalization.
3.2. Carbonfunctionalization
Fig.5demonstratestheRamanspectrafortheelectrodesafter
CVscansandH2SO4immersion.Asshown,bothsamplesrevealed
characteristicpeakswhichweredefinedasD-band(1310cm−1)
andG-band(1596cm−1),respectively.TheD-bandrepresentedthe
presenceofdefectsanddisorderinthecarbonstructurewhilethe
G-bandreflectedthegraphiticin-planevibrationswithE2g
sym-metry[17].Hence,theratioofD/Gsignalssuggestedthedegree
ofcrystallinityinthecarbonstructure.Asmentionedearlier,the
electrodeundergoingtheH2SO4immersionwasselectedfor
com-parisonpurposeanditexhibitedaD/Gvalueof2.57.Incontrast,
thesampleafterCVscansrevealedaD/Gvalueof2.67.This
mod-eratevariationintheD/Gratioinferredthatthecarbonstructure
wasreasonablymaintainedafterCVtreatmentsandtheformation
2000 1800 1600 1400 1200 1000 G-band CV scans with O2 Intensity (a.u.) Raman shift (cm-1) H 2SO4 immersion D-band
Fig.5.RamanspectraforelectrodesafterCVscanswithambientoxygenandH2SO4
immersiononly.Theseelectrodescontaincarboncloth,XC72R,andNafionionomer.
0 200 400 600 800 F1s O1s (b) (c) Intensity (a.u.)
Binding energy (eV)
(a) C1s
F
KLLFig.6.XPSsurveysfor(a)as-preparedelectrode,aswellaselectrodesafterCVscans
(b)withoutambientoxygenand(c)withambientoxygen.Theseelectrodescontain
carboncloth,XC72R,andNafionionomer.
ofoxygenatedfunctionalgroupswascausedbythedecomposition
ofNafionionomer.
TheXPSwasadoptedtoobtainvariationsonthesignalsfrom
carbon,oxygen,andfluorinefortheelectrodesunderCVscanswith
andwithoutthesupplyofambientoxygen.Wealsoperformedthe
XPSanalysisontheas-preparedelectrodewithoutCVscansfor
comparison.AsshowninFig.6,relevantpeaksontheXPSprofiles
(resolutionin1eV)werelabeledproperlyandtheywere
identi-fiedasF1s,FKLL,O1s,andC1s,respectively.Table1liststheir
respectiveatomicratios.Itcanbeseenthattherewasnegligible
differenceintheatomicratiosbetweensamplesintheas-prepared
stateandafterCVscanswithoutthesupplyofambientoxygen.
However,thesampleafterCVscanswiththesupplyofambient
oxygenrevealedasimilarcarbonamountbutitsatomicratioforthe
oxygenwasincreasedconsiderablyinconjunctionwithanotable
reductioninthefluorinecontent.Theseresultssuggestedthatthe
CVscanscoupledwiththesupplyofambientoxygenwereable
toproduceoxygen-richfunctionalgroupsontheelectrodesurface
whiletheNafionionomerwaspartiallydecomposed.
Fig.7(a)presentstheC1sXPSprofiles(resolutionin0.1eV)for
theas-preparedelectrodeandelectrodesafterCVscanswithand
withoutthesupplyofambientoxygen,respectively.Apparently,
theas-preparedsample andtheoneafterCVscanswithoutthe
supplyofambientoxygendisplayedsimilarpatternsasexpected.In
contrast,thesampleafterCVscanswiththesupplyofambient
oxy-gendemonstratedanotablepeakaround286–288eVinaddition
tothetypicalC1ssignalat284.5eV.Tounderstanditsnature,this
C1sprofilewassubjectedtocurvefittingwithknownfunctional
groupstodeterminetheirrelativeamounts.Fig.7(b)illustratesthe
curvefittingresultsandtheatomicratiosforindividualfunctional
groupsarelistedinTable2.Thesefunctionalgroupswereselected
fromearlierliteraturereportsandwerepresumedtobepresentin
thefunctionalizedelectrodes[23,25–27].FromTable2,the
sam-pleafterCVscanswiththesupplyofambientoxygenrevealeda
Table1
Theatomicratiosforcarbon,oxygen,andfluorinefromXPSprofilesforas-prepared
electrode,aswellaselectrodesafterCVscanswithandwithoutthesupplyof
ambi-entoxygen.
C(at%) O(at%) F(at%)
As-prepared 61 4.3 34.7
CVscanswithoutO2 62 3.5 34.5
Table2
TheatomicratiosfortheC–C,–OH,–C O,–COOH,andC–FfromXPScurvefittingforas-preparedelectrode,aswellaselectrodesafterCVscanswithambientoxygenand withoutambientoxygen.
C–C(at%)(backbone) –OH(at%)(a) –C O(at%)(b) –COOH(at%)(c) C–F(at%) (a+b+c)/C–C
As-prepared 63.7 14 6.3 5.7 10.3 40.8%
CVscanswithoutO2 63.7 14 6.3 5 11 39.7%
CVscanswithO2 48 14 20.6 9.1 8.1 91%
notablereductionintheamountforC–Fgroup.Inaddition,the
oxi-dized–C Oand–COOHgroupsweresubstantiallyincreasedalong
withconsiderablereductionintheC–Cbackbone.
Sofar,ourresultsindicatedthatthedecompositionofNafion
ionomerwasinitiated by theambientoxygenand this process
resultedintheformationofoxygenatedfunctionalgroupsonthe
carbonsurface.Tovalidateourpremise,weattemptedtoobtainthe
S2psignalbutthe0.5MH2SO4aqueoussolutionprovided
unnec-essarybackgroundnoises.Hence,wepreparedseveralelectrodes
(carboncloth/XC72R/Nafionionomer)andsubjectedthemtoCVs
in0.1MHClaqueoussolutioninstead.ThepurposefortheseCV
scanswastodecomposetheNafionionomersotheHClsolution
withconcentratedresidueswasformed.AccordingtoTeranishiet
al.,thedegradationofNafionproducedF−,SO32−,CO2,SO2,and
somefluorocarbons[43].Subsequently,weimmersedtheelectrode
madeofXC72RandcarbonclothintheHClsolutioncontaining
con-280 285
290 295
Intensity (a.u.)
Binding energy (eV) as-prepared CV scans without O2 CV scans with O2
a
284 288 292 Intensity (a.u.)Binding energy (eV)
b
C-C sp2/sp3 C-OH C=O C-OOH C-FFig.7.(a)C1sXPSprofilesforas-preparedelectrode,aswellaselectrodesafterCV
scanswithoutambientoxygenandwithambientoxygen.(b)CurvefittingfortheC
1sXPSprofilefromelectrodeafterCVscanswithambientoxygen.Theseelectrodes
containcarboncloth,XC72R,andNafionionomer.
Fig.8.IonchromatogramforNafionionomerdegradationin0.1MHClaqueous
solution.
centratedNafionionomerresiduestoallowsufficientadsorptionof
thedecomposedspecies.AsshowninFig.8,signalsfromtheion
chromatographywereattributedtoSO42−andF−indifferent
inten-sities.SimilarconstituentswereobservedinearlierworkbyChen
andFullerforNafionmembranedegradation[44].Intheirwork,
aratherstrongCF3COO−peakwasidentifiedonthecathodeside
associatedwiththeoxygenreductionreaction.Unfortunately,in
ourcasetheamountofCF3COO− wasbelowthedetectionlimit.
ThisnotableabsenceofCF3COO− waspossiblyduetoits
imme-diatereadsorptionontothecarbonsurfaceafterdetachmentfrom
theNafionbackbone.
To monitor the extent of Nafion ionomer degradation, we
recordedthesignalfor SO42− uponCVcyclesandtheresulting
dataaredisplayedinFig.9.Thevalueforthe0cyclewasobtained
fromthesamplewithimmersioninthe0.1MHClaqueous
solu-tionfor17min,whichservedasthereferencebecausethesample
100 80 60 40 20 0 0 1 2 3 4 5 2rd cycle Sulfate concentration (ppm) CV cycle numbers Immersion in HCl 1st cycle
Fig.9.VariationofsulfateconcentrationasafunctionofCVscanswithambient
oxygen.Thedataat0thcycleisobtainedfromtheelectrodeimmersedin0.1MHCl
Table3
TheatomicratiosforC–C,–OH,–C O,–COOH,andC–FfromC1sXPScurvefittingforas-preparedelectrode,aswellaselectrodesmadeofXC72R/carbonclothwithand
withoutimmersioninHClsolutioncontainingconcentratedresiduesfromNafionionomerdecomposition.
C–C(at%)(backbone) –OH(at%)(a) –C O(at%)(b) –COOH(at%)(c) C–F(at%) (a+b+c)/C–C
As-prepared 63.7 14 6.3 5.7 10.3 40.8%
XC72R/carbonclothwithoutimmersion 63.7 17.2 8.3 7 3.8 51%
XC72R/carbonclothwithimmersion 46.5 11.6 8.2 27.9 5.8 102.5%
of20CVcyclesexperiencedthesameamountoftimeinthe0.5M
H2SO4aqueoussolution.Asshown,thereferencesamplerevealed
sulfateconcentrationof0.35ppm.Thisreducedamountwasnot
unexpectedastheNafionionomerlikelymaintainedreasonable
chemicalstabilityagainstthe0.1MHClaqueoussolutionat26◦C.
However,onceCVscanswereapplied,thesulfateanion
concentra-tionsbecamelargerconsiderablyreachingaplateauafter20cycles
at4.3ppm.Apparently,withinthefirst20cycles,thereappeared
alinearincreaseinthesulfateconcentrationwithcyclingnumber.
ThisindicatedthatadesirableamountofNafiondecompositionand
itssubsequentcarbonfunctionalizationwaspossiblebyselecting
appropriateCVscans.
Fig.10providestheC1sXPSprofiles(resolutionin0.1eV)for
theas-prepared electrode(carboncloth/XC72R/Nafion ionomer)
as well as electrodes (carbon cloth/XC72R) with and without
immersionintheHCl solutioncontainingconcentratedresidues
fromNafionionomerdecomposition.Apparently,theelectrodeof
XC72R/carbonclothdemonstrated a singleC 1speak at 284eV
whiletheas-preparedelectrodeexhibitedanadditionalC–Fpeak
around 291eV. However, the electrode of XC72R/carbon cloth
immersed in the HCl solution with concentrated decomposed
Nafionionomerresiduesrevealedastrongsignalaround289eV
whichwasattributedtotheoxygenatedgroupsonthecarbon
sur-280 284 288 292
(c)
(b)
Intensity (a.u.)Binding energy (eV)
(a)
Fig.10.C1sXPSprofilesfor(a)as-preparedelectrode(carboncloth/XC72R/Nafion
ionomer),aswellaselectrodes (carboncloth/XC72R)(b)before and(c)after
immersioninHClsolutioncontainingconcentratedresiduesfromNafionionomer
decomposition.
face.Table3liststheatomicratiosfortheindividualfunctional
groupsfromthecurvefittingresultsofFig.10.Apparently,the
sam-pleafterimmersingintheHClsolutionshowedalargeamount
of oxygenatedfunctional groups. We surmised that theNafion
ionomerresidueintheHClsolutionwaslikelypresentasCF3COO−.
Afterchemicaladsorption,theseresiduesweretransformedtothe
oxygenatedfunctionalgroupsonthecarbonsurface.
ThechemicaladsorptionofNafionionomerresiduescanalso
beconfirmedfromtheS2pXPSprofile(resolutionin0.1eV)
dis-playedinFig.11.TheelectrodeofXC72R/carbonclothrevealeda
characteristicS2psignalnear164eVwhichwasattributedtothe
impurityintrinsictothecarbonmaterial.However,theelectrode
ofXC72R/carboncloth/Nafionionomerdemonstratedanadditional
peakaround168eVwhichwascausedbytheHSO3fromtheNafion
ionomer.Interestingly,theXC72R/carbonclothsampleimmersed
intheHClsolutionwithconcentratedNafionionomerdecomposed
residuesalsoexhibitedtheHSO3signalinadditiontotheS2pfrom
impurity.Thisfurthersupportedourpremisethatthedecomposed
Nafionionomerresidueswereabletochemicallyadsorbontothe
carbonsurface. 150 155 160 165 170 175 180
Binding energy (eV)
(c)
(b)
(a)
Intensity (a.u.) S HSO3Fig.11.S2pXPSprofilesfor(a)as-preparedelectrode(carboncloth/XC72R/Nafion
ionomer),aswellaselectrodes(carboncloth/XC72R)(b) beforeand(c) after
immersioninHClsolutioncontainingconcentratedresiduesfromNafionionomer
Table4
ElectrochemicalparametersobtainedfromCVprofilesonfunctionalizedandbaselineelectrodesformethanolelectro-oxidation.
PtaLoading(gcm−2) Anodicscan Cathodicscan
Vab iac iad iae Vcf icg ich ici
mV mAcm−2 mAPt−1mg−1 mAPt−1cm−2 mV mAcm−2 mAPt−1mg−1 mAPt−1cm−2
Functionalizedelectrode 511 729 38.9 76.1 0.47 502 43.7 85.5 0.53
Baselineelectrode 301 677 20.4 67.7 0.33 428 19.7 65.4 0.32
atotalweightofPtasdeterminedbyICP-MS.
bpeakpotentialinanodicscan.
c peakapparentcurrentdensityinanodicscan.
d peakmassactivityinanodicscan.
epeakPtsurfaceactivityinanodicscan.
f peakpotentialincathodicscan.
gpeakapparentcurrentdensityincathodicscan.
h peakmassactivityincathodicscan.
i peakPtsurfaceactivityincathodicscan.
Fig.12.TEMimagesfordepositedPtnanoparticleson(a)functionalizedand(b)
baselineelectrodes.
3.3. Methanolelectro-oxidation
Fig. 12(a)demonstrates theTEMimage for Pt nanoparticles
deposited onthe functionalized electrode followed by
electro-chemicalreduction.Asshown,therewereplentyPtnanoparticles
uniformly distributed with notable aggregations. The primary
particlesizefromtheimageanalysissoftwarewas2.68±1.62nm.
TheTEMimageonthebaselineelectrode(simpleH2SO4immersion
followedbyelectrochemicalreduction)ispresentedinFig.12(b).
Apparently, the amount of Pt nanoparticles was substantially
reduced, a fact consistent with earlier findings from ICP-MS.
In addition, their size was slightly smaller at 2.20±1.45nm.
These results confirmed that the functionalized electrode
enabled a larger amount of Pt deposits, albeit with moderate
coalescence.
Fig.13 presentsthe CVprofiles ofhydrogen desorptionand
adsorptionforthefunctionalizedandbaselineelectrodes,
respec-tively.Asexpected,thereappearedstrongerresponsesinhydrogen
desorptionandadsorptionforthefunctionalizedelectrodebecause
of its relativelylargeramount of Ptdeposit. Estimationon the
ECSA wasconductedby theintegral areafor hydrogen
desorp-tion in the anodic scan and the resulting ECSA values were
82.2 and 60.9cm2 for the functionalized and baseline
elec-trodes, respectively. This ratio of 1.35 was smaller to that of
1.70for thePtloadingfromICP-MS.We attributedthereduced
ESCAratiototheobservedPtaggregationonthefunctionalized
electrode.
Fig. 14(a) provides the CV profiles for methanol
electro-oxidationinapparentcurrentdensityforthefunctionalizedand
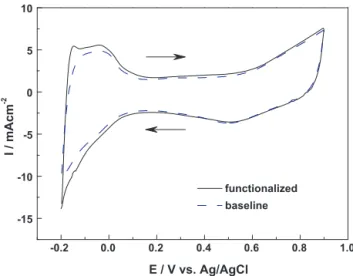
1.0 0.8 0.6 0.4 0.2 0.0 -0.2 -15 -10 -5 0 5 10 functionalized baseline I / mAcm -2 E / V vs. Ag/AgCl
1.0 0.8 0.6 0.4 0.2 0.0 -0.2 -10 0 10 20 30 40 functionalized baseline I / m A c m -2 E / V vs. Ag/AgCl
(a)
1.0 0.8 0.6 0.4 0.2 0.0 -0.2 -20 0 20 40 60 80 functionalized baseline I / mA mg -1 E / V vs. Ag/AgCl(b)
1.0 0.8 0.6 0.4 0.2 0.0 -0.2 -0.2 0.0 0.2 0.4 0.6 functionalized baseline I / mA Pt -1 surf ace cm -2 E / V vs. Ag/AgCl(c)
Fig.14.CVprofilesforfunctionalizedandbaselineelectrodesonmethanol
electro-oxidationin (a)apparent currentdensity, (b) mass activity, and (c) unit Pt
electrochemicalsurfacearea.
baselineelectrodes,respectively.Relevantelectrochemical
param-etersfromthesecurvesarelistedinTable4.Accordingtoliterature,
intheseprofilestheanodicpeak(ia)isattributedtotheoxidation
ofmethanolwhilethecathodicpeak(ic)correspondstothe
oxi-dationof carbonaceousspeciesproduced fromearliermethanol
oxidation[45–47].Inaddition,theratio(ia/ic)indicatesthe
elec-trocatalyticabilitytoremoveCO.Hence,anelectrodewithahigher
1800 1200 600 0 4 8 12 16 20 functionalized baseline I / mAmg -1 Time (sec)
Fig. 15.Chronoamperograms for functionalized and baseline electrodes on
methanolelectro-oxidationat0.5Vfor30mininmassactivity.
apparentcurrentandalargeria/icratioisalwaysdesirable.Notably,
thefunctionalizedelectrode demonstrateda substantialcurrent
incrementoverthatofbaselineelectrode.Thisnotable
improve-mentwaspartiallycausedbyalargerPtdepositswhichledtoa
highernominalcurrent.However,theia/icratioforthe
function-alizedelectrodewas0.89,whichwasslightlysmallerthan1.03of
baselineelectrode.Tocomparefairly,itisnecessarytoreplotthe
CVprofilesinmassactivityandunitPtECSA,asshowninFig.14(b)
and(c).Apparently,thereappearedaconsistenttrendinwhichthe
functionalizedelectroderevealedsignificantenhancementsover
thatofbaselineelectrode.Theseresultssuggestedthatthe
oxy-genatedfunctionalgroupswerelikelycontributingtothemethanol
electro-oxidation.Similarphenomenawerealsoreported
previ-ouslyinwhichtheoxidizedfunctional groupswerebelievedto
provideoxygen-richspeciestofacilitateCOoxidationonPtsurface
[26,48,49].
AfterconfirmingenhancementsinCVsforthefunctionalized
electrode, itis necessarytoevaluateitschronoamperogram for
lifetimedetermination.Fig.15providesthechronoamperograms
forthefunctionalizedandbaselineelectrodesat0.5Vfor30min
inmassactivity.Apparently,bothelectrodesdisplayedanotable
currentdecayinthefirst20min.However,theamountofcurrent
decaywasrelativelyconstantforbothelectrodesandthe
function-alizedelectrodewasconsistentlybetterthanthebaselineelectrode.
SincethePtwasusedinourstudy,theseperformance
degrada-tionswerenotunexpectedaspoisonousintermediateswereable
toadsorbonthePtsurfacecompromisingitscatalyticabilityfor
methanoloxidation.Itisnotedthatsimilarbehaviorswerealso
observedbyMaetal.intheirstudyofPt–Ru(OxHy)m
electrocata-lysts[50].
Sofar,ourworkdemonstratesafacileapproachto
functional-izecatalystsupportswithoutinvolvinghightemperatureandlarge
anodicpotentials.SincetheNafionionomeritselfisoftenusedas
abinderinelectrodefabrication,asimpleCVwithdissolved
oxy-gennearbycoulddecomposetheNafionionomerpartiallyresulting
intheformationofoxygenatedfunctionalgroups.Thesefunctional
groupsareactiveinassistingthePtformethanolelectro-oxidation.
It is noted that the enhancement effect observed in this work
is differentfrom conventional approaches in which the role of
NafionionomeristoextendtheinterfacialareabetweentheNafion
ionomerandelectrocatalyst[51].Moreover,thefunctionalgroups
couldpotentiallyenablealargerelectrocatalystimpregnation
lead-ingtoanimprovedstability[52].Furtherstudiesareunderwayto
exploretheanchoringeffectforthefunctionalizedgroupsandlife
4. Conclusions
WeconductedmultipleCVscansinanacidicelectrolyteonthe
electrodescontainingcarboncloth,XC72R,and Nafionionomer.
Withthesupplyofambientoxygen,theNafionionomer
experi-encedchemicalattacksleavingdecomposedresidueswhichwere
abletoadsorbontothecarbonsurfaceleadingtoanaccelerated
formationofoxygenatedfunctionalgroups.Thedecompositionof
Nafionionomerwasattributedtothehydrogenperoxideproduced
fromtheoxygenreductionreactionduringCVscans.Raman
analy-sisonthecarbonelectrodesrevealedminorstructuralmodification
afterCVscans.ResultsfromXPSsurveysindicated a significant
increaseoftheoxygenatedfunctionalgroupsonthecarbon
sur-faceinconjunctionwithanotablereductioninfluorinecontent.
The functionalized electrode was determined to allowa larger
amountofPtionadsorptionascomparedtothebaselineelectrode.
Afterelectrochemicalreduction,thePtnanoparticleswereevenly
formedonthecarbonsupports.Electrochemicalanalysisonthe
methanolelectro-oxidationwasperformedandweobserved
sig-nificantincrementsinapparentcurrentdensity,massactivity,as
wellasunitPtESCAforthefunctionalizedelectrode.
Acknowledgement
TheauthorsaregratefultoProfessorChuen-JinnTsaiandMiss
Yi-LinLiufromtheInstituteofEnvironmentalEngineeringfortheir
kindassistancewiththelaboratoryequipment.Financialsupports
fromNationalScienceCouncilofTaiwan(NSC-96-2221-E-009-110)
andNationalSynchrotronRadiationResearchCenter
(2009-2-063-1)areacknowledged.
References
[1] A.L.Dicks,J.PowerSources156(2006)128–141.
[2]M.S.Saha,A.Kundu,J.PowerSources195(2010)6255–6261.
[3]J.F.Drillet,H.Bueb,R.Dittmeyer,U.Dettlaff-Weglikowska,S.Roth,J.
Elec-trochem.Soc.156(2009)F137–F144.
[4]J.H.Kim,B.Fang,S.B.Yoon,J.S.Yu,Appl.Catal.B88(2009)368–375.
[5]J.F.Lin,V.Kamavaram,A.M.Kannan,J.PowerSources195(2010)466–470.
[6]A. Guha, T.A. Zawodzinski, D.A. Schiraldi, J. Power Sources 195 (2010)
5167–5175.
[7]N.Spataru,X.T.Zhang,T.Spataru,D.A.Tryk,A.Fujishima,J.Electrochem.Soc.
155(2008)B264–B269.
[8]V.Baglio,A.DiBlasi,C.D’Urso,V.Antonucci,A.S.Arico,R.Ornelas,D.
Morales-Acosta,J.Ledesma-Garcia,L.A.Godinez,L.G.Arriaga,L.Alvarez-Contreras,J.
Electrochem.Soc.155(2008)B829–B833.
[9]N.Y.Hsu,C.C.Chien,K.T.Jeng,Appl.Catal.B84(2008)196–203.
[10]S.Maass,F.Finsterwalder,G.Frank,R.Hartmann,C.Merten,J.PowerSources
176(2008)444–451.
[11]W.Phompan,N.Hansupalak,J.PowerSources196(2011)147–152.
[12] Y.F.Hsieh,Y.C.Hsieh,P.W.Wu,C.H.Liao,Y.M.Chang,J.Electrochem.Soc.157
(2010)B39–B44.
[13]J.R.C.Salgado,F.Alcaide,G.Alvarez,L.Calvillo,M.J.Lazaro,E.Pastor,J.Power
Sources195(2010)4022–4029.
[14]Y.M.Chang,Y.C.Hsieh,P.W.Wu,DiamondRelat.Mater.18(2009)501–504.
[15]R.I.Jafri,T.Arockiados,N.Rajalakshmi,S.Ramaprabhu,J.Electrochem.Soc.157
(2010)B874–B879.
[16]B.Liu,S.Creager,J.PowerSources195(2010)1812–1820.
[17]G.X.Zhang,S.H.Sun,D.Q.Yang,J.P.Dodelet,E.Sacher,Carbon46(2008)
196–205.
[18]P.L.Antonucci,F.Romeo,M.Minutoli,E.Alderucci,N.Giordano,Carbon26
(1988)197–203.
[19]N.Giordano,P.L.Antonucci,E.Passalacqua,L.Pino,A.S.Arico,K.Kinoshita,
Electrochim.Acta36(1991)1931–1935.
[20]E.Passalacqua,P.L.Antonucci,M.Vivaldi,A.Patti,V.Antonucci,N.Giordano,K.
Kinoshita,Electrochim.Acta37(1992)2725–2730.
[21]S.I.Pyun,E.J.Lee,T.Y.Kim,S.J.Lee,Y.G.Ryu,C.S.Kim,Carbon32(1994)
155–159.
[22] A.M.Puziy,O.I.Poddubnaya,V.N.Zaitsev,O.P.Konoplitska,Appl.Surf.Sci.221
(2004)421–429.
[23]Z.R.Yue,W.Jiang,L.Wang,S.D.Gardner,C.U.Pittman,Carbon37(1999)
1785–1796.
[24]R.Berenquer,J.P.Marco-Lozar,C.Quijada,D.Cazorla-Amoros,E.Morallon,
Car-bon47(2009)1018–1027.
[25]K.H.Kangasniemi,D.A. Condit,T.D.Jarvi, J.Electrochem.Soc.151 (2004)
E125–E132.
[26]S.Stevanovic,V.Panic,D.Tripkovic,V.M.Jovanovic,Electrochem.Commun.11
(2009)18–21.
[27]J.J.Wang,G.P.Yin,Y.Y.Shao,S.Zhang,Z.B.Wang,Y.Z.Gao,J.PowerSources171
(2007)331–339.
[28]C.Chen,T.F.Fuller,J.Electrochem.Soc.156(2009)B1218–B1224.
[29] C.Chen,G.Levitin,D.W.Hess,T.F.Fuller,J.PowerSources169(2007)288–295.
[30]F.A.deBruijn,V.A.T.Dam,G.J.M.Janssen,FuelCells8(2008)3–22.
[31]D.A.Schiraldi,Polym.Rev.46(2006)315–327.
[32]D.E.Curtin,R.D.Lousenberg,T.J.Henry,P.C.Tangeman,M.E.Tisack,J.Power
Sources131(2004)41–48.
[33]G.Hubner,E.Roduner,J.Mater.Chem.9(1999)409–418.
[34]T.Xie,C.A.Hayden,Polymer48(2007)5497–5506.
[35]H.P.Boehm,Carbon32(1994)759–769.
[36] A.Bravo,H.R.Bjorsvik,F.Fontana,L.Liguori,A.Mele,F.Minisci,J.Org.Chem.
62(1997)7128–7136.
[37]M.Sansotera,C.L.Bianchi,G.Lecardi,G.Marchionni,P.Metrangolo,G.Resnati,
W.Navarrini,Chem.Mater.21(2009)4498–4504.
[38] J.Xie,D.L.Wood,K.L.More,P.Atanassov,R.L.Borup,J.Electrochem.Soc.152
(2005)A1011–A1020.
[39]T.Morimoto,K.Hiratsuka,Y.Sanada,K.Kurihara,J.PowerSources60(1996)
239–247.
[40] C.T.Hsieh,H.Teng,Carbon40(2002)667–674.
[41] K.Kinoshita,Oxygenelectrochemistry,in:ElectrochemicalOxygenTechnology,
JohnWiley&Sons,NewYork,1992,pp.21–26.
[42]C.Chen,T.F.Fuller,Electrochim.Acta54(2009)3984–3995.
[43] K.Teranishi,K.Kawata,S.Tsushima,S.Hirai,Electrochem.Solid-StateLett.9
(2006)A475–A477.
[44]C.Chen,T.F.Fuller,Polym.Degrad.Stab.94(2009)1436–1447.
[45]T.C.Deivaraj,J.Y.Lee,J.PowerSources142(2005)43–49.
[46] Z.B.Wang,G.P.Yin,J.Zhang,Y.C.Sun,P.F.Shi,Electrochim.Acta51(2006)
5691–5697.
[47]Z.L.Liu,X.Y.Ling,B.Guo,L.Hong,J.Y.Lee,J.PowerSources167(2007)272–280.
[48]J.H.Chen,M.Y.Wang,B.Liu,Z.Fan,K.Z.Cui,Y.Kuang,J.Phys.Chem.B110
(2006)11775–11779.
[49]M.A.Scibioh,I.H.Oh,T.H.Lim,S.A.Hong,H.Y.Ha,Appl.Catal.B77(2008)
373–385.
[50]J.H.Ma,Y.Y.Feng,J.Yu, D.Zhao,A.J.Wang,B.Q.Xu,J. Catal.275(2010)
34–44.
[51]C.H.Park,M.A.Scibioh,H.J.Kim,I.H.Oh,S.A.Hong,H.Y.Ha,J.PowerSources
162(2006)1023–1028.
[52]W.M.Chen,Q.Xin,G.Q.Sun,Q.Wang,Q.Mao,H.D.Su,J.PowerSources180