國 立 交 通 大 學
材料科學與工程學系
博 士 論 文
Synthesis of Supramolecular Side-Chain Conjugated
Polymers Containing Pyridyl Group: Applications in
Chemosensor, Liquid Crystal and Polymeric Light-Emitting
Devices
含超分子作用力之吡啶基側鏈共軛高分子在化學感測、液晶
及高分子發光二極體元件之應用
研 究 生 : 楊博仁
指導教授 : 林宏洲 博士
中華民國九十八年十一月
含超分子作用力之吡啶基側鏈共軛高分子在化學感測、液晶及高分子
發光二極體元件之應用
Synthesis of Supramolecular Side-Chain Conjugated Polymers
Containing Pyridyl Group: Applications in Chemosensor, Liquid
Crystal and Polymeric Light-Emitting Devices
研究生:楊博仁 Student: Po-Jen Yang
指導教授:林宏洲 博士 Advisor: Dr. Hong-Cheu Lin
國立交通大學 材料科學與工程學系
博士論文
A Thesis
Submitted to Department of Materials Science and Engineering College of Engineering
National Chiao Tung University In Partial Fulfillment of the Requirement For the Degree of Doctor of Philosophy of Science
In Materials Science and Engineering November 2009
Hsinchu, Taiwan
ACKNOWLEDGEMENTS
本論文承蒙指導教授林宏洲博士的協助下才得以順利完成,非常感謝老師這 些年來對我的照顧,使我在實驗方面有足夠的經費能夠將理想付諸實行。感謝中 研院化學所林建村和孫世勝老師、清大化學系韓建中老師、交大應用化學系許千 樹老師和材料系劉典膜老師,在百忙之中審核論文並給予寶貴的建議及指正,使 本論文能更趨完善。 研究所六年的時光,很幸運也很快樂地在我們實驗室度過這些日子,相信以 後我會一直記得這段難忘時光。感謝實驗室的學長姐:孟丹、孝先、建民、昇璋 及幫我許多忙的金賢和偉綜學長,在這些年的求學生涯中讓我感到許多的歡笑與 溫暖,非常懷念大家一起結伴出遊的時光。也感謝實驗室的學弟妹玄之、仁甫、 竟軒、世賢、威宏、曉萍、彥興、鈞傑、守仁、怡婷、裕證、沛霖、老魏、世傑、 春吉,因為有你們讓我增添了許多快樂的回憶,讓我論文得以順利的完成。 最後由衷地感謝一直栽培我的父母親,讓我能夠衣食無缺的度過十多年的求 學生涯和長久以來的照顧和關懷,以及謝謝我的家人們一路上的扶持,使我能夠 順利地完成學位。含超分子作用力之吡啶基側鏈共軛高分子在化學感測、液晶
及高分子發光二極體元件之應用
研究生:楊博仁 指導教授:林宏洲 博士
國立交通大學材料科學與工程研究所
博士班
中 文 摘 要
本論文研究方向為探討一系列包含吡啶基側鏈共軛高分子,且利用超分子作 用力作為自組裝之橋樑,並發展在化學感測、液晶及高分子發光二極體元件之應 用。 第一個部份,一系列發光質子受體包含三各共軛環單體,包括一個末端吡啶 基和二個側邊取代之甲氧基,是利用 Horner-Wadsworth-Emmons (HWE) 和 Sonogashira 偶合反應。然後,在以自由基聚合反應,依不同莫爾比和具有傳電 洞能力的咔唑共聚。這些質子受體之共聚物和不同代數具有傳電子能力的噁二唑 樹枝狀結構,利用氫鍵自組裝去形成超分子之側鏈共聚物。當引進咔唑基團在質 子受體共聚物中,是有效的增加玻璃轉移溫度和有較小分子鏈的作用力,在這些 發光質子受體之間,且相似的效應也發生在氫鍵樹枝狀錯合物中。另外,氫鍵樹 枝狀錯合物螢光放光可以調控 61 nm 的紅位移,且激發咔唑基團可以獲得較強的螢光強度比激發質子受體。電化學方面,引進噁二唑樹枝狀結構在氫鍵樹枝狀 錯合物裡,可以獲得較低的最低未占分子軌道的能階和一個好的電子注入性質。 質子受體高分子和它的氫鍵樹枝狀錯合物,在電致發光發光放光範圍為 464 到 519 nm,從藍光到綠光。元件方面,氫鍵樹枝狀錯合物顯示一個 519 nm 放光, 驅動電壓為 6.5 V,一個最大發光 408 cd/m2 在 18 V 和發光效率 0.39 cd/A 在 100 mA/cm2。 第二部份,一系列側鏈型氫鍵液晶共聚高分子網路,包含不同共聚比之發光 的質子受體和質子予體,是被成功的合成且利用自由基聚合方式。氫鍵共聚高分 子網路擁有較高的玻璃轉移溫度比它們個別的均聚物。氫鍵共聚物和均聚物兩者 都表現層列 A 液晶相。氫鍵共聚高分子網路隨著質子予體含量增加,澄清溫度 也隨之增加且有利於穩定層列 A 液晶相。另外,氫鍵共聚高分子網路螢光放光 可以調控 39 nm 的紅位移比照它的均聚物時,且在電致發光元件和螢光放光的 光色可以被調控,從496-500 nm 到 531-537 nm 為藍綠到綠光。 最後一部份,開發發光質子受體包含三各共軛環單體,包括一個末端吡啶基 和二個側邊取代之甲氧基及二各雙鍵之共軛三環結構,是利用 Wittig and Pd-catalyzed Heck 偶合反應和聚合方式為自由基聚合。均聚物表現一個玻璃轉移 溫度為 60 °C 和 融點為 150 °C。在醋酸溶液中,螢光放光最大紅位移可以從 460 到 605 nm。這均聚物具有高的感測和選擇性,對於鎳離子比其他過渡金屬 離子,這是歸因於它具有較強的鍵結且焠熄常數為 5.65 × 106 M-1。另外,當加 入 PMDTA 到高分子和銅離子的錯合物,螢光的 ON-OFF-ON 切換行為可被發 現,這未來利用在化學感測可以回收利用的價值。
Synthesis of Supramolecular Side-Chain Conjugated
Polymers Containing Pyridyl Group: Applications in
Chemosensor, Liquid Crystal and Polymeric Light-Emitting
Devices
Student: Po-Jen Yang Advisor: Dr. Hong-Cheu Lin
Department of Materials Science and Engineerong National Chiao Tung
University
Abstract
First, a novel light-emitting hydrogen-bonded acceptor containing three conjugated aromatic rings, including one pyridyl terminus and two lateral methoxyl groups, was successfully synthesized via Horner-Wadsworth-Emmons (HWE) olefination and Sonogashira coupling reaction. Moreover, different molar ratios of light-emitting H-acceptor monomer and hole-transporting monomer bearing a carbazole unit were copolymerized through free radical polymerization to obtain light emitting and hole-transporting H-acceptor copolymers. H-acceptor copolymers were complexed with different generations of dendritic H-donors bearing 1,3,4-oxadiazole (OXD) dendrons and terminal benzoic acids via H-bonded self-assembly to form supramolecular side-chain copolymers. In contrast to H-acceptor homopolymer, H-acceptor copolymers incorporated with carbazole moieties effectively enhance the
light-emitting H-acceptor units, and similar effects occur in their H-bonded dendritic complexes. In addition, red shifts of photoluminescence (PL) emissions in H-bonded dendritic complexes can be tuned up to 61 nm. Furthermore, H-bonded dendritic complexes excited OXD absorption can create a stronger fluorescence than that excited at acceptor absorption. The OXD dendritic wedges in H-bonded dendritic complexes can lower the LUMO energy levels and provide a better electron injection property. H-acceptor polymer and its H-bonded dendritic complexes showed electroluminescence (EL) emissions in the range of 464-519 nm from blue to green. In addition, a PLED device containing H-bonded dendritic complex showed an EL emission of 519 nm under a turn-on voltage of 6.5 V, with a maximum luminance of 408 cd/m2 at 18 V and a luminance efficiency of 0.39 cd/A at 100 mA/cm2, respectively.
Second, a series of H-bonded side-chain mesogenic copolymer networks
containing different molar ratios of light-emitting proton acceptor and proton donor were synthesized via free radical polymerization. The H-bonded copolymer networks have higher glass transition temperatures (Tgs) than the individual hompolymers. Both H-bonded copolymer and homopolymer complex networks show mesomorphic behavior with the smectic A phase. The isotropization temperatures (Tis) and SA phase stabilities of the H-bonded copolymer networks increase as the molar ratios of H-donor unit increase. Furthermore, the red-shifts of PL emissions in H-bonded copolymer and homopolymer complex networks can be tuned up to 39 nm in contrast to H-acceptor homopolymer. The electroluminescence (EL) and photoluminescence (PL) results of H-acceptor homopolymer and its fully H-bonded cross-linking copolymer show emission colors varying from c.a. 496-500 nm (greenish-blue) to 531-537 nm (green), respectively.
rings, including one pyridyl terminus and two lateral methoxyl groups (on the middle ring), was successfully synthesized via Wittig and Pd-catalyzed Heck coupling reactions. Homopolymer shows a Tg of 60 °C and Ti up to 150 °C. In CH3COOH solution, homopolymer exhibits a pH-tunable photoluminescence with emission maximum varies from 460 to 605 nm. Homopolymer exhibits an extraordinary sensory selectivity for Ni2+ over other transition metal ions as a result of the stronger binding ability of the Ni2+ onto Homopolymer than other transition metals ions. Stern-Volmer constant for the Ni2+ ion sensing was determined through concentration dependent studies as 5.65 × 106 M-1. In addition, the ON-OFF-ON fluorescent switch behavior upon the addition of PMDTA to the polymer-Cu2+ complexes demonstrates a superior reusability of this chemosensor which is important for the practical use.
Table of Contents
Acknowledgements ···II 中文摘要 ···III Abstract ···V Table of Contents ···VIII Table Lists ···XII Figure Lists ···XIV
Chapter 1. Introduction ···1
1.1 Introduction to Supramolecular chemistry ···1
1.2 Self-assembled liquid crystalline polymers ···3
1.3 Self-assembled π-conjugated systems polymers ···6
1.4 Sensors and switches from supramolecular chemistry ···10
1.5 AIM ···14
Chapter 2. Study of Supramolecular Side-Chain Copolymers Containing Light-Emitting H-Acceptors and Electron-Transporting Dendritic H-Donors ···17
2.1 Abstract ···17
2.2 Introduction ···19
2.3 Experimental Section ···23
2.3.2 Materials ···27
2.4 Results and Discussion ···34
2.4.1 Syntheses and Characterization of Polymers ···34
2.4.2 Thermal Properties ···39
2.4.3 Optical Properties ···43
2.4.4 Electrochemical Properties ···55
2.4.5 Electroluminescence (EL) Properties ···57
2.5 Conclusion ···61
Chapter 3. H-Bonded Liquid Crystalline Polymer Networks Self-Assembled from Side-Chain Copolymers and Homopolymer Complexes Containing Fluorescent H-Acceptor and Non-Photoluminescent H-Donor Pendants ···63
3.1 Abstract ···63
3.2 Introduction ···64
3.3 Experimental Section ···68
3.3.1 Measurements and Characterization ···68
3.3.2 Materials ···72
3.4 Results and Discussion ···76
3.4.1 Synthesis and Properties of Polymers ···76
3.4.3 XRD Studies ···84 3.4.4 FTIR Studies ···90 3.4.5 Optical Properties ···92 3.4.6 Electrochemical Properties ···97 3.4.7 Electroluminescence Properties ···99 3.5 Conclusion ···104
Chapter 4. Novel chemosensory materials based on Side-Chain Polymer Containing Fluorescent Receptor Pendants Functionalized with Pyridyl Groups ···106
4.1 Abstract ···106
4.2 Introduction ···107
4.3 Experimental Section ···109
4.3.1 Measurements and Characterization ···109
4.3.2 Materials ···112
4.3.3 Metal ion titration ···117
4.4 Results and Discussion ···118
4.4.1 Syntheses and Characterization of Polymers ···118
4.4.2 Thermal Properties ···120
4.4.4 UV-visible and Photoluminescence Titration ···123 4.5 Conclusions ···132 Chapter 5. Conclusions ···134 References ···137 Appendix ···150 學經歷資料 ···158 List of Publications ···159
Table Lists
Table 2.1 Composition, Yields, Molecular Weights, and Degradation Temperatures of
Polymers P1-P5 ···37
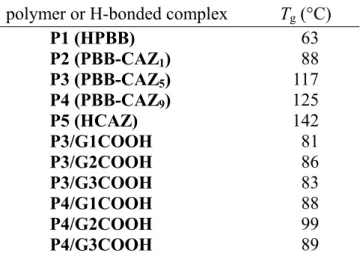
Table 2.2 Glass Transition Temperatures of Polymers (P1–P5) and H-Boned
Side-Chain Dendritic Complexes Containing H-Acceptor Polymers P3 and P4 ···40
Table 2.3 Absorption and PL Emission Spectral Data of Polymers (P1–P5) in THF
Solutions and Solid Films ···44
Table 2.4 Photophysical Properties of H-Boned Side-Chain Dendritic Complexes
Containing H-Acceptor Polymers P3 and P4 ···48
Table 2.5 Electrochemical Properties of H-Acceptor Polymer P4 and Its H-Bonded
Side-Chain Dendritic Complexes ···56
Table 2.6 Electroluminescence (EL) Device Performance Characteristics of
H-Acceptor Polymer P4 and Its H-Boned Side-Chain Dendritic Complexes ···61
Table 3.1 Compositions, Yields, Molecular Weights, Polydispersity Index (PDI), and
Degradation Temperatures of Homopolymers and Copolymers ···78
Table 3.2 Thermal Properties of Polymers P1−P6 and Fully H-Bonded Homopolymer
Complex P1/P5 ···80
P 1 − P 5 a n d F u l l y H - B o n d e d H o m o p o l y m e r C o m p l e x P 1 / P 5
···86
Table 3.4 Absorption and PL Emission Spectral Data of Polymers P1−P4, P6, and
Fully H-Bonded Homopolymer Complex P1/P5 in THF Solutions and Solid Films ···97
Table 3.5 Electrochemical Properties of H-Acceptor Homopolymer P1 and Its Fully
H-Boned Copolymer P2 and Homopolymer Complex P1/P5 ···99
Table 3.6 Electroluminescence (EL) Device Performance Characteristics of
H-Acceptor Homopolymer P1 and Its Fully H-Boned Copolymer P2 and Homopolymer Complex P1/P5 ···102
Table 4.1 Absorption and PL emission of P1 for Different Solvents and Film ····121 Table 4.2 KSV Value of P1 for Different Metal Ions ···127
Figure Lists
Figure 1.1.1 Organization of non-covalent interactions by bond strength ···2 Figure 1.2.1 Schematic illustration of the structural components used to fabricate
simple LCs, SCLPs, to self-assembled SCLPs from rodshaped mesogens ···4
Figure 1.2.2 Molecular components used to prepare the first examples of
self-assembled SCLPs ···5
Figure 1.2.3 Complexes formed as a result of hydrogen bonding and proton transfer,
which are joined at each repeat unit of poly(4-vinylpyridine) ···6
Figure 1.3.1 Rod like π-conjugated oligomers 5-10 ···7 Figure 1.3.2 OPV derivatives 11-13 are able to gelate in hexane monitored by
temperature-dependent absorption and emission spectra of 11. For comparison, these spectra are also given in a good solvent like chloroform and as a film from hexane 8
Figure 1.3.3 Conjugated oligomers provided with strong hydrogen-bonding
interacting units ···10
Figure 1.4.1 Designing a receptor for which the substrate has a specific affinity may
not suffice; something should signal to the operator that recognition has occurred. Assembling a receptor and a signalling unit makes a sensor ···11
Figure 1.4.2 Fluorescence response of tmeda-PPETE/Cu2+ (white) or tmeda- PPETE (black) to various 10 μM cations in room-temperature solution; the
concentrations of tmeda-PPETE (with respect to the repeat unit) and Cu2+ were fixed at 5 μM ···12
Figure 1.4.3 Schematic diagram of the excited states deactivation processes on the
surfaces of the nanoparticles ···13
Figure 2.1 Different generations of dendritic H-donors (G1COOH–G3COOH) used
in H-bonded side-chain dendritic complexes ···22
Figure 2.2 Schematic representation of H-acceptor copolymers and H-bonded
side-chain dendritic complexes bearing different generations of dendritic H-donors (G1COOH–G3COOH) ···22
Figure 2.3 FTIR spectra of (a) H-acceptor polymer P3 (PBB-CAZ5), (b) dendritic H-donor G1COOH, and (c) H-bonded side-chain dendritic complexes P3/G1COOH at room temperature ···38
Figure 2.4 Absorption spectra of H-acceptor polymers P1–P4 in THF solutions,
normalized at the maximum absorption peak of the light-emitting PBB segments at 385 nm ···44
Figure 2.5 Normalized PL spectra of H-acceptor polymers P1–P4 excited at the
maximum absorption (397 nm) of the light-emitting PBB segments in solid films 46
Figure 2.6 Absorption spectra of H-acceptor polymer P3 and its H-bonded side-chain
the light-emitting PBB cores along with model compound 1 (containing an OXD unit with the maximum absorption around 305 nm in THF solution) ···48
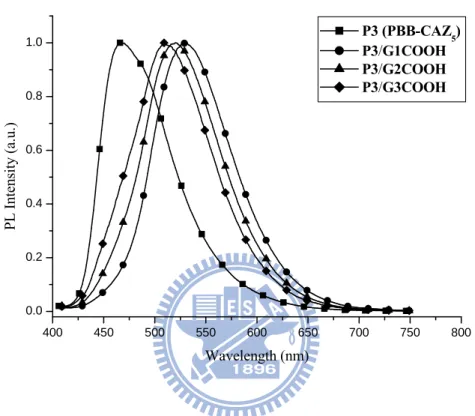
Figure 2.7 Normalized PL spectra of H-acceptor polymer P3 and its H-bonded
side-chain dendritic complexes excited at the maximum absorption (397 nm) of the light-emitting PBB cores in solid films ···50
Figure 2.8 UV spectra and PLE spectra of H-acceptor polymer P4 and its H-bonded
side-chain dendritic complexes in solid films normalized at the light-emitting PBB units (397 nm), where PLE spectra were monitored at the corresponding maximum PL emissions ···52
Figure 2.9 PL spectra of H-acceptor polymer P4 and its H-bonded side-chain
dendritic complexes in solid films, which were excited at the dendritic peripheral OXD units (at 305 nm for open symbols) and at the maximum absorption of the light-emitting PBB cores in H-bonded side-chain dendritic complexes containing dendritic H-donors (ca. 397 nm for solid symbols) ···54
Figure 2.10 Normalized EL spectra of PLED devices with the configuration of
ITO/PEDOT:PSS/polymer (P4 or its H-bonded side-chain dendritic complexes)/BCP/Alq3/LiF/Al ···58
Figure 2.11 (a) Current density-voltage (I-V) curves and (b) luminance-voltage (L-V)
its H-bonded side-chain dendritic complexes)/BCP/Alq3/LiF/Al ···60
Figure 3.1 Simplified schematic illustration of idealized H-bonded liquid crystalline
polymer networks self-assembled from side-chain copolymers and homopolymer complexes containing fluorescent H-acceptor PBB and non-photoluminescent H-donor BA pendants ···68
Figure 3.2 Optical micrographs of liquid crystalline textures (from POM with crossed
polarizers): (a) H-acceptor homopolymer P1 at 90 °C (cooling), (b) H-donor homopolymer P5 at 135 °C (cooling), (c) fully H-bonded copolymer P2 at 95 °C (cooling), and (d) fully H-bonded homopolymer complex P1/P5 at 115 °C (cooling). The scale bars all correspond to 100 μm ···83
Figure 3.3 One-dimensional powder X-ray diffraction (1D-XRD) patterns (intensity
against angle profiles) obtained in the SA phase of the fully H-bonded copolymer P2 and fully H-bonded homopolymer complex P1/P5 at 85 °C (cooling) ···85
Figure 3.4 (a) Two-dimensional X-ray diffraction (2D-XRD) pattern obtained from
the as-drawn fiber of H-bonded copolymer P2 at room temperature and (b) azimuthal scan of low and high angle reflections. Fiber direction is horizontal axis ···87
Figure 3.5 Temperature-dependent one-dimensional powder X-ray diffraction
(1D-XRD) pattern in the low angle region of H-boned copolymer P4 recorded during the cooling process from the isotropic state to room temperature ···89
Figure 3.6 FT-IR spectra recorded for (a) pure H-acceptor homopolymer P1 and (b)
pure H-donor homopolymer P5 at room temperature, and (c) fully H-bonded copolymer P2 at (c) room temperature (glassy state), (d) 100 °C (SA phase), and (e) 170 °C (isotropic state) ···91
Figure 3.7 Absorption spectra of polymers P1−P4, P6, and fully H-bonded
homopolymer complex P1/P5 in solid films ···94
Figure 3.8 Normalized PL spectra of polymers P1−P4, P6, and fully H-bonded
homopolymer complex P1/P5 were excited at the maximum absorption of light-emitting H-acceptor PBB segments in solid films ···95
Figure 3.9 Normalized EL spectra of PLED devices with configurations of
ITO/PEDOT:PSS/polymer (H-acceptor P1 or its fully H-bonded cross-linking copolymer P2 and homopolymer complex (P1/P5))/BCP/Alq3/LiF/Al ···100
Figure 3.10 (a) Current density-voltage (I-V) curves and (b) luminance-voltage (L-V)
curves of PLED devices with configurations of ITO/PEDOT:PSS/polymer (H-acceptor P1 or its fully H-bonded cross-linking copolymer P2 and homopolymer complex (P1/P5))/BCP/Alq3/LiF/Al ···103
Figure 4.1 Absorption spectra of M1 in THF solution and P1 in various solvents and
solid film were normalized to the maximum absorption ···122
film were normalized to the maximum PL intensity (excited at the maximum absorption) ···122
Figure 4.3 PL titration spectra of polymer P1 (5 × 10-6 M) in THF before and after
adding Ni2+ with versus Ni2+. Excitation wavelength is at 400 nm ···125
Figure 4.4 PL emission response profiles of polymer P1 to various metal ions in THF
solution ···127
Figure 4.5 PL titration spectra of polymer P1 (5 × 10-6 M) in THF before and after
adding Cu2+ with versus Cu2+. Excitation wavelength is at 400 nm ···129
Figure 4.6 PL titration spectra of polymer M1 (5 × 10-6 M) in THF before and after
adding Cu2+ with versus Cu2+. Excitation wavelength is at 400 nm ···130
Figure 4.7 PL spectra of polymer M1 (5 × 10-6 M) in THF before and after the
addition of Cu2+, and recovered the addition of PMDTA. Excitation wavelength is at 400 nm ···131
Figure 4.8 The speculated conversion cycle of P1 in the presence of Cu2+ and PMDTA ···132 Figure A1 1H-NMR spectra of monomers PBB (M1) and CAZ (M2), and polymers P1–P5 in DMSO-d6 ···151
Figure A2 Polarized optical micrograph (POM) image of homopolymer P1 exhibited
Figure A3 Powder X-ray diffraction (XRD) intensity against angle profiles obtained
in the nematic phase of homopolymer P1 at 90 °C (heating) ···153
Figure A4 Absorption spectra of polymers P1−P4 are normalized at the maximum
absorption of light-emitting PBB segments in solid films ···154
Figure A5 (a) Chemical structure of model compound 1 and (b) the spectral overlap
in the emission of model compound 1 and homopolymer P5 and the absorption of homopolymer P1 in THF solutions. Note: excited at 305 nm for model compound 1 and homopolymer P5 ···155
Figure A6 1H-NMR spectra of monomers PBB (M1) and BA (M2), and BAMe (M3)
in DMSO-d6 ···156
Chapter 1
Introduction
1.1 Introduction to Supramolecular chemistry
From before Linus Pauling’s ground breaking work on the hydrogen bond in the 1930’s1 to Jean Marie Lehn’s ‘Chemistry beyond the molecule’ that led to the term ‘supramolecular chemistry’, the nature of non-covalent bonds has fascinated chemists for over a century. In particular, the last thirty years have been exceptionally fruitful for scientists from a variety of disciplines who have made enormous advances in exploiting the non-covalent bond to construct sophisticated architectures.2,3 As macromolecular structures and functional materials have continued to evolve with higher degrees of complexity and function, traditional covalent-based synthetic strategies have become increasingly difficult to employ. Accordingly, many scientists have begun to replace traditional polymer synthesis with self-assembly in order to overcome a variety of synthetic hurdles and to exploit the dynamic nature of the noncovalent bond. Tremendous growth and elegant advances in polymer science have taken place as supramolecular science, self-assembly, and polymer chemistry continue to converge.3 This review explores the design principles and functionalization strategies inherent to one class of supramolecular polymers, side-chain functionalized polymers (SCFPs), and will high light the advances that
have given rise to the sophisticated non-covalent functionalization methods of today. A large variety of recognition motifs and non-covalent forces have been reported for supramolecular polymers ranging from π–π interactions and hydrogen bonding to metal coordination and electrostatic interactions. Non-covalent interactions and forces can fall into three major classes: i) weak interactions (0–15 kcal mol-1 bond strength), ii) medium interactions (15–60 kcal mol-1 bond strength) and iii) strong interactions (above 60 kcal mol-1 bond strength).4 Due to their high dependence on external influences such as pressure, solvent, and temperature, most non-covalent forces do not fall into a single category. Figure 1.1.1 describes the general division of noncovalent interactions based on bond strength. In general, π-π, cation-π, hydrophobic, or van der Waals interactions or hydrogen bonds are very weak. In contrast some metalcoordination complexes (strongly dependent on the ligand system and the metal used) and electrostatic interactions have very strong bond strength. Multiple hydrogen bonds and some metal-coordination complexes can be categorized as medium strength.
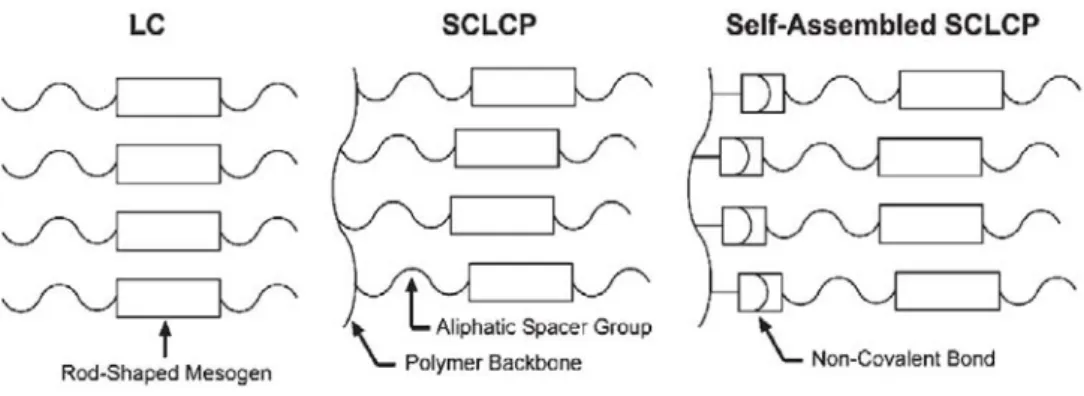
1.2 Self-assembled liquid crystalline polymers
Non-covalent side-chain functionalization strategies were first reported for the synthesis of liquid crystalline materials.5,6 Liquid crystals (LCs) possess orientational or weak positional ordering that give rise to materials with important characteristic of crystals but flow behaviour similar to liquids. Formation of this unique phase of matter is a direct consequence of the anisotropic alignment of small molecules or mesogens via non-covalent forces such as π-π stacking or hydrophobic interactions. Polymeric liquid crystals employ a variety of self-organizational processes to achieve long-range order. Conventional side-chain liquid crystalline polymers (SCLPs) are typically prepared via covalent tethering of mesogenic entities, structurally similar to low molecular weight LCs mesogens, with long, flexible aliphatic chains.7 This spacer group, situated between the polymer backbone and the mesogen, decouples the motion of the polymer from the sidechain giving flexibility to the molecular orientation of the mesogenic components. In the field of self-assembled SCLPs, the principles described above for SCLPs are generally followed, yet covalent attachment of the mesogen is replaced with non-covalent bonds (Figure 1.2.1).
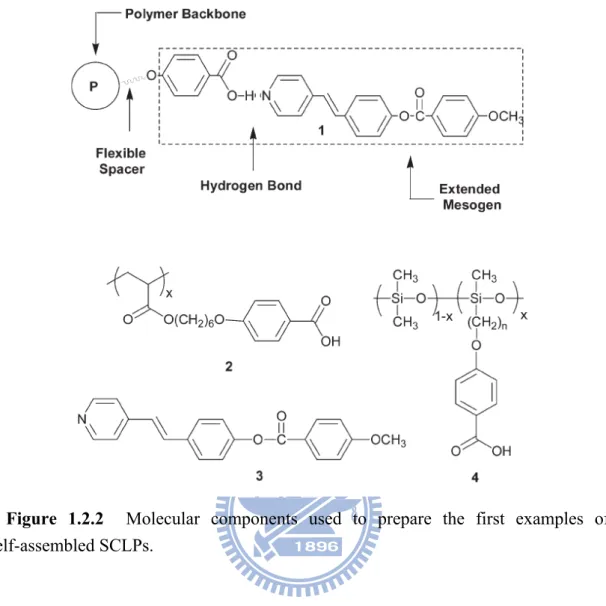
Kato and Fréchet were the first to explore the non-covalent attachment of traditional liquid crystalline components to a variety of polymer backbones.8 In 1989 they disclosed their report describing binary mixtures of 2 and 3 to form thermotropic
complexes (1) (Figure 1.2.2). In this example, each component independently shows liquid crystalline behaviour. However, when 1:1 mixtures of 2 and 3 or 4 and 3 are prepared by slow evaporation from pyridine solutions, nematic mesophases with higher transition temperatures than those of the individual components are observed.
This mesophase stabilization is attributed to the formation of extended mesogenic units involving the hydrogen-bond bonded complex 1 shown in Figure 1.2.2. While Kato and Fréchet’s novel class of liquid crystalline polymers was based on a single weak non-covalent bond, this report held much significance not only for the field of selfassembled SCLPs, but also established fundamental design strategies for the preparation of supramolecular SCFPs in general. Following their original report, much effort was directed toward examining self-assembled SCLPs engineered with a variety of structural configurations.
Figure 1.2.1 Schematic illustration of the structural components used to fabricate
Figure 1.2.2 Molecular components used to prepare the first examples of
self-assembled SCLPs.
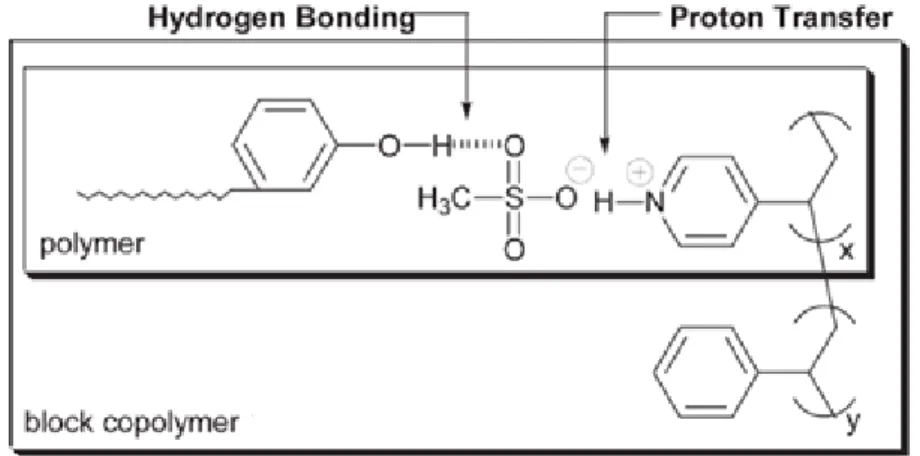
Ikalla and co-workers have employed multiple interactions to fabricate supramolecular nanostructures with microsturctural control on two different length scales.9 Here, more than one type of non-covalent bond is joined at a single anchoring site, thereby allowing for multi-functionalization at each repeat unit. The motivation for forming such complexes lies mainly in the ease of synthesis, allowing one to rapidly tailor the properties of the resultant materials. In one example, poly(4-vinylpyridine) was functionalized via proton transfer using methane sulfonic acid (see Figure 1.2.3).
Figure 1.2.3 Complexes formed as a result of hydrogen bonding and proton
transfer, which are joined at each repeat unit of poly(4-vinylpyridine).
1.3 Self-assembled π-conjugated systemspolymers
π-Conjugated (semiconducting) polymers are by far the most promising functional polymers in view of applications in less expensive and flexible electronic devices. Prototype field-effect transistors (FET)s,10 light-emitting diodes (LED)s,11 photovoltaic cells, and related devices have already been fabricated, and Philips introduced the first commercial LED based on polymer technology in 2002. Nowadays a plethora of conjugated polymers exists having a base structure of alternating single and double/triple bonds of which some parent structures are shown.
In in oligo(p-phenylenevinylene)s was studied as a function of the conjugation length (5-7). It was found that only strong π-π interactions in the longest oligomer could phase separate from the tridodecyl chains. Columnar order in OPV derivatives was also reached using hydrogen-bonded dimers between carboxylic acids aided by
strong phase separation of the tridodecyloxyphenyl groups (8, 10). Also, dimerization of OPVs by coordination (9), ionic, and fluorophilic interactions have led to discrete liquid-crystalline supramolecular structures that further organize into columnar mesophases (see Figure 1.3.1).12
Figure 1.3.1 Rod like π-conjugated oligomers 5-10.
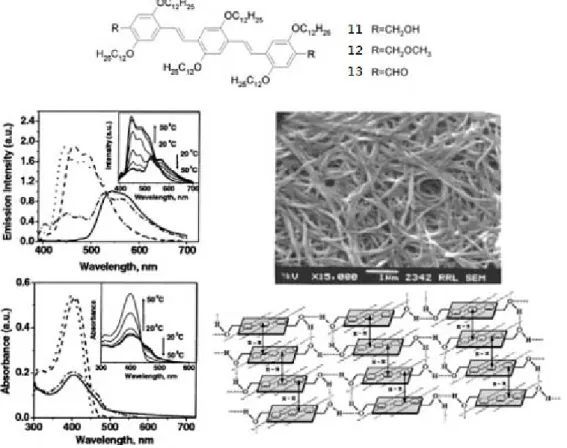
Ajayaghosh et al. serendipitously extended this concept to OPV derivatives (11-13) and reported a completely thermoreversible self-assembly process in a series of hydrocarbon solvents from single OPV molecules to fibers and ultimately to an entangled network structure. The absorption and emission properties showed dramatic changes during gelation, which is an indication of strong intermolecular π electronic coupling of the ordered OPV segments. In a comparative study it was shown that gelation was cooperative and strongly dependent on the choice of hydrogen-bonding
motif, alkoxy side-chain length, and conjugation length. An alcohol OPV trimer (11) equipped with six lateral dodecyl chains easily forms a gel at moderate concentrations in hexane, whereas the aldehyde analogue (13) could not gelate the solvent or a methyl ester analogue (12) under extreme conditions. SEM, X-ray diffraction, and IR on the gel from 11 revealed an entangled network of fibers up to micrometer lengths and 100-150 nm in width consisting of well-ordered lamellae of stacked molecules positioned by hydrogen bonds (Figure 1.3.2).13
Figure 1.3.2 OPV derivatives 11-13 are able to gelate in hexane monitored by
temperature-dependent absorption and emission spectra of 11. For comparison, these spectra are also given in a good solvent like chloroform and as a film from hexane.
The SEM picture of a dried gel of 11 shows an entangled fibrillar network. The self-assembly of the gel is proposed in the scheme.
Supramolecular polymers are constructed from monomeric units that are glued together by reversible noncovalent hydrogen-bond interactions and as such comprise a special class of self-assembled systems. Recently Meijer et al. reported on supramolecular hydrogen-bonded OPV dimers (14) and polymers (15) in which the specific electronic and optical properties of conjugated OPV oligomers were combined with the material properties of polymers.14,15 These polymeric systems are based on the dimerization of strong quadruple hydrogen-bonding ureidopyrimidinone units, resulting in a random coil polymer in solution lacking higher mesoscopic order. The supramolecular polymer could easily be processed, revealing smooth films, and photoinduced electron transfer was observed when blended with a C60 derivative. This blend could be successfully incorporated in a photovoltaic device. Hydrogen bonds have also been used to obtain liquid-crystalline phases. When nucleobases such as adenine and thymine are attached to known mesogens such as alkoxyphenylethynylenes 16-19, no liquid crystal phase could be observed. However, the 1:1 blends of the complementary nucleobase derivatives resulted in formation of fairly stable lyotropic liquid-crystalline phases, Figure 1.3.3.
Figure 1.3.3 Conjugated oligomers provided with strong hydrogen-bonding
interacting units.
1.4 Sensors and switches from supramolecular chemistry
Molecular recognition is one of the corner stones of supramolecular chemistry. Given any substrate (molecule, cation, or anion), the supramolecular approach is that an appropriate receptor, possessing structural and chemical features suitable for substrate recognition, can be designed. The keyword is shape. This concept is illustrated in Figure 1.4.1.16
Figure 1.4.1 Designing a receptor for which the substrate has a specific affinity
may not suffice; something should signal to the operator that recognition has occurred. Assembling a receptor and a signalling unit makes a sensor.
Most literature reports use fluorescence quenching as the readout mechanism for the sensor response. Very few involve a fluorescence “turn-on” response. Birck et. al. recently published an elegant example involving small molecule sensors that selectively identified iron cations by amplified fluorescence. The greatest advantage of fluorescence “turn-on” sensors related to “turn-off” sensors is the ease of measuring low-concentration contrast relative to a “dark” background. This reduces the likelihood of false positive signals and increases the sensitivity, as demonstrated by numerous studies. Poly[p-(phenyleneethynylene)-alt-(thienyleneethynylene)] (PPETE) with a N,N,N-trimethylethylenediamino receptor loaded on the thienylene ring (tmeda-PPETE) was synthesized on the basis of a strategy we advanced for a series of fluorescence “turn-on” chemosensors (see Figure 1.4.2).17
Figure 1.4.2 Fluorescence response of tmeda-PPETE/Cu2+ (white) or tmeda-
PPETE (black) to various 10 μM cations in room-temperature solution; the
concentrations of tmeda-PPETE (with respect to the repeat unit) and Cu2+ were fixed at 5 μM.
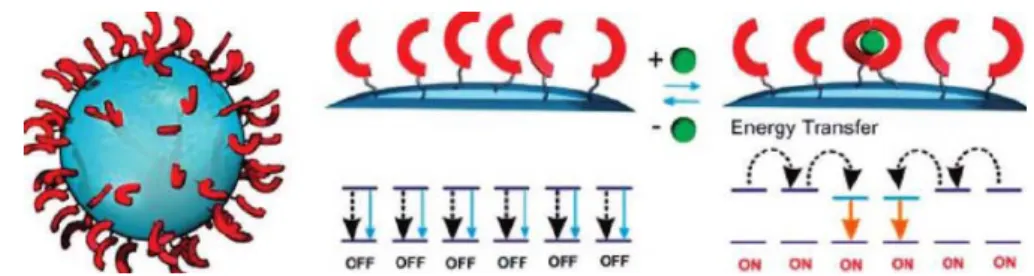
In conventional fluorescent chemosensors, the recognition of the target by the receptor unit affects the fluorescence properties of a single covalently coupled fluorescent moiety. As shown in Figure 2.9, a suitable TSQ derivative is densely
grafted onto the surface of preformed silica nanoparticles electronic interactions between the individual chemosensor units enable the free units to recognize the state of the surrounding complexed ones. As a result, the fluorescence transduction is not limited to the local site where binding occurs, but it involves a wider region of the fluorophore network that is able to transfer its excitation energy to the complexed units. Such behavior leads to an amplification of the fluorescence signal. What they report here is the first example of amplification in the case an off-on chemosensor due to its organization onto the surface of silica nanoparticles. They also describe a simple general model to approach amplification in multifluorophoric systems based on the localization of the excited states, which is valid for assemblies such as the supramolecular ones where molecular interactions are weak and do not significantly perturb the individual electronic states. The introduction of an amplification factor in particular allows for a simple quantitative estimation of the amplification effects (see Figure 1.4.3).18
1.5 AIM
In the present study, our strategy is to extend the π-conjugated system from H-acceptor emitters of small molecules to light-emitting H-acceptor polymers. A series of novel homopolymeric and random-copolymeric H-acceptor emitters containing carbazole moieties (to increase Tg values and hole-transporting properties of PLEDs) and three-conjugated aromatic rings, including one pendent pyridyl terminus (as the H-acceptor site) and two lateral methoxyl groups on the middle rings (to increase solubility after polymerization), were successfully synthesized. By incorporating with different generations of OXD dendritic H-donors bearing benzoic acids, the supramolecular side-chain copolymers (i.e., H-bonded dendritic complexes) were consecutively constructed. Hopefully, supramolecular dendrimers bearing light-emitting H-acceptor polymers (in comparison with small molecular emitters) will have a better film-forming property by the spin-coating process, which may eventually be more useful in PLED device applications.
In our previous work, the mesomorphic and photophysical properties of the H-bonded trimers and polymer networks can be easily adjusted by tuning the non-photoluminescent H-donor acids (or H-donor polymers) and fluorescent H-acceptors (small molecules) in the H-bonded complexes. Unique mesomorphic properties can be introduced to these supramolecular structures containing
non-mesogenic H-acceptor emitters. In the meanwhile, the emission properties of bis-pyridyl H-acceptor emitters can be manipulated by their surrounding non-photoluminescent proton donors. Moreover, new light-emitting H-bonded side-chain dendritic complexes containing side-chain H-acceptor copolymers and electron-transporting H-donor dendrimers have been developed recently, where side-chain H-acceptor copolymers were composed of light-emitting H-acceptor and hole-transporting (carbazole) moieties. In this report, H-bonded side-chain copolymer and homopolymer complex networks containing non-photoluminescent H-donor (benzoic acid) and fluorescent H-acceptor pendants, which involve three-conjugated aromatic rings with pyridyl terminus and lateral methoxyl groups on the middle rings (to increase solubility after polymerization), were successfully synthesized. The copolymerization of H-donor (benzoic acid) and fluorescent H-acceptor monomers with different molar ratios is to avoid the spontaneous aggregation of the π-conjugated H-acceptor emitters and further to tune their emission colors. In addition, the processes of blending H-donor and H-acceptor homopolymers to form H-bonded homopolymer complexes are prepared for comparative purposes. Accordingly, H-bonded effects on the mesomorphic, photophysical, and electro-optical properties of these H-bonded polymer networks are investigated in the present work. Moreover, herein we design and synthesize a side-chain conjugated polymer with pyridyl
pendant group as receptors for analyte matirals. The selectivity and sensitivity of polymer sensor, titration experiments were conducted with an addition of various metal ions. In addition, compare the sensitivity of polymer with its complementary monomer, PL-quenching characteristics toward Cu2+ ion were investigated.
Chapter 2
Study of Supramolecular Side-Chain Copolymers Containing
Light-Emitting H-Acceptors and Electron-Transporting Dendritic
H-Donors
2.1 Abstract
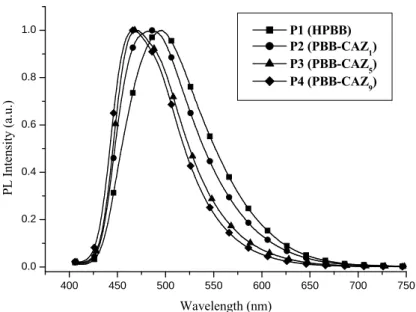
A novel light-emitting hydrogen-bonded (H-) acceptor PBB (M1) containing three conjugated aromatic rings, including one pyridyl terminus and two lateral methoxyl groups (on the middle ring), was successfully synthesized via Horner-Wadsworth-Emmons (HWE) olefination and Sonogashira coupling reaction. Moreover, different molar ratios of light-emitting H-acceptor monomer PBB (M1) and hole-transporting monomer CAZ (M2) bearing a carbazole unit were copolymerized through free radical polymerization to obtain light emitting and hole-transporting H-acceptor copolymers (P1-P5). H-acceptor copolymers P3 and P4 were complexed with different generations of dendritic H-donors (G1COOH-G3COOH) bearing 1,3,4-oxadiazole (OXD) dendrons and terminal benzoic acids via H-bonded self-assembly to form supramolecular side-chain copolymers (i.e., H-bonded dendritic complexes). In contrast to H-acceptor homopolymer P1 (HPBB), H-acceptor copolymers P2-P4 incorporated with
carbazole (CAZ) moieties effectively enhance the glass transition temperatures (Tgs) and minimize the interchain interations of the light-emitting H-acceptor units, and similar effects occur in their H-bonded dendritic complexes. In addition, red shifts of photoluminescence (PL) emissions in H-bonded dendritic complexes can be tuned up to 61 nm. Furthermore, H-bonded dendritic complexes excited at 305 nm of OXD absorption can create a stronger fluorescence than that excited at 397 nm of PBB absorption, indicating that the intensity of the sensitized emission of PBB (by energy transfer from OXD absorption at 305 nm) is even stronger than that of a direct emission of PBB (by merely PBB absorption at 397 nm). The OXD dendritic wedges in H-bonded dendritic complexes can lower the LUMO energy levels and provide a better electron injection property. H-acceptor polymer P4 and its H-bonded dendritic complexes showed electroluminescence (EL) emissions in the range of 464-519 nm from blue to green. In addition, a PLED device containing H-bonded dendritic complex P4/G1COOH showed an EL emission of 519 nm under a turn-on voltage of 6.5 V, with a maximum luminance of 408 cd/m2 at 18 V and a luminance efficiency of 0.39 cd/A at 100 mA/cm2, respectively.
2.2 Introduction
In recent years, polymeric materials based on spontaneous formation of supramolecular architectures by self-assembly of various organic molecular components have attracted great attention in areas ranging from chemistry to materials science.1,2 Simple association of two complementary compounds through specific noncovalent interactions, such as hydrogen-bonded (H-bonded),2,19-26 ionic,27,28 and metal-coordinative29-32 interactions between molecular components, can give rise to unique properties and phase structures, which are not possessed by the individual components. Intensive researches have been directed toward functional supramolecular systems to control the dimensionality and shape of self-assembled structures through molecular design, but it remains a challenge driven by a wide variety of potential applications in the fields of catalyzes, microelectronics, nonlinear optics, sensors, and display technologies. Since the first polymeric light-emitting diode (PLED) based on poly(p-phenylenevinylene) (PPV) was reported by Burroughes et al.,33 various kinds of conjugated main-chain and side-chain polymers have been developed for electroluminescent (EL) devices.34-37 Future applications of PLEDs to full-color and large-area flat panel displays become possible due to their high luminescence efficiency, low cost, high flexibility, and easy fabrication of spin-coating technique.38 However, the most important problem with the π-conjugated
systems is their tendency to form aggregates/excimers via π-π interactions in the solid state, which will lead to red shifts or low-energy band gaps of emission spectra, self-quenching of excitons, and reduction of fluorescence quantum efficiencies. To overcome this problem, one of the approaches is to introduce dendritic architectures into the π-conjugated systems so as to prevent close chains from packing and thus to increase the polymer luminescent efficiency and reduce the tendency of aggregation. For instance, Fréchet-type poly(aryl ether) dendrons attached to fluorene units were reported by Carter et al.39 to demonstrate the shielding effect by attaching dendritic side chains to the conjugated polyfluorene backbones, which improved the luminescence properties of these materials due to the reduction of both aggregates/excimers in solution and solid states. Müllen et al.40 prepared polyfluorene-based conjugated polymers with bulky polyphenylene dendritic substituents at C-9 position, which suppressed the formation of aggregates with long wavelength emissions, and thus a pure blue emission was acquired. More recently, a number of dendrimers have been reported for a wide variety of applications in such EL device41-51 and photovoltaic (PV) cell52,53 materials. In our previous work,54 H-donor dendrimers with a benzoic acid terminus were singly/doubly H-bonded to mono/bis-pyridyl H-acceptor emitters to form several series of novel supramolecular dendrimers, whose emission colors could be easily adjusted by their
non-light-emitting H-donors. Moreover, the higher generation of dendritic sizes could afford stronger siteisolation and dendron-dilution effects, so better energy transfer along with higher fluorescence quantum efficiencies were achieved.
In the present study, our strategy is to extend the π-conjugated system from H-acceptor emitters of small molecules to light-emitting H-acceptor polymers. According to Scheme 1, a series of novel homopolymeric and random-copolymeric H-acceptor emitters containing carbazole moieties (to increase Tg values and hole-transporting properties of PLEDs) and three-conjugated aromatic rings, including one pendent pyridyl terminus (as the H-acceptor site) and two lateral methoxyl groups on the middle rings (to increase solubility after polymerization), were successfully synthesized. By incorporating with different generations of OXD dendritic H-donors bearing benzoic acids (see Figure 2.1), the supramolecular side-chain copolymers (i.e., H-bonded dendritic complexes) were consecutively constructed as shown in Figure 2.2. Hopefully, supramolecular dendrimers bearing light-emitting H-acceptor polymers (in comparison with small molecular emitters) will have a better film-forming property by the spin-coating process, which may eventually be more useful in PLED device applications. Accordingly, H-bonded effects on the thermal, photophysical, and photo-/electro-luminescent properties of these supramolecules in the solid state will be illustrated.
COOH O O N N O N N O O O COOH O O O O N N O N N O O O O O N N O N N O O O G1COOH G2COOH O O O O N N O N N O O O O O N N O N N O O O COOH O O O O O O N N O N N O O O O O NN O NN O O O G3COOH
Figure 2.1 Different generations of dendritic H-donors (G1COOH–G3COOH)
used in H-bonded side-chain dendritic complexes.
Figure 2.2 Schematic representation of H-acceptor copolymers and H-bonded
side-chain dendritic complexes bearing different generations of dendritic H-donors (G1COOH–G3COOH).
2.3 Experimental Section
2.3.1 Measurements and Characterization
1H NMR spectra were recorded on a Varian Unity 300 MHz spectrometer using
CDCl3 and DMSO-d6 as solvents. Elemental analyses were performed on a HERAEUS CHN-OS RAPID elemental analyzer. High resolution electron impact mass data were obtained on a Finnigan-MAT-95XL. Phase transition temperatures were determined by differential scanning calorimetry (DSC, model: Perkin Elmer Diamond) under N2 with a heating and cooling rate of 10 °C/min and polarizing optical microscope (POM, model: Leica DMLP) equipped with a hot stage. Thermogravimetric analyses (TGA) were carried out on a TA Instruments Q500 thermogravimetric analyzer at a heating rate of 20 °C/min under nitrogen. Gel permeation chromatography (GPC) analyses were conducted on a Water 1515 separation module using polystyrene as a standard and THF as an eluant. Fourier transform infrared (FTIR) spectra of samples (dispersed in KBr disks) were recorded on a Perkin-Elmer Spectrum 100 Series. Synchrotron powder X-ray diffraction (XRD) measurements were performed at the beamline BL17A of the National Synchrotron Radiation Research Center (NSRRC), Taiwan (for details of the XRD installation, see Supporting Information). UV-vis absorption spectra were recorded on a HP G1103A spectrophotometer, and photoluminescence (PL) spectra were obtained on a Hitachi
F-4500 spectrophotometer in dilute THF solutions (10-6 M). The PL quantum yields (ΦPL) of polymers were measured with 9,10-diphenylanthracene as a reference (in cyclohexane, ΦPL = 0.9).55 Thin films in UV-vis and PL measurements were prepared by spin-coating of THF solutions (with a concentration of 10 mg/mL) at 3000 rpm on a quartz substrate. Cyclic voltammetry (CV) measurements were performed at a scanning rate of 100 mV/s in a solution of 0.1 M tetrabutylammonium hexafluorophosphate (Bu4NPF6) dissolved in acetonitrile at room temperature using an Autolab PGSTAT30 potentiostat/galvanostat with a standard three-electrode electrochemical cell. A platinum disk working electrode, a Pt wire counter electrode, and an Ag/AgCl reference electrode were used. The sample films were coated on the surface of a platinum disk by the solution-dipping process from THF solutions.
A series of electroluminescence (EL) devices with the configuration of ITO/PEDOT:PSS/polymer (P4 or its H-bonded dendrimers
complexes)/BCP/Alq3/LiF/Al were made, where BCP (i.e.,
2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline) was used as a hole-blocking layer and Alq3 (i.e., tris(8-hydroxyquinoline)aluminium) was used as an electron-transporting layer. ITO substrates, where glass substrates were coated with indium-tin oxide (ITO) having a sheet resistance of ~20 Ω/square and an effective individual device area of 3.14 mm2, were routinely cleaned by ultrasonic treatments in
detergent solutions and diluted water, followed by rinsing with acetone and then ethanol. After drying, ITO substrates were kept in oxygen plasma for 4 min before being loaded into the vacuum chamber. The poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS) films were first deposited on pre-cleaned ITO substrates by spin-coating at 6000 rpm for 1 min and subsequently cured in an oven at 120 °C for 1 h. Then, light-emitting polymers (P4 or its H-bonded side-chain dendritic complexes) in THF solutions (10 mg/mL) were spin-coated onto the PEDOT:PSS layer at 4000–5500 rpm. The thicknesses of PEDOT:PSS and LED polymers were measured by an Alfa Step 500 Surface Profiler (Tencor). BCP and Alq3 were thermally deposited at a rate of 1–2 Å/s under a pressure of ~2 × 10-5 torr in an Ulvac Cryogenic deposition system. Under the same deposition condition, one layer of LiF was thermally deposited as a cathode at a rate of 0.1–0.2 Å/s, which was followed by capping with aluminum. The current-voltage-luminescence characteristics were measured in ambient condition by Keithley 2400 source meter and Newport 1835C optical meter equipped with 818ST silicon photodiode.
Scheme 2.1 Synthetic Routes of H-Acceptor Monomer and Polymers (P1-P5)a I H3CO OCH3 O H I H3CO OCH3 OH I Cl OCH3 H3CO I PO(OEt)2 OCH3 H3CO N H3CO OCH3 I Br HO HO(CH2)10O Br HO(CH2)10O OH HO(CH2)10O HO(CH2)10O N H3CO OCH3 O(CH2)10O N H3CO OCH3 O N (CH2)2 O O (1) (2) (3) (4) (5) (6) (7) (8) (9) (M1) CAZ (M2) N H3CO OCH3 (5) + P1 (HPBB), x/y = 1/0 P2 (PBB-CAZ1), x/y = 1/1 P3 (PBB-CAZ5), x/y = 1/5 P4 (PBB-CAZ9), x/y = 1/9 P5 (HCAZ), x/y = 0/1 PBB (M1)
(i) (ii) (iii)
(iv) (v) (vi) (vii) (viii) (ix) (x) + O N (CH2)2 O (CH2)10 O O C CH3 CH2 O C CH3 co CH2 x y
a Reagents and conditions: (i) NaBH
4, MeOH/THF, room temperature, 1 h; (ii) conc. HCl, dioxane, reflux, 10 h; (iii) P(OEt)3, reflux, 12 h; (iv) pyridine-4-carboxaldehyde,
t-BuOK, THF, room temperature, 12 h; (v) 10-bromodecanol, K2CO3, KI, acetone, reflux, 48 h; (vi) 2-methyl-3-butyn-2-ol, Pd(PPh3)2Cl2, CuI, PPh3, Et3N, 70 °C, 12 h; (vii) KOH, dioxane, reflux, 3 h; (viii) Pd(PPh3)2Cl2, CuI, PPh3, Et3N/THF, 50 °C, overnight; (ix) vinyl methacrylate, 1,3-dichloro-1,1,3,3-tetrabutyldistannoxane, 2,6-di-tert-butyl-4-methylphenol, THF, 50 °C, 48 h; (x) AIBN, THF, 60 °C, 24 h.
2.3.2 Materials
Chemicals and solvents were reagent grades and purchased from Aldrich, ACROS,
TCI, and Lancaster Chemical Co. Tetrahydrofuran (THF) and triethylamine (Et3N) were distilled to keep anhydrous before use. Azobisisobutyronitrile (AIBN) was recrystallized from methanol before use. The other chemicals were used without further purification. Different generations of dendritic H-donors (G1COOH–G3COOH), as shown in Figure 1, used in H-bonded side-chain dendritic complexes were reported in our previous work.54 10-Bromodecanol,56 1,3-dichloro-1,1,3,3-tetrabutyldistannoxane,57 and monomer CAZ (M2)58 were prepared by following the already published procedures.
4-Iodo-2,5-dimethoxybenyl alcohol (2). To a stirred solution of
4-iodo-2,5-dimethoxybenzaldehyde 1 (8.0 g, 27.4 mmol) in 200 mL of THF/MeOH (1:1), NaBH4 (0.52 g, 13.7 mmol) was added very slowly to react at room temperature. After 1 h, the solution was cooled to 0 °C by ice bath, acidified with dilute HCl solution, and extracted with ethyl acetate. The resulting materials in organic phase were combined and washed with water. Afterward, the organic extracts were dried over Na2SO4 and evaporated. The purified residue was recrystallized from dichloromethane/2-propanol to give a colorless crystal. Yield: 7.6 g (95%). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.25 (s, 1H), 6.85 (s, 1H), 4.65 (d, J = 6.3 Hz, 2H), 3.85
(s, 3H), 3.82 (s, 3H), 2.22 (t, J = 6.6 Hz, 1H).
1-Iodo--4-chloromethyl-2,5-dimethoxybenzene (3). To a stirred solution of 2 (7.0
g, 23.8 mmol) in 1,4-dioxane (200 mL), concentrated HCl (20 mL) was added to reflux for 10 h. After the reaction was completed, the crude mixture was added with water. The organic layer was extracted with ethyl acetate, dried over Na2SO4, and evaporated. The residual product was purified by flash column chromatography (silica gel, n-hexane/ethyl acetate 40:1) to give a white solid. Yield: 6.8 g (92%). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.29 (s, 1H), 6.86 (s, 1H), 4.60 (s, 2H), 3.85 (s, 3H), 3.83 (s, 3H).
4-Iodo-2,5-dimethoxybenzyldiethylphosphonate (4). Compound 3 (6.0 g, 19.2
mmol) was mixed with an excess of triethyl phosphite (20 mL), and the mixture was heated to reflux and reacted for 12 h. The excess of triethyl phosphite was removed under reduced pressure, and the crude product was purified by washing with hot hexane to give a white solid. Yield: 7.2 g (90%). 1H NMR (300 MHz, CDCl
3): δ (ppm)
7.25 (s, 1H), 6.9 (s, 1H), 4.08−3.99 (m, 4H), 3.83 (s, 3H), 3.80 (s, 3H), 3.20 (d, J = 21.6 Hz, 2H), 1.24 (t, 6.9 Hz, 6H).
1-Iodo-2,5-dimethoxy-4-[2-(4-pyridyl)ethenyl]benzene (5). To a solution of
pyridine-4-carboxaldehyde (1.86 g, 17.4 mmol) in anhydrous THF (10 mL), a suspension of 4 (6.0 g, 14.5 mmol) and t-BuOK (2.93 g, 26.1 mmol) in anhydrous
THF (60 mL) under nitrogen was slowly added. The mixture was stirred to react at room temperature for 12 h. After the reaction was completed, it was quenched with water and extracted with dichloromethane. After that, the organic layer was dried over Na2SO4 and evaporated. The crude product was purified by column chromatography (silica gel, dichloromethane/acetone 30:1) to give a yellow solid. Yield: 3.2 g (60%). 1H NMR (300 MHz, CDCl
3): δ (ppm) 8.55 (d, J = 4.5 Hz, 2H), 7.57 (d, J = 16.5 Hz,
1H), 7.37 (d, J = 4.5 Hz, 2H), 7.30 (s, 1H), 7.01 (d, J = 16.5 Hz, 1H), 7.00 (s, 1H), 3.88 (s, 3H), 3.84 (s, 3H).
10-(4-Bromophenoxy)-decan-1-ol (6). To a stirred solution of 4-bromophenol (4.0
g, 23.1 mmol) in acetone (200 mL), potassium carbonate (9.6 g, 69.3 mmol), 10-bromodecanol (6.6 g, 27.7 mmol), and a few amounts of potassium iodide (ca. 10 mg) were added to reflux for 48 h under nitrogen. After cooling to room temperature, the solvent was removed under reduced pressure, and the residue was taken up in water and extracted with ethyl acetate. Next, the organic layer was dried over Na2SO4 and evaporated. The crude product was purified by column chromatography (silica gel, n-hexane/ethyl acetate 3:1) to give a white solid. Yield: 6.1 g (80%). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.33 (d, J = 9.0 Hz, 2H), 6.75 (d, J = 9.0 Hz, 2H), 3.89 (t, J = 6.3 Hz, 2H), 3.62 (t, J = 6.6 Hz, 2H), 1.78−1.69 (m, 2H), 1.59−1.50 (m, 2H), 1.41−1.29 (m, 12H).
4-[4-(10-Hydroxy-decyloxy)-phenyl]-2-methyl-3-butyn-2-ol (7). A solution of 6
(4.0 g, 12.1 mmol), PPh3 (63.1 mg, 0.24 mmol), and CuI (45.6 mg, 0.24 mmol) in dry Et3N (80 mL) was degassed with nitrogen for 5 min. Then, the solution was added with 2-methyl-3-butyn-2-ol (2.0 g, 24.2 mmol) and Pd(PPh3)2Cl2 (84.1 mg, 0.12 mmol) at room temperature, and the reaction mixture was stirred to react at 70 °C for 12 h. The mixture was filtered and the solvent was removed in vacuum. The crude mixture was extracted using ethyl acetate, and the extract was washed with water, dried over Na2SO4, and then evaporated. Subsequently, the crude product was purified by column chromatography (silica gel, n-hexane/ethyl acetate 1:1) to give a light yellow solid. Yield: 3.14 g (78%). 1H NMR (300 MHz, CDCl
3): δ (ppm) 7.31 (d, J =
9.0 Hz, 2H), 6.79 (d, J = 9.0 Hz, 2H), 3.92 (t, J = 6.6 Hz, 2H), 3.62 (t, J = 6.6 Hz, 2H), 1.77−1.70 (m, 2H), 1.60 (s, 6H), 1.58−1.50 (m, 2H), 1.42−1.29 (m, 12H).
4-Ethynyl-1-(10-hydroxydecan-1-yloxy)benzene (8). A stirred solution of 7 (2.5 g,
7.5 mmol) and finely powdered KOH (1.26 g, 22.5 mmol) in 1,4-dioxane (80 mL) was refluxed under nitrogen for 3 h. After cooling to room temperature, the solvent was removed under reduced pressure and the residue was taken up in water, and then the mixture was extracted with ethyl acetate and acidified with 3 N HCl (150 mL). The organic solution was washed with water, dried over Na2SO4, and then evaporated. Afterward, the crude product was purified by column chromatography (silica gel,
n-hexane/ethyl acetate 5:1) to give a light yellow solid. Yield: 1.75 g (85%). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.39 (d, J = 9.0 Hz, 2H), 6.80 (d, J = 9.0 Hz, 2H), 3.92 (t,
J = 6.6 Hz, 2H), 3.62 (t, J = 6.6 Hz, 2H), 2.97 (s, 1H), 1.80−1.70 (m, 2H), 1.57−1.50
(m, 2H), 1.42−1.29 (m, 12H).
1-{[4-(10-Hydroxy-decyloxy)-phenyl]-ethynyl}-2,5-dimethoxy-4-[2-(4-pyridyl)e thenyl]benzene (9). A solution of 5 (1.0 g, 2.72 mmol), 8 (0.78 g, 2.85 mmol), and
PPh3 (14.1 mg, 0.054 mmol) in 80 mL of dry Et3N/THF (1:1) was degassed with nitrogen for 5 min. Then, the solution was added with CuI (10.3 mg, 0.054 mmol) and Pd(PPh3)2Cl2 (19.1 mg, 0.027 mmol) at room temperature, and it was stirred to react at 50 °C overnight. The mixture was filtered and the solvent was removed in vacuum. The crude mixture was extracted using dichloromethane, and the extract was washed with water, dried over Na2SO4, and then evaporated. After that, the crude product was purified by column chromatography (aluminum oxide, dichloromethane/acetone 40:1) to give a yellow solid. Yield: 1.28 g (92%). 1H NMR (300 MHz, CDCl
3): δ (ppm) 8.56 (d, J = 4.5 Hz, 2H), 7.66 (d, J = 16.5 Hz, 1H), 7.50 (d, J = 9.0 Hz, 2H), 7.39 (d, J = 4.5 Hz, 2H), 7.11 (s, 1H), 7.04 (d, J = 16.5 Hz, 1H), 7.04 (s, 1H), 6.87 (d, J = 9.0 Hz, 2H), 3.99 (t, J = 6.6 Hz, 2H), 3.97 (s, 3H), 3.89 (s, 3H), 3.62 (t, J = 6.6 Hz, 2H), 1.81−1.72 (m, 2H), 1.57−1.51 (m, 2H), 1.41−1.30 (m, 12H). 1-{[4-(10-Methacryloyloxy-decyloxy)-phenyl]-ethynyl}-2,5-dimethoxy-4-[2-(4-p
yridyl)ethenyl]benzene, PBB (M1). To a Schlenk tube, compound 9 (1.0 g, 1.95
mmol), vinyl methacrylate (0.55 g, 4.88 mmol), 1,3-dichloro-1,1,3,3-tetrabutyldistannoxane (43.12 mg, 0.078 mmol), and 2,6-di-tert-butyl-4-methylphenol (25.78 mg, 0.117 mmol) in dry THF (2 mL) were purged with nitrogen for 15 min at room temperature. The tube was sealed and heated with stirring at 50 °C for 2 days. After cooling to room temperature, the reaction mixture was extracted using dichloromethane, and the extract was washed with water, dried over Na2SO4, and then evaporated. The crude product was purified by column chromatography (aluminum oxide, n-hexane/dichloromethane 1:1) and then washed with hexane to give a light yellow solid. Yield: 0.97 g (85%). 1H NMR (300 MHz, CDCl3): δ (ppm) 8.57 (d, J = 4.5 Hz, 2H), 7.66 (d, J = 16.5 Hz, 1H), 7.50 (d, J = 9.0 Hz, 2H), 7.39 (d, J = 4.5 Hz, 2H), 7.11 (s, 1H), 7.04 (d, J = 16.5 Hz, 1H), 7.04 (s, 1H), 6.87 (d, J = 9.0 Hz, 2H), 6.10 (s, 1H), 5.55 (s, 1H), 4.14 (t, J = 6.6 Hz, 2H), 3.97 (t, J = 6.6 Hz, 2H), 3.96 (s, 3H), 3.89 (s, 3H), 3.62 (t, J = 6.6 Hz, 2H), 1.95 (s, 3H), 1.81−1.75 (m, 2H), 1.58−1.53 (m, 2H), 1.42−1.30 (m, 12H). HRMS (EI): calcd for C37H43NO5, 581.3141; found 581.3146. Anal. Calcd for C37H43NO5: C, 76.39; H, 7.45; N, 2.41. Found: C, 76.15; H, 7.37; N, 2.44.
General Procedure for the Syntheses of Homopolymers and Copolymers (P1-P5). All polymerization procedures were carried out according to the free radical
polymerization described by the following steps. To a Schlenk tube, 1.5 g of monomers M1, M2, or the mixture of M1 and M2 were dissolved in dry THF (7.5 mL) with 20 wt% of monomer concentration and AIBN (2 mol% of total monomer concentration) as an initiator. The solution was degassed by three freeze-pump-thaw cycles and then sealed off. The reaction mixture was stirred and heated at 60 °C for 24 h. After polymerization, the polymer was precipitated into diethyl ether. Then, the precipitated polymer was collected, washed with diethyl ether, and dried under high vacuum. P1 (HPBB). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 8.44 (br, 2H), 7.53−6.77 (br, 10H), 3.80 (br, 10H), 1.66−1.24 (br, 21H). P2 (PBB-CAZ1). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 8.46 (br, 2H), 7.97−6.75 (br, 18H), 3.88 (br, 14H), 1.43−1.08 (br, 26H). P3 (PBB-CAZ5). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 8.48 (br, 2H), 7.92−6.76 (br, 50H), 4.33−3.83 (br, 30H), 1.43−-0.06 (br, 46H). P4 (PBB-CAZ9). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 8.48 (br, 2H), 7.88−6.76 (br, 80H), 4.34−3.83 (br, 45H), 1.43−-0.16 (br, 65H). P5 (HCAZ). 1H NMR (300 MHz, DMSO-d6): δ (ppm) 7.89 (br, 2H), 7.32−7.00 (br, 6H), 4.34−3.94 (br, 4H), 0.99−-0.15 (br, 5H).
H-Bonded Side-Chain Dendritic Complexes). H-bonded side-chain dendritic
complexes were made of appropriate (fully H-bonded) molar ratios of H-acceptor copolymers (P3 and P4) and dendritic H-donors (G1COOH–G3COOH) in THF solutions, and then the solvent was removed by slow evaporation and followed by drying under vacuum at 50 °C.
2.4 Results and Discussion
2.4.1 Syntheses and Characterization of Polymers
The syntheticroutes of monomer PBB (M1) and polymers P1-P5 are shown in Scheme 1. The starting material 1 (i.e., 4-iodo-2,5-dimethoxybenzaldehyde) was synthesized by following a reported procedure59 via iodination of 2,5-dimethoxybenzaldehyde with iodine and silver nitrate in the presence of methanol. The aldehyde group of compound 1 was further reduced to a benzyl alcohol and then was transformed into a benzyl chloride group with hydrochloric acid in the presence of 1,4-dioxane to givecompound 3, which was converted to the corresponding phosphonate ester 4 by Michaelis-Arbuzov reaction under the triethyl phosphite treatment with a yield of 90%.60 Compound 5 was prepared by means of Horner-Wadsworth-Emmons (HWE) olefination reaction between compound 4 and pyridine-4-carboxaldehyde using potassium tert-butoxide as a base in THF to give 60% yield.61 Compound 6 in 80% yield was obtained by reaction of 4-bromophenol
with 10-bromodecanol under Williamson etherification condition (K2CO3/acetone). The latter Sonogashira (Pd-catalyzed) coupling reaction of compound 6 with 2-methyl-3-butyn-2-ol afforded compound 7 in the presence of a catalytic amount (1 mol %) of Pd(PPh3)2Cl2 in Et3N with a yield of 78%,62 which was then deprotected with KOH/1,4-dioxane to acquire compound 8 in 85% yield. Subsequently, the three-conjugated rings of compound 9 with a yield of 92% was prepared through a second Sonogashira coupling reaction between compounds 8 and 5 in Et3N/THF (1:1). Finally, monomer PBB (M1) with a high yield of 85% was obtained by transesterification reaction63 between compound 9 and an excess amount (2.5 equiv) of vinyl methacrylate in the presence of a higher concentration of 1,3-dichloro-1,1,3,3-tetrabutyldistannoxane as a catalyst and 2,6-di-tert-butyl-4-methylphenol as an inhibitor in THF. The yield is much better than that reported64 by our previous esterification reaction. The final chemical structure of monomer PBB (M1) was confirmed by 1H NMR spectroscopy, HRMS, and elemental analysis (see Experimental Section and Appendix A1).
The polymerization reactions with good yields (ranging 73-88%) were carried out in THF at 60 °C through free radical polymerization in the presence of azobis(isobutyronitrile) (AIBN) as an initiator. The feeding ratios of monomers PBB (M1) to CAZ (M2) were 1:0, 1:1, 1:5, 1:10, and 0:1, respectively, to acquire
H-acceptor polymers P1-P5. The chemical structures of polymers P1-P5 in DMSO-d6 were verified by 1H NMR spectroscopy in comparison with those of their monomers
PBB (M1) and CAZ (M2) (see Appendix A1). In the 1H NMR spectra of polymers, the disappearance of proton peaks in the region of vinyl (methacrylate) groups with chemical shifts at 5.4-6.1 ppm indicated that all monomers were reacted. The copolymer compositions were estimated by comparing the relative integration areas of the peaks at 8.5 ppm (corresponding to two protons of α-pyridyl groups in PBB) and 6.7-7.9 ppm (corresponding to the other overlapped aromatic protons of PBB and
CAZ units), respectively. Regarding the compositions of copolymers P2-P4, the
actual compositions of P2 and P3 are very close to the feeding ratios of monomers, except that P4 has a slightly lower molar ratio than that of feeding. All polymers are soluble in common organic solvents, such as THF, DMSO, and DMF. The weightaverage molecular weights (Mw) and polydispersity indexes (PDI) of polymers
P1-P5, determined by gel permeation chromatography (GPC) with THF as the eluting
solvent and polystyrene as standards, are in the range 14400-53100 g/mol and 1.72-3.69, respectively. The compositions (input and output molar ratio), molecular weights (Mw), PDI values, and yields of polymers P1-P5 are summarized in Table 2.1.
Table 2.1 Composition, Yields, Molecular Weights, and Degradation Temperatures of Polymers P1-P5 polymer molar ratio yield (%) Mw (g/mol)b PDI (Mw/Mn)b Td (oC)c feed monomer
(PBB/CAZ) polymers(x/y)a
P1 (HPBB) 1/0 1/0 73 14400 1.72 357 P2 (PBB-CAZ1) 1/1 1/1 88 38100 3.24 323 P3 (PBB-CAZ5) 1/5 1/5 85 45600 2.99 305 P4 (PBB-CAZ9) 1/10 1/8.75 81 53100 3.69 300 P5 (HCAZ) 0/1 0/1 75 47800 3.65 276 a Determined by 1H NMR spectra.
b Weight-average molecular weight (M
w) and polydispersity index (PDI) determined by GPC in THF using polystyrene as a standard.
c Temperature (°C) at 5% weight loss measured by TGA at a heating rate of 20 °C
min-1 under nitrogen.
The existence of hydrogen bonds in the H-bonded complexes can be confirmed by FTIR spectroscopy. Therefore, the IR spectra (at room temperature) of H-acceptor polymer P3, H-donor G1COOH, and H-bonded side-chain dendritic complexes (side-chain copolymers with pendent dendrimers) P3/G1COOH shown in Figure 2.3 are compared to analyze the formation of hydrogen bonds between H-acceptor polymer P3 and H-donor dendrimer G1COOH. In contrast to the O-H bands of pure H-donor (H-bonded dendritic dimer) G1COOH at 2627 and 2562 cm-1, the weaker O-H bands observed at 1925 and 2500 cm-1 in the H-bonded side-chain dendritic complex P3/G1COOH are indicative of stronger hydrogen bonding between the

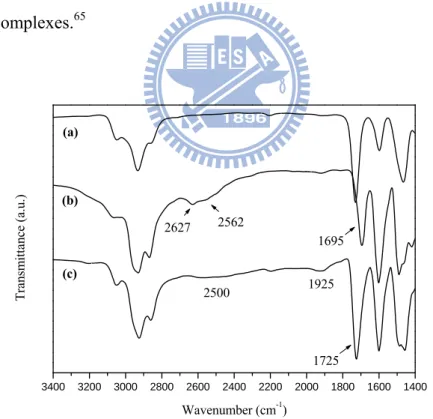
![HPSH [ 分子間作用力 - 凡得瓦力 ]](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)