The influence of washing and calcination condition on urea-derived
ceria-yttria-doped tetragonal zirconia powders
Jyung-Dong Lin
a, Jenq-Gong Duh
a,∗, Bi-Shiou Chiou
baDepartment of Materials Science and Engineering, National Tsing Hua University, Hsinchu 300, Taiwan, ROC bDepartment of Electronics Engineering and Institute of Electronics, National Chiao Tung University, Hsinchu 300, Taiwan, ROC
Received 10 October 1999; received in revised form 18 March 2000; accepted 28 March 2000
Abstract
The influence of washing and calcination conditions on the powder characteristics of urea-derived ceria-yttria-doped tetragonal zirconia powders (Ce-Y-TZP) ceramics are studied. The use of urea hydrolysis followed by acetone–toluene–acetone (ATA) washing can produce a finer and more sinterable Ce-Y-TZP powder as compared to that washed by alcohol. It is observed that as the calcination temperature is raised to 800◦C for 2 h, the sintered density achieves the highest value regardless of the increase of monoclinic content in as-derived powders. The higher the calcination temperature, the more the monoclinic content. The transformability of tetragonal phase is related to the
Astemperature and tetragonality. The monoclinic content in as-derived powders after calcination and ball-milling processes is proportional
to the Astemperature and tetragonality of tetragonal phase. © 2001 Elsevier Science B.V. All rights reserved.
Keywords: Calcination; ATA washing; Ce-Y-TZP powders
1. Introduction
The mechanical properties and reliability of ceramic ma-terials are governed invariably by processing flaws [1]. An appropriate powder preparation is necessary to improve both mechanical properties and reliability of ceramics. Recently, the hydrolysis method has been extensively employed in the fabrication of fine ceramics due to its convenience and con-trollability of processes [2–8]. Urea hydrolysis is one exam-ple, which gives a relatively pure precipitate, and the degree of supersaturation is greatly decreased during precipitation [9]. In contrast, the fast co-precipitation, as with ammonia co-precipitation, of what is performed under high pH value (>10.7) and often merges a few impurities. These impu-rities may deteriorate the mechanical properties due to an amorphous layer formed in the grain boundary [10,11]. In addition, according to our previous results [7], the zirconia powder synthesised by urea hydrolysis can achieve 100% tetragonal owing to its moderate precipitation reaction.
The degree of supersaturation during precipitation deter-mines the number of nuclei formed and thus controls the particle size of the precipitate. Thus, the urea hydrolysis usually yields a coarse powder due to its low supersatura-tion and the higher temperature used (∼80–100◦C) [6,9]. A suitable crystallisation treatment will significantly affect the characteristics of the resultant powders. It is thus worth
fur-∗Corresponding author.
ther studying the characteristics of the urea-derived powders followed by subsequent crystallisation treatment.
Although acetone–toluene–acetone (ATA) washing has been successfully employed to produce sinterable ZrO2 or HfO2 powders [3,12], many investigators have also argued its effect to yield active powders as compared to alcohol washing in cases of CaO–ZrO2 [13] or Y2O3–ZrO2 [14]. This would be due to the nature of gels since various pre-cipitation processes and dopants are used. It is also likely that the processing of ATA washing is not optimised such that the powder characteristics could not be improved.
The agglomeration state of powder significantly in-fluences the sintering behaviour. A preferential intra-agglomerate sintering predominates over inter-intra-agglomerate sintering and pulls away from neighbouring agglomerate, leaving large lenticular voids which are difficult to close [15]. Alternatively, the calcination treatment would mod-ify the size, morphology, the crystallographic defective configuration and the surface chemistry [16] of powder particles. Recently, Montanaro et al. [17] stated that the higher calcination temperature (∼1000◦C) can prevent the abnormal grain growth of cubic phase and a higher flex-ural strength for a pure coprecipitated 6 mol% YO1.5-TZP powder is achieved. As a result, one can expect that the suitable calcination condition will alter the porous and de-fective agglomerates into a denser structure and thus the intra-agglomerate sintering is eliminated.
0254-0584/01/$ – see front matter © 2001 Elsevier Science B.V. All rights reserved. PII: S 0 2 5 4 - 0 5 8 4 ( 0 0 ) 0 0 2 9 0 - X
J.-D. Lin et al. / Materials Chemistry and Physics 68 (2001) 42–55 43 This work is one of our series of tasks in the development
of high strength and thermally stable CeO2–Y2O3–ZrO2 ceramics [7,8,18–21]. In the current study, the influences of organic medium washing and calcination condition on urea-derived powder characteristics, especially, particle size distribution, sinterability and monoclinic content in as-derived powders, will be explored in detail. In addition, the grain morphology of sintered body is also studied.
2. Experimental procedure
A urea-hydrolysis process was applied to produce fine powders. Starting materials of ZrOCl2·8H2O, Ce(NO3)3·
Fig. 1. The experimental flowchart for studying the effect of organic washing on powder characteristics.
6H2O, and Y(NO3)3·5H2O (Merck & Co. Inc., Darmstadt, Germany) with various compositions were used to prepare the stock solution with a cation concentration of 0.1 M. An excess addition of urea (0.42 M) was introduced for en-hancing the hydrolysis reaction. The solutions were boiled continuously for 5 h in a flask and then a white colloidal suspension was formed after 50 min, as previously reported [7]. Usually the post-precipitation washing with organic solvent significantly improves the sinterability of powders. In this study, comparison between the ATA washing and ethanol washing was made. The precipitate produced by urea hydrolysis was washed by these two methods and then calcined at 500◦C for 0.5 h. The detailed procedure of
organic washing is shown in Fig. 1. After organic washing was carried out, calcination was employed to control the particle size and size distribution of the powder. The cal-cined powders were then wet-milled in ethanol for 36 h and subsequently dried by infrared lamp.
The effect of calcination on the sintered density was further studied. Once ATA washing was finished, the pow-der was calcined at 500◦C for 0.5, 2, 4, and 8 h, and at 700, 800, 850, and 1000◦C for 2 h. The powders were uniaxially pressed at ∼80 MPa and were then isostatically cold-pressed at 200 MPa. Finally, the powder compacts were sintered at 1300, 1400, and 1500◦C for 2 h. The density of samples was measured to explore the optimum calcination condition for urea-derived and ATA-washed ceria-yttria-doped tetragonal zirconia powders (Ce-Y-TZP).
The as-derived powders composition were measured by inductively coupled plasma (ICP, GVM-1000P, Shimadzu, Japan). The particle size distribution was measured by a SediGraph 5100 particle size analysis system (Model 5100, Micrometrics Instrument Corp., USA). The particles de-posited on copper grid were examined by transmission elec-tron microscopy (JEM-100cxII, Jeol, Japan), and the size of agglomerate was also probed.
The volume fraction of monoclinic phase, Vm, was eval-uated by the following equation [22]:
Vm= 1.311Xm
1+ 0.311Xm (1)
Xm= I(¯1 1 1)m+ I(1 1 1)m
I(1 1 1)t + I(¯1 1 1)m+ I(1 1 1)m
(2)
where Xmis the integrated intensity ratio and the subscripts ‘m’ and ‘t’ of I represent the intensity of K␣1for monoclinic and tetragonal phase, respectively, after the peak separation and fitting.
The crystallite size and microstrain of ultrafine zirco-nia powders were determined through an X-ray diffrac-tion line-broadening method. Through the software based on the Warren–Averbach procedure, the crystallite size and microstrain of both t and m phases were thus deter-mined. A detailed analysis procedure has been reported elsewhere [21]. The density of powders and bulk den-sity were measured by a gas pycnometry (AccuPyc 1330, Micrometrics Instrument Corp., USA) and Archimedes technique, respectively, with water as the immersion medium.
Samples were sintered at 1500◦C for 2 h and were subse-quently polished and thermally etched at 1500◦C for 10 min. Field emission scanning electron microscopy (FESEM, Hi-tachi S-4000, Japan) was used to examine the morphology of polished and thermally etched surface. The grain size, ¯D, of the sintered samples was estimated using the linear inter-cept technique [23].
3. Results and discussion
3.1. Urea hydrolysis
Firstly, the pH value of the solution in terms of boiling time is shown in Fig. 2. The pH value of suspension solution reaches a stable value of 8.2 after 4 h boiling. The precipita-tion process begins at pH 4 and finishes at pH 8.1. Compared with ammonia coprecipitation, precipitation formed by urea hydrolysis processes slowly under the lower pH environ-ments during a period of∼3 h [7]. For a batch of 10 g pow-ders yielded, Fig. 3a–c show the particle size distribution of powders synthesised by ammonia coprecipitation plus cal-cination, urea hydrolysis plus hydrothermal crystallisation (HTX), and urea hydrolysis plus calcination, respectively. Except for the HTX-treated powders, the other two pow-ders are ball-milled in alcohol for 36 h (the milling media is zirconia). The powders produced by ammonia coprecipi-tation and calcination at 500◦C for 0.5 h has the finest size. The HTX-treated powder exhibits a wide distribution of par-ticle size ranged from 10 to 1m and shows the coarsest mean particle size than others. The larger particle size of HTX-treated powders may be due to the process without ball-milling and/or the use of high concentration gels in the HTX treatment resulting from the restriction of vessel vol-ume [7].
Similar to binary systems [8], the actual compositions of coprecipitated powders are nearly identical to the nominal compositions, as indicated in Fig. 4a and b. The relation be-tween measured compositions and nominal compositions of Ce-Y-TZP ternary ceramics can be expressed by the follow-ing equations:
Fig. 2. The pH value of the stock solution in terms of boiling time at 100◦C during hydrolysis.
J.-D. Lin et al. / Materials Chemistry and Physics 68 (2001) 42–55 45
Fig. 3. The particle size distribution of 5.5 mol% CeO2–2 mol% YO1.5–ZrO2 powders produced by various methods: (a) ammonia coprecipitation and
calcination; (b) urea hydrolysis and HTX; (c) urea hydrolysis and calcination.
mol% Ymeasured= 0.0423 + 0.9143 mol% Ynominal, R2= 0.998, S.D. = 0.092 (3)
mol% Cemeasured= 0.1147 + 0.9206 mol% Cenominal, R2= 0.993, S.D. = 0.288 (4) where R2 represents square of correlation coefficient and S.D. is the standard deviation. The relationships between measured compositions and nominal compositions derived from linear regression of data are nearly identical regardless the addition of another stabiliser contents. Furthermore, un-like the case of binary system [8], the deviation from nom-inal composition of cerium in these Ce-Y-TZP ceramics is comparable to that of yttrium. Alternatively, standard
devi-ation of measured cerium amount with respect to the nom-inal one is larger than that in yttrium. This implies that in the fabrication of Ce-Y-TZP powders, the control of cerium content is relatively difficult as compared to that of yttrium.
3.2. Organic medium washing
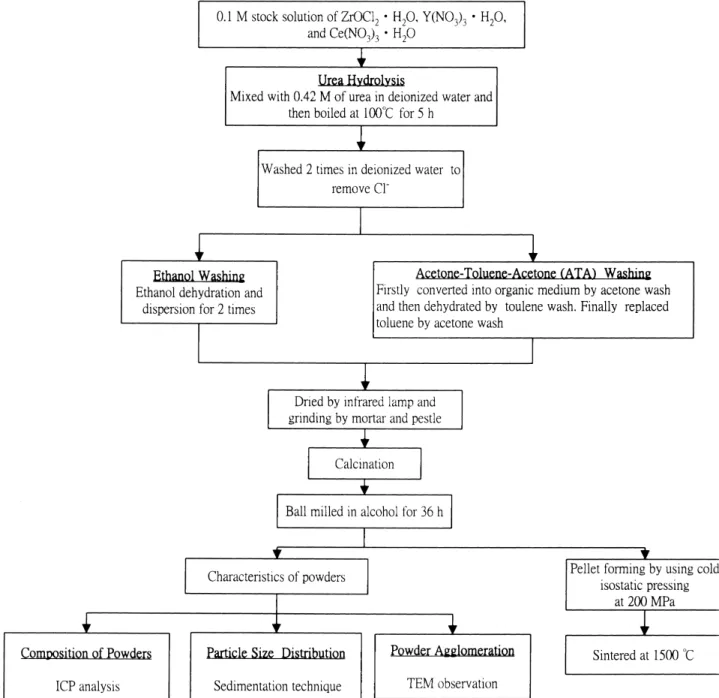
Fig. 5 compares the particle size distribution of the un-calcined powders after alcohol washing with that of ATA washing. Powders treated by ATA washing exhibits rela-tively narrow distribution, being mostly around, 1–10m. whereas alcohol-derived powders show a rather wide dis-tribution, ranging from 20 to 1m. The TEM micrograph also provides this evidence, as shown in Fig. 6, in which the size of uncalcined alcohol-derived powder is smaller than
Fig. 4. The relation between measured composition and nominal compo-sition in urea-derived powders: (a) cerium; (b) yttrium.
that of ATA-derived powder. After calcination at 500◦C for 0.5 and 36 h ball-milling in alcohol, the ATA-derived amor-phous powder is converted into a finer powder as compared to alcohol-derived one (Fig. 3c). According to TEM obser-vation, the calcined alcohol-derived powder often exhibits a degree of large agglomerates (Fig. 6b). Similarly, on the ba-sis of sintering experiments, the ATA-derived powders can reach higher density and shows larger shrinkage at the same sintering temperatures (Fig. 7). In summary, the ATA wash-ing is more effective than alcohol washwash-ing to obtain active and sinterable urea-derived powders.
Alternatively, ATA washing can inhibit the formation of m phase after calcined at 500◦C for 0.5 h and ball-milled in ethanol for 36 h (Table 1). Since the ball-milling can enhance the occurrence of t→m phase transformation in the TZP ceramic powder [24–26], the ball-milling step
Fig. 5. Particle size distribution of urea-derived amorphous powder after ATA washing and alcohol washing and infrared drying.
raises the amount of m phase in milled powders. After a 36 h ball-milling, the alcohol-derived powders contain a small amount of m phase, and the presence of m phase in the powders depends on the amount of stabiliser added (Table 1). ATA-washed powders still exhibit pure tetrago-nal phase even after 36 h ball-milled in ethanol regardless of stabiliser content. The presence of monoclinic phase in alcohol-derived powder would be related to its agglom-eration state of powders. Since the alcohol-derived pow-ders usually contain larger agglomerates after calcination (∼4 m, as shown in Fig. 6b), the ball-milling process can stress-induce these larger particles into m phase.
Table 2 summaries powder characteristics and sintered density of powders prepared by three synthesis methods. The urea-derived powder exhibits the highest powder den-sity, while the HTX-treated one shows the highest surface area, highest green density, and smallest powder density. This implies that the HTX-treated powder is very porous or post-ultrasonic dispersion does not break the agglomer-ates of powders. In addition, ATA washing can significantly improve the sinterability of urea-derived powders as com-pared to alcohol washing. It is argued that ethanol wash-ing only eliminates the H2O adsorbed on the surface and terminal hydroxyl of the hydroxide. But the hydroxyl lig-and within the structure lig-and inside the hydroxide particles would still exist. To obtain an uniform and more active pow-der, the majority of OH− ligand inside hydroxide should be replaced by organic medium using ATA procedure. Ac-cording to the result of DTA, obscure peaks related to hy-drogen bond interaction between gel and water are not ob-served in the urea-derived and ATA washed powders [7,20]. This implies that reduction in agglomeration reaction occurs in these ATA washed powders due to the absence of OH− ligand.
J.-D. Lin et al. / Materials Chemistry and Physics 68 (2001) 42–55 47
Fig. 6. TEM micrographs of urea-derived 5.5 mol% CeO2–2 mol% YO1.5–ZrO2powders produced by various methods: (a) alcohol washing and infrared
drying; (b) alcohol washing and calcination 500◦C for 0.5 h; (c) ATA washing and infrared drying; (d) ATA washing and calcination 500◦C for 0.5 h.
Table 1
The compositions, characteristics, and monoclinic content of Ce-Y-TZP powders were treated by alcohol and ATA washing after calcination at 500◦C for 0.5 h then followed by 36 h ball-milling in ethanol
Sample designation Cea (mol%) Ya(mol%) Crystallite size
of t phasehDiab(nm) Microstrain hε2i1/2 L=(1/2)hDia b (×102) Monoclinic content Vm (vol.%) Alcohol washing 5.5–2-C36 5.29 1.92 5.26 2.15 22.4 5.5–3-C36 5.30 2.77 5.26 2.13 12.7 5.5–4-C36 5.23 3.62 5.36 2.52 0 8–2-C36 7.26 1.86 5.46 2.11 12.1 8–3-C36 7.48 2.36 5.53 2.12 9.4 8–4-C36 7.53 3.74 5.31 2.53 0 ATA washing 5.5–1.5-ATA 5.11 1.51 4.85 2.18 0 5–2-ATA – – 5.43 2.13 0 5.5–2.5-ATA – – 5.64 2.33 0 6–2-ATA – – 5.55 2.29 0 8–1.5-ATA 7.71 1.50 6.04 2.32 0
aMeasured by ICP technique.
bArea-weighted mean crystallite sizehDi
a and microstrainhε2i1/2
L=(1/2)hDia calculated from Warren–Averbach method [21].
3.3. The effects of calcination time and temperature
The choice of calcination condition is important to achieve high density. In fact, this requires a compromise between green density and fineness of powders. The higher the green density, the higher the sintered density. For example, the use of CIP tends to enhance the density of green body and results in an increase of sintered density at lower temper-ature (≤1400◦C), as shown in Fig. 8. The agglomeration state of the powders will influence the sintering behaviour [16,27].
After the calcination process, ball-milling in alcohol is necessary to deagglomerate the calcined powders. In fact, the powder density and green density of calcined powders at lower temperature (at 500◦C for 0.5 h) are around 83 and 38.6% T.D., respectively. To improve the density, the time and temperature of calcination is tentatively raised in the
Table 2
Powder characteristics of 5.5 mol% CeO2–2 mol% YO1.5–ZrO2 powders prepared by three kinds processes
Powder synthesis crystallization treatment with washing method Ammonia coprecipitation Urea hydrolysis Calcinationd Hydrothermal crystallisatione Calcinationa
Alcohol washing – ATA washing Alcohol washing
Phase t and m t t t and m
surface area (m2g−1) 88–108.7 200–125 108c –
Particle sizea (m) 0.53 6.39c 0.76 1.65
Powder densityb (apparent density) (g cm−3) 5.04 4.18 5.22 –
Green density (g cm−3) 2.24 2.70–2.97 2.36 2.5
Sintered density (% T.D.) 97.5 97.7 97.6 95.4
aModal diameter.
bMeasured by gas pycnometer. cWithout ball-milled.
dCalcination at 500◦C for 0.5 h followed by 36 h ball-milling in ethanol. eAt 200◦C for 5 h.
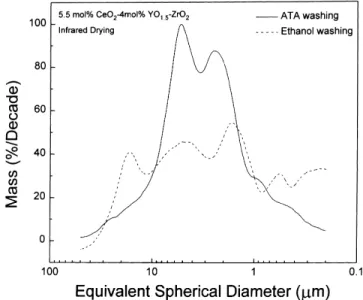
present study. When the calcination time is increased from 2 to 8.6 h at 500◦C, the sintered density of samples is indicated in Fig. 9. It appears that the increase of calcination time at 500◦C fails, however, to significantly enhance the green den-sity of samples with a green denden-sity around 35% T.D. after formed by uniaxial cold-pressing. As the sintering temper-ature is raised beyond 1400◦C, the final density of sintered samples is reduced with calcination time. As the calcination temperature is raised, both the mean particle size and green density increase, as shown in Figs. 10 and 11, respectively. Fig. 10 indicates that the increment of mean particle size of the calcined powders is much pronounced as calcination temperature is raised up to 1000◦C. For the calcination temperature in the range of 500–850◦C, the mean particle size is around 1 mm. Once the calcination temperature is raised to 1000◦C, the particle significantly grows to a size around 2m. Besides, the shape of particle size distribution
J.-D. Lin et al. / Materials Chemistry and Physics 68 (2001) 42–55 49
Fig. 8. The comparison of relative density for the 5.5 mol% CeO2–4 mol% YO1.5–ZrO2 powders formed by CIP and uniaxial press.
remains nearly unchanged regardless of calcination temper-ature, while the width of particle size distribution becomes slightly broadened with calcination temperature as tempera-ture is<850◦C. In contrast, further increase of temperature up to 1000◦C reduces the width of particle size distribution.
Fig. 9. The relative density of 5.5 mol% CeO2–1.5 mol% YO1.5–ZrO2 ceramics. Powders were calcined at 500◦C for 2, 4, and 8.6 h. It appears that as the calcination temperature is at 800◦C, the sintered density stands the highest. The crystallite size of the t phase increases with the calcination temper-ature, whereas the microstrain,hε2i1L=(1/2)hDi/2
a, of t phase
Fig. 10. The particle size distribution of 5.5 mol% CeO2–2 mol% YO1.5–ZrO2 powders after calcination at various temperatures.
J.-D. Lin et al. / Materials Chemistry and Physics 68 (2001) 42–55 51 Table 3
The effect of calcination temperature on the crystallite size and microstrain of t phase and monoclinic content of the urea-derived 5.5 mol% CeO2–2 mol%
YO1.5–ZrO2 powders Calcinationa temperature (◦C) Crystallite size of t phasehDia (nm) Microstrainhε2i1/2 L=(1/2)hDia (×102) Monoclinic contentc Vm(vol.%) Secondary particle sizeb(m) 500 5.26 2.66 0 0.76 750 6.66 2.08 28.5 0.96 850 8.80 1.98 40.9 0.93 1000 12.88 1.46 43.5 1.59 aFor 2 h.
bModal particle size, indicating particle size at peak of particle size distribution. cAs-calcined powders.
Table 4
The characteristics of ternary Ce-Y-TZP ceramic powders produced by urea hydrolysis, ATA washing, and calcination at 800◦C for 2 h Sample designation Cea (mol%) Ya (mol%) Crystallite size of t phasehDia (nm) Microstrain hε2i1/2 L=(1/2)hDia (×102) Monoclinic content Vm (vol.%) 3.5–x (x=2, 3, 4) 3.5–2 3.20 1.96 9.28 0.85 73 3.5–3 3.18 2.91 9.74 1.06 54.3 3.5–4 3.13 3.99 9.46 1.24 36.1 5.5–x (x=1.5, 2, 2.5, 3, 4) 5.5–1.5 5.03 1.48 9.98 1.11 56 5.5–2 5.00 1.98 10.09 1.09 50.9 5.5–2.5 4.84 2.47 9.77 1.21 42.1 5.5–3 4.93 2.92 9.60 1.16 38.9 5.5–4 4.81 3.97 9.98 1.26 16.3 8–x (x=1.5, 2, 2.5, 3, 4) 8–1.5 7.20 1.45 9.98 1.17 33.9 8–2 7.35 2.06 11.02 1.24 29 8–2.5 7.48 2.45 12.11 1.28 13.3 8–3 7.63 2.97 10.20 1.26 17.1 8–4 7.53 3.95 10.78 1.28 0.9 References 0–6 0.02 6.10 10.69 1.26 25 12–0 11.11 0.03 15.61 0.83 17.4
aMeasured by ICP technique.
Table 5
The lattice parameters, theoretical density, and tetragonality of Ce-Y-TZP ceramics sintered at 1500◦C for 2 h
Ce (mol%) Y (mol%) a (nm) c (nm) Theoreticala density (g cm−3) Tetragonality (c/a)
3.18 2.91 0.51066 0.51945 6.11 1.0172 3.13 3.99 0.51100 0.51929 6.09 1.0162 4.51 2 0.51091 0.52010 6.13 1.0180 5.11 1.5 0.51118 0.52056 6.13 1.0183 5.09 2.03 0.51123 0.52036 6.13 1.0179 5.11 2.5 0.51143 0.52029 6.13 1.0173 6.22 2.79 0.511472 0.52019 6.15 1.0170 6.09 3.77 0.51180 0.52001 6.14 1.0160 6.06 4.4 0.51166 0.51965 6.14 1.0156 7.71 1.5 0.51198 0.52120 6.17 1.0180 7.35 2.06 0.51211 0.52087 6.16 1.0171 8.47 2.79 0.51209 0.52084 6.18 1.0171 8.20 3.56 0.51248 0.52061 6.16 1.0159 8.16 4.4 0.51265 0.52040 6.16 1.0151
Table 3 (column length (L), a multiple of the interplanar spacing). Additionally, as the calcination temperature is raised, the m content in powders also increases. As indi-cated in Table 3, when the powder is calcined at 1000◦C for 2 h, the m content is 43.5 vol.%. The presence of m phase may affect the sintering state of powders due to the formation of m→t phase transformation during sintering. However, according to the result of the current study, the effect of these large amounts of m phase on the sintered density and shrinkage of sintered bulk is not apprecia-ble for powders sintered at 1500◦C for 2 h, as shown in Fig. 11.
The crystallite size of these ternary powders is indepen-dent of the composition and exhibits a value around 5.4 nm after calcined at 500◦C for 0.5 h, as indicated in Table 1. Once the calcination is conducted at 800◦C for 2 h, the crys-tallite size increases into 10 nm. For powders calcined at 800◦C for 2 h, the m content in powders decreases with de-creasing YO1.5 content. Only the powders with composi-tions of 8.0 mol% CeO2and 4 mol% YO1.5possess 100% t phase, as indicated in Table 4.
It is interesting to probe the relation of the monoclinic con-tent and the powder synthesis process employed. Firstly, as mentioned in Section 3.2, the urea-derived powders washed by ATA procedure also yield 100% tetragonal phase when calcined at low temperature (500◦C). In addition, for the highly transformable powder with composition of 5.5 mol% CeO2–2 mol% YO1.5–ZrO2, the gels derived by ammonia coprecipitation can transform into fully tetragonal phase by HTX, while no tetragonal is obtained by calcination alone, as previously reported [7].
Besides, as indicated in Tables 1, 3 and 4, the microstrain of powders atL = (1/2)hDiadecreases from 2.2×10−2to 1.2×10−2as the calcination temperature is raised from 500 to 800◦C. This implies that the higher calcination tempera-ture reduces the defects in the crystal, and the crystallinity of lattice is enhanced. This result is in agreement with the work of Morterra et al. [16], in which detailed FTIR and, CO and CO2adsorption spectrums of the Y-TZP powders calcined at various temperatures reveal a much more defective and reactive surface for powders calcined at lower temperature (500◦C).
3.4. Lattice parameters and tetragonality
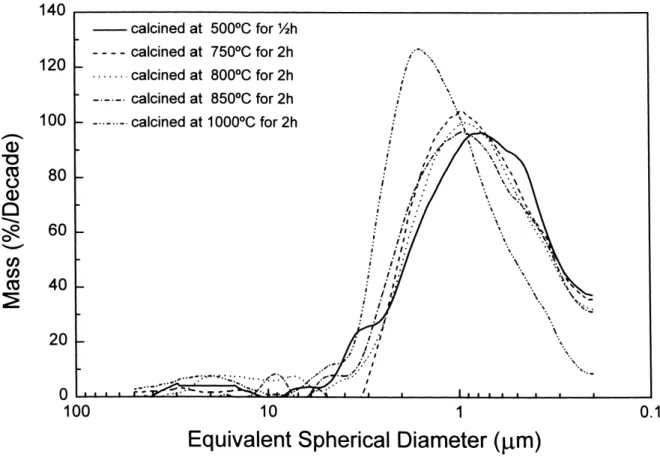
The lattice parameters of t phase in the Ce–Y–TZP ternary ceramic system have been measured by X-ray pow-der diffraction. Table 5 represents the lattice parameters of t phase for Ce–Y–TZP ceramics sintered at 1500◦C for 2 h. The tetragonality c/a can be used to evaluate the transformability of t phase [28]. As the tetragonality of t phase approaches 1, the stability of t phase is raised. Fig. 12 shows the contour of tetragonality for Ce-Y-TZP ceramics as functions of mol% CeO2 and mol% YO1.5. Although the tetragonality in the current study is slightly larger than that in the study of Urabe et al. [29], the
Fig. 12. The tetragonality contour as the function of mol% CeO2 and
mol% YO1.5. The tetragonality values are calculated according to Eq. (5).
trend of tetragonality with respect to compositions is iden-tical. The transformability of the t phase under an ap-plied stress will influence the fracture toughness of TZP materials.
In addition to tetragonality of t phase, the transforma-bility of t phase is also related to the Ms temperature (the starting temperature of t→m transformation) [30–32]. This means that the Ms temperature of high toughened TZP ma-terials should be close to room temperature. Mstemperature is usually dependent on the dopant amount, grain size and shape of t phase [30–32]. Urabe et al. [29] reported, however, that the As temperature (the starting temperature of m→t transformation) of Ce-Y-TZP ceramics can be expressed by
As(◦C) = 1195.5 − 170.7 YO1.5(mol%)
−72.2 CeO2(mol%) (5)
In general, Astemperature is sensitive to the dopant content, yet is less influenced by the grain size as compared to Ms temperature. Bastide et al. [33] reported that the difference between Ms and As temperatures for (1–x)[ZrO2–2 mol% YO1.5]xCeO2 (x=0, 1.3, 2.1 and 3 mol%) ceramics with grain size of∼1 m is around 130◦C regardless of stabiliser contents. Thus, Ms temperature is replaced by As tempera-ture in further discussion. On the basis of Eq. (5), the ceria exhibits a weaker effect than the yttria in depressing Ms or
Astemperatures.
As discussed in the previous section, the amount of m phase in the calcined and ball-milled powders depends on the calcination temperature and dopant amount. For the same yttrium doping level, the more the ceria content, the less the content of m phase in powders. The increase of calcination temperature will raise the crystallite size of t phase and the increase in stabiliser contents tend to de-crease the transformability of the t phase [8]. In fact, the amount of m phase in milled powders is dependent on the transformability of t phase. Hence, it may be related
J.-D. Lin et al. / Materials Chemistry and Physics 68 (2001) 42–55 53
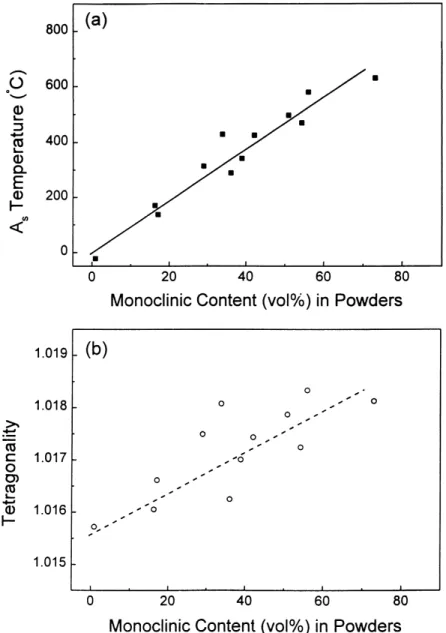
Fig. 13. (a) The Astemperature and (b) tetragonality plotted with respect to monoclinic amounts in Ce-Y-TZP powders after calcination at 800◦C for 2 h.
to the As temperatures or tetragonality of t phase. The
As temperature and tetragonality as a function of mono-clinic amount after 36 h ball-milling are thus plotted in Fig. 13a and b. It is clear that the monoclinic amount in-creases with the As temperature or the tetragonality of t phase.
3.5. Microstructure
For the powder compacts sintered at 1500◦C for 2 h, the morphologies of the thermally etched surface is examined. The average grain size of samples 5.5–3, 5.5–4, 3.5–3 and 3.5–4 is smaller than 1m. For samples doped with the same mol% of YO1.5, the higher the mol% CeO2, the larger the average grain size. In literature, it is well-documented that the average grain size of Ce-TZP is generally larger than
of Y-TZP [19,34–36]. Theunissen et al. [37] proposed that when Ce-TZP was doped with yttrium, the smaller grain sizes was obtained because of the segregation of yttrium to grain boundary and the grain growth of Ce-Y-TZP is de-pressed due to impurities drag [37,38]. Results of SEM ob-servation for Ce-TZP samples containing about 7–16 mol% CeO2 revealed that the shape and size of grains remain unchanged regardless of CeO2 content [39,40]. Similar to Ce-TZP, the shape and the average grain size of Ce-Y-TZP ceramics are insensitive to the contents of CeO2as the added amount of YO1.5is<2.5 mol%, as shown in Fig. 14. This is in agreement with the work by Boutz et al. [38,41] and Hi-rano and Inada [42], who reported that an specific amount of yttrium concentration is necessary to inhibit the grain growth of Ce-Y-TZP. This yttrium amount is found to be around 2.5 mol% in the current study.
Fig. 14. Correlation of average grain size and mol% YO1.5 for Ce-Y-TZP ceramics after sintered at 1500◦C for 2 h.
4. Conclusions
1. At a suitable amount of urea and reaction time, the com-position of derived powder is nearly identical to that of stock solution. The relation of measured composition and nominal compositions have been reported in the current study.
mol% Ymeasured= 0.0423 + 0.9143 mol% Ynominal mol% Cemeasured= 0.1147 + 0.9206 mol% Cenominal 2. The use of urea hydrolysis followed by ATA washing can
produce a finer and more sinterable Ce-Y-TZP powder as compared to that washed by alcohol after calcined at 500◦C for 0.5 and 36 h ball-milled in ethanol. Besides, the urea-derived powders washed by ATA procedure also yield 100% tetragonal phase when calcined at low tem-perature (500◦C).
3. The monoclinic content in urea-derived and ATA washed Ce-Y-TZP powder depends on the stabiliser amount and calcination temperature after 36 h ball-milling in alcohol. As the stabiliser content is increased, the monoclinic con-tent decreases. The higher the calcination temperature, the more the monoclinic content. The transformability of tetragonal phase is further related to the As temperature and tetragonality, the monoclinic content in as-derived powders after calcination and ball-milling processes is proportional to the As temperature and tetragonality of t phase.
4. To raise the sintered density of Ce-Y-TZP powders, the increase of calcinaton temperature is more effective than the prolongation of calcination time. When powders are calcined at 800◦C for 2 h, the sintered density achieves the highest. The grain size of Ce-Y-TZP ceramics de-creases with yttria content, while the addition of ce-ria increases grain size. The grain shape and grain size of Ce-Y-TZP ceramics are insensitive to the content of CeO2as the YO1.5amount in Ce-Y-TZP is<2.5 mol%.
Acknowledgements
The authors are grateful for the financial support from National Science Council, Taiwan, under contracts no. NSC82-0425-E-007-188 and NSC83-0405-E-007-017.
References
[1] A.G. Evans, in: G.L. Messing, E.R. Fuller Jr., H. Hausner (Eds.), Ceramic Transactions, Vol. 1, Ceramic Powder Science II, American Ceramic Society, Westerville, OH, 1989, pp. 989–1010.
[2] B. Aiken, W.P. Hsu, E. Matijevic, J. Am. Ceram. Soc. 71 (10) (1988) 845–853.
[3] E. Luccini, S. Meriani, O. Sbaizero, Int. J. Mater. Prod. Tech. 4 (2) (1989) 167–175.
[4] M. Hirano, H. Inada, J. Ceram. Soc. Jpn. 99 (1) (1991) 23–29. [5] Y.X. Huang, G.J. Gao, Powder Technol. 72 (1992) 101–104. [6] M. Dechamps, B. Djuricic, S. Pickering, J. Am. Ceram. Soc. 78 (11)
(1995) 2873–2880.
J.-D. Lin et al. / Materials Chemistry and Physics 68 (2001) 42–55 55 [8] J.D. Lin, J.G. Duh, J. Am. Ceram. Soc. 81 (4) (1998) 853–860.
[9] P.K. Gallagher, in: S.J. Schneider Jr., J.R. Davis, G.M. Davidson, S.R. Lampman, M.S. Woeds, T.B. Zorc (Eds.), Engineered Materials Handbook, Vol. 4, Ceramics and Glasses, ASM International, Metals Park, OH, 1991, pp. 52–64.
[10] M.L. Mecarthney, J. Am. Ceram. Soc. 70 (1) (1987) 54–58. [11] A. Krell, P. Blank, J. Eur. Ceram. Soc. 9 (1992) 309–322. [12] S.L. Dole, R.W. Scheidecker, L.E. Shiers, M.F. Berand, O. Hunter
Jr., Mater. Sci. Eng. 32 (1978) 277–281.
[13] B.S. Chiou, W.Y. Hsu, J.G. Duh, J. Mater. Sci. Lett. 5 (1986) 931– 934.
[14] M.A.C.G. Van de Graaf, A.J. Burggraaf, in: N. Claussen, M. Rüle, A.H. Heuer (Eds.), Advance in Ceramics, Vol. 12, Science and Technology of Zirconia II, The American Ceramic Society, Columbus, OH, 1983, pp. 744–765.
[15] F.F. Lange, J. Am. Ceram. Soc. 67 (2) (1984) 83–89.
[16] C. Morterra, G. Gerrato, L. Ferroni, L. Montanaro, Mater. Chem. Phys. 37 (1994) 243–257.
[17] L. Montanaro, L. Ferroni, S. Pagliolico, M.V. Swain, T.J. Bell, J. Am. Ceram. Soc. 79 (4) (1996) 1034–1040.
[18] J.G. Duh, H.T. Dai, B.S. Chiou, J. Am. Ceram. Soc. 71 (10) (1988) 813–819.
[19] J.G. Duh, J.U. Wan, J. Mater. Sci. 27 (1992) 6197–6203. [20] J.D. Lin, J.G. Duh, J. Mater. Sci. Lett. 16 (1997) 843–845. [21] J.D. Lin, J.G. Duh, J. Mater. Sci. 32 (1997) 5779–5790.
[22] H. Toraya, M. Yoshimura, S. Somiya, J. Am. Ceram. Soc. 67 (6) (1984) C119–C121.
[23] M.I. Mendelson, J. Am. Ceram. Soc. 52 (8) (1969) 443–446. [24] J.E. Bailey, D. Lewis, Z.M. Librant, L.J. Portor, Trans. J. Br. Ceram.
Soc. 71 (1) (1972) 25–30.
[25] R. Ge, Z. Li, H. Chen, D. Zhang, T. Zhao, Ceram. Int. 22 (2) (1996) 123–130.
[26] Y. Murase, E. Kato, J. Am. Ceram. Soc. 62 (9/10) (1979) 527. [27] W.H. Rhodes, J. Am. Ceram. Soc. 64 (1) (1981) 19–22. [28] M. Yoshimura, Am. Ceram. Bull. 67 (12) (1988) 1950–1955. [29] K. Urabe, K. Ogata, H. Ikawa, S. Udagawa, Mater. Sci. Forum.
34–36 (1988) 1147–1152.
[30] P.F. Becher, G. Bogun, E.F. Funkenbusch, in: S. Somiya, N. Yamamoto, H. Yanagida (Eds.), Advances in Ceramics, Vol. 24, Science and Technology of Zirconia III, The American Ceramic Society, Westerville, OH, 1988, pp. 645–651.
[31] P.F. Becher, M.V. Swain, M.K. Ferber, J. Mater. Sci. 22 (1987) 76– 84.
[32] P.F. Becher, M.V. Swain, J. Am. Ceram. Soc. 75 (3) (1992) 493– 502.
[33] B. Bastide, P. Canale, P. Odier, J. Eur. Ceram. Soc. 5 (1989) 289– 293.
[34] T. Sato, M. Shimada, Am. Ceram. Soc. Bull. 64 (10) (1985) 1382– 1384.
[35] T. Masaki, J. Am. Ceram. Soc. 69 (8) (1986) 638–640.
[36] K. Tsukuma, T. Takahata, M. Shiomi, in: S. Somiya, N. Yamamoto, H. Yanagida (Eds.), Advances in Ceramics, Vol. 24, Science and Technology of Zirconia III, The American Ceramic Society, Westerville, OH, 1988, pp. 721–728.
[37] G.S.A.M. Theunissen, A.J.A. Winnubst, A.J. Burggraaf, J. Eur. Ceram. Soc. 9 (1992) 251–263.
[38] M.M.R. Boutz, A.J.A. Winnubst, A.J. Burggraaf, J. Eur. Ceram. Soc. 13 (1994) 89–102.
[39] K. Tsukuma, Am. Ceram. Soc. Bull. 65 (10) (1986) 1386–1389. [40] K. Tsukuma, M. Shimada, J. Mater. Sci. 20 (1985) 1178–1184. [41] M.M.R. Boutz, A.J.A. Winnubst, B. Van Langerak, R.J.M. Olde
Scholtenhuis, K. Kreuwell, A.J. Burggraaf, J. Mater. Sci. 30 (7) (1995) 1854–1862.