The Journal of Physical Chemistry A is published by the American Chemical Society. 1155 Sixteenth Street N.W., Washington, DC 20036
Article
Experimental and Theoretical Investigation of
High-Power Laser Ionization and Dissociation of Methane
†M. Sharifi, F. Kong, S. L. Chin, H. Mineo, Y. Dyakov, A. M. Mebel, S. D. Chao, M. Hayashi, and S. H. Lin
J. Phys. Chem. A, 2007, 111 (38), 9405-9416 • DOI: 10.1021/jp074053fDownloaded from http://pubs.acs.org on December 21, 2008
More About This Article
Additional resources and features associated with this article are available within the HTML version: • Supporting Information
• Links to the 1 articles that cite this article, as of the time of this article download • Access to high resolution figures
• Links to articles and content related to this article
Experimental and Theoretical Investigation of High-Power Laser Ionization and
Dissociation of Methane
†M. Sharifi,‡F. Kong,§S. L. Chin,‡H. Mineo,*,#Y. Dyakov,# A. M. Mebel,⊥S. D. Chao,|
M. Hayashi,∇and S. H. Lin#,O
Department of Physics, Engineering Physics and Optics, LaVal UniVersity, Quebec City, Quebec, GIK 7P4, Canada, Institute of Chemistry, Chinese Academy of Science, Beijing 100080, People’s Republic of China, Institute of Atomic and Molecular Science, Academia Sinica, P.O. Box 23-166, Taipei, Taiwan,
Department of Chemistry and Biochemistry, Florida International UniVersity, Miami, Florida 33199, Institute of Applied Mechanics, National Taiwan UniVersity, Taipei 106, Taiwan,
Center for Condensed Matter Sciences, National Taiwan UniVersity, Taipei 106, Taiwan, and Department of Applied Chemistry, National Chiao-Tung UniVersity, Hsin-chu, Taiwan ReceiVed: May 25, 2007; In Final Form: July 30, 2007
The main purpose of this paper is to report the high-power laser ionization-dissociation of CH4at various
femtosecond (fs) laser intensities (from 1× 1014W/cm2to 2× 1015W/cm2) with a laser pulse duration of
48 fs. The generalized molecular Keldysh theory has been applied to calculate the ionization yields for CH4
+
and CH4
++
. Outside the influence of the fs intense laser, we propose to calculate the mass spectra due to the decomposition of CH4+ and CH4++, using the Rice-Ramsperger-Kassel-Marcus (RRKM) theory. The
agreement between the experimental mass spectra and calculated mass spectra seems to be reasonable.
1. Introduction
The earliest experiment on the fragmentation of energy-selective methane (CH4) ions was that of von Koch in 1964,1
in which the ions were generated by charge exchange. Later, the photoelectron photoion coincidence (PEPICO) techniques2,3
s particularly, the form that uses zero energy or threshold electrons (TREPICO)shas been used to examine the fragmentation of energy-selective CH4ions.4-6 The ground-state case has been
studied in detail, and the Rice-Ramsperger-Kassel-Marcus (RRKM) theory has been used successfully to explain the experimental results.4
Recently, the application of an intense ultrashort laser to ionize and dissociate molecules has attracted considerable attention.7-14 The conventional knowledge of the high-power
laser ionization-dissociation of molecules is that the molecule is first ionized in the intense laser field, followed by fragmenta-tion of the molecular ions.7-18
In this paper, we shall report the experimental results of the high-power laser ionization-dissociation of CH4 performed
using the 800-nm laser with a pulse duration of 48 fs and with a laser intensity that varies from 1.5× 1014W/cm2to 1.9×
1015W/cm2. Depending on the laser intensity, both CH 4+and
CH4++and their fragmented daughter ions can be observed.
Recently, the fragmentation pattern of CH4was
experimen-tally studied at an intensity of ∼1014W/cm2, with the laser
duration varying from 8 fs to 110 fs. For the case in which the laser duration was 8 fs, only the primarily fragmental CH3+
ion was observed, in addition to the parent CH4+ion. When
the laser duration was increased to 30 fs, CH2+and H+began
to appear, and when the laser duration reached 110 fs, some doubly charged ions were also observed, in addition to the abundant singly charged ions.19,20The field-assisted dissociation
(FAD) model and quasi-classical trajectory (QCD) calculation were applied to predict the dissociation probability of CH4+.19-21
The calculated probability was corrected with the molecular orientation effect and the spatial distribution of laser intensity. The modified results indicated that the dissociation would require at least 23 fs and was saturated with long pulse widths (∼100 fs).
The present paper is organized as follows. In section 2, the experimental setup and experimental results will be presented, which will be followed by the theoretical treatment of the high-power laser ionization of CH4(section 3). We will discuss the
calculation of the dissociation patterns of CH4+and CH4++in
section 4. Discussion and the conclusion will be presented in section 5.
2. Experiment
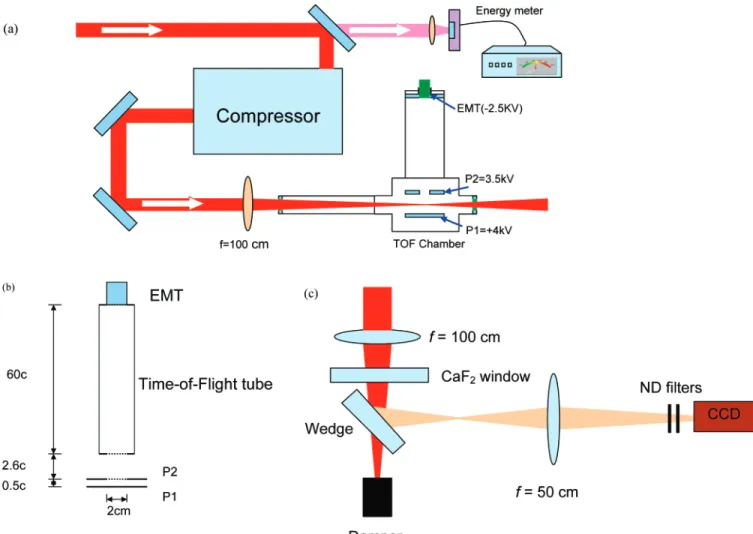
2.1. Experimental Setup. Figure 1a schematically shows the
experimental setup. We have used a commercial laser system (built by Spectra Physics) that consists of a Ti:sapphire oscillator (Tsunami), followed by a regenerative amplifier (spitfire). The 10 Hz seed from the spitfire passes through a two-pass Ti:sapphire amplifier and compressor. The laser pulses have a central wavelength of 800 nm and the compressed pulse has a pulse duration of 48 fs (fwhm), as measured by a single-shot auto-correlator (made by Positive Light), and the maximum output energy is 12 mJ/pulse.
* Author to whom correspondence should be addressed. Tel.: 886 2 2364 4261. Fax: 886 2 2362 0200. E-mail: mineo@gate.sinica.edu.tw.
†Part of the “Sheng Hsien Lin Festschrift”.
‡Department of Physics, Engineering Physics and Optics, Laval
University.
§Institute of Chemistry, Chinese Academy of Science. #Institute of Atomic and Molecular Science, Academia Sinica. ⊥Department of Chemistry and Biochemistry, Florida International
University.
|Institute of Applied Mechanics, National Taiwan University. ∇Center for Condensed Matter Sciences, National Taiwan University. ODepartment of Applied Chemistry, National Chiao-Tung University.
9405
J. Phys. Chem. A 2007, 111, 9405-9416
10.1021/jp074053f CCC: $37.00 © 2007 American Chemical Society Published on Web 09/01/2007
The linearly polarized laser pulses are focused into the interaction chamber using a f ) 100 cm focusing lens. A pyroelectric energy meter (Molectron EPM 1000) is placed behind the last mirror in the laser path before the compressor, to monitor the laser energy by detecting the leak from the mirror.
CH4molecules are injected into the high-vacuum chamber
of the time-of-flight (TOF) mass spectrometer through a variable-leak valve. The background pressure was 5 × 10-9 Torr, whereas the pressure of CH4molecules was set at different
pressures, from 1× 10-8Torr to 5× 10-6Torr, depending on the intensity used. Higher pressures are used at lower intensities,
Figure 1. Experimental setup for (a) laser and time-of-flight (TOF) mass spectrometer, (b) ion detection, and (c) measurement of the focal point.
to avoid the saturation of the ion detector (electron multiplier tube (EMT)). The CH4molecules interact with the laser pulses,
and the parent and fragment ions are accelerated by a static field of 500 V/cm toward the ion detector through an opening 2 cm in diameter, followed by a field-free flight path of 60 cm. As shown in Figure 1b, there are two openings. One is located at the site marked P2 in Figure 1 and one is located at the entrance of the TOF tube. A fine metallic mesh is welded at both openings, to ensure a uniform electric field. The transmis-sion of the mesh is∼99%. An EMT (Thorn EMI electron tube, type 105 EM) with a detection area of diameterφ ) 21.6 mm
was used to detect the ions. The signals at different laser energies were recorded using a 500 MHz oscilloscope (Tektronix TDS 504A). Measurements of the focal spot area and the averaged
laser intensity have been performed outside the chamber, as shown in Figure 1c.
For the focal spot measurement, the focus is imaged onto a charge-coupled device (CCD) camera (Cohu 4810) after the wedge. In this measurement, low pulsed energy is used while the wedge with good optical surface quality further reduces the energy of the pulse at the focus in air, so that ionization is avoided. The laser intensity at the focus was calculated from the laser pulse energy, the pulse duration, and the measured focal area.
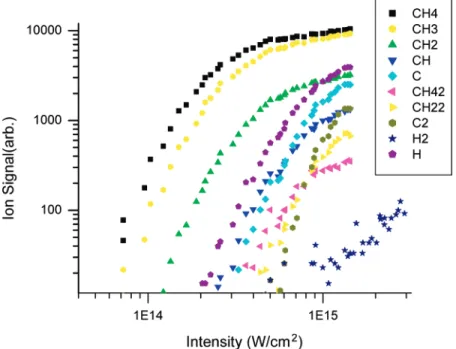
2.2. Experimental Results. Figure 2 shows the evolution of
mass spectrum of CH4 at different laser intensities. Some of
the ions peaks are saturated (flat-top peaks), to enhance the weaker peaks for inspection. In the quantitative measurement
Figure 3. Ion signal versus intensity for the CH4parent ion and fragments of CH4.
Figure 4. Mass spectra of CH4at a pressure of P ) 2× 10-7Torr and intensity of 1.9× 1015W/cm2.
(see discussion below), such saturation is avoided. Figure 3 shows the ion signal versus intensity for parent ion and fragments of CH4, and a particular case of mass spectra at the
laser intensity of 1.9× 1015W/cm2is shown in Figure 4. The
assignment of the mass spectra for the ionization-dissociation of CH4is shown in Figure 4; from this figure, we can see that
the impurity ions (such as H2O+and OH+) can be observed.
3. Theoretical Treatments of High-Power Laser Ionization of Molecules
In this section, we will show how to compute the molecular cations using the generalized Keldysh theory,22 which is
developed from the original Keldysh theory23by combining the
molecular orbital (MO) theory and the Born-Oppenheimer (BO) approximation. The wave function for the electronic ground state of the molecules and molecular cations are obtained from ab initio calculations. For the ionized state, the ionized electron wave function is described by the Volkov continuum state,
where e is the charge of the electron (e ) -1), pb is the momentum of the ionized electron, and, under the dipole approximation, the vector potential AB(t) is given by AB(t) ) -FB sin(ωt)/ω for the linearly polarized laser field FB(t) ) FB
cos-(ωt).
The total electronic wave function of the molecule and molecular cation is expressed as
where Neis the number of electrons and
with
Therefore, by following the approach of Keldysh,22-24 the
ionization rate constant k(FB) can be written as
where cjrepresents the coefficients of the linear combination
of atomic orbitals (LCAO) MO, I0is the ionization potential of
molecule, and U is the ponderomotive energy (U ) (eF)2/
(4mω2)). Here, d is the degeneracy of energy states, S ) x2
for the closed shell, and S ) 1 for the open shell. Using the analytical continuation to the complex plane,24,25L
j(pb) is given
by
where
Here we have
where
Note that Vj(pb + eFb/ωu) has two poles, at u ) u+and u ) u-,
where
andγ is the Keldysh parameter, which is given as
There are some possible choices of contour C, and the details of the calculations for eq 3-6 are given in the work by Mineo et al.24,25
For the multi-ionization of molecules, we consider the following sequential ionization process:
where kiis the photoionization rate constant of the Mi+cation.
The rate equation for eq 3-9 then can be written as
with ψpb( rb,t) ) exp
{
i[
(pb - ebA(t))‚ rb - 1 2me∫
-∞ t dt′(pb - eAB(t′))2]
}
(3-1) ΨM( rb1,..., rbN e,t) )Ψg( r b 1,..., rbNe,t) +∫
d3p (2π)3cbp(t)ΨI,pb( r b 1,..., rbNe,t) (3-2) cbp(t) ) -i∫
-t∞ dt'〈ΨI,pb( rb1,..., rbNe,t)|VL( rb1,..., rbNe,t)|Ψg( rb1,..., rbNe,t)〉 (3-3) VL( rb1,..., rbN e,t) ) -∑
i)1 Ne e rbi‚FB cos(ωt) (3-4) k(FB) )∑
j)1 d 2πS d |cj| 2∫
d 3p (2π)3 |Lj(pb)| 2∑
N)-∞ ∞ δ(
I0+ U + p 2 2m- Npω)
(3-5) Lj(pb) ) 1 2π∫
CduVj[
b +p(
eFB ω)
u]
exp(
ij(u) ω)
(3-6) j(u) ) Nω(
1 i)
log[iu + (-ixu - 1xu + 1)] -ξ(-ixu - 1xu + 1) - Uu(-ixu - 1xu + 1) (3-7) Vj(pb) ) -K[
FB‚pb(pb)j m(I0+ p2/(2m))4 - (FB)j 3(I0+ p2/(2m))3]
(3-8) u ) sin(ωt) K ) 96ex
πa5I03 ξ ) epb‚ FB mω u+) γ(
p cosθx
2mI0 + ix
1 +p 2 sin2θ 2mI0)
) u* -γ )ωx
|e|F2mI0 M 98 k1 M+98 k2 M2+‚‚‚98 kn Mn+ (3-9) d dtA(t) ) -k1A(t) (3-10) d dtA i+ (t) ) kiA(i-1)+(t) - ki+1Ai+(t) (3-11) d dtA n+ (t) ) knA(n-1)+(t) (3-12) A(t0) ) [M]0, Ai+(t0) ) 0 (for i ) 1, 2, ..., n) (3-13)where Ai+(t) (for i ) 0, 1, ..., n) represents the ionization yields
of the molecule and molecular cations. Here, we remark that the photoionization rate constant is assumed to be almost independent of the laser time t during the pulse duration, which is checked in our previous work18by taking into account the
laser pulse for the generalized Keldysh theory. In ref 18, the laser has a Gaussian shape,
where the FB0is the peak amplitude and∆tFis the full width at
half-maximum (fwhm). We note that the pulse laser of a Gaussian shape is not used to calculate the rate constants.
As an outlook, it is very important to consider the time dependence of the pulse laser in solving the rate equations, because, in the actual ionizations, the single- and double-ionization processes will occur sequentially with some time difference, where the time dependence of the rate constant might have an important role.
By solving eqs 3-10 through 3-13, we obtain
It is also important to consider the spatial distribution of the laser pulse,26and, similar to ref 18, we assume that the laser
has a Gaussian shape of the spatial distribution with a width ∆R, i.e.,
The ionization yield then is dependent on time and R (i.e., Ai+
-(t,R)), and we define the spatial averaged ionization yield as
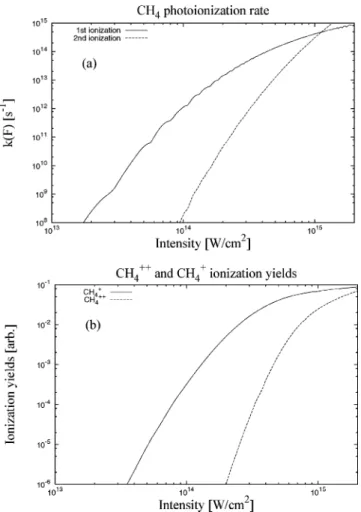
In Figure 5a, we have plotted the photoionization rate constants of the first and second ionizations for CH4, using the
generalized Keldysh theory. The ionization potentials of mol-ecule and molecular cation are calculated using the GAUSSI-AN03 package27and adopt the B3LYP method with STO-3G
basis sets. The calculated ionization potentials are I0 )
12.08 eV for the first ionization and I0) 19.76 eV for the second
ionization. The laser wavelength is fixed atλ ) 800 nm, and
we set the time duration as∆tF) 48 fs and the space width as
∆R ) 30 µm.26If the laser intensity is I < 1014W/cm2, the
first ionization rate is much faster than the second one by a factor of more than 104; on the other hand, if the laser intensity
is close to I ) 1015 W/cm2, both the first and the second
ionization rate constants are almost comparable. In Figure 5b,
we also find that, similar to the rate constants, if the laser intensity is I < 1014W/cm2, the ionization yields of CH
4+are
much larger than CH4++by a factor of more than 105, whereas
if the laser intensity is close to I ) 1015W/cm2, the ionization
yields of CH4+and CH4++are comparable. This fact indicates
that possible fragmentations of CH4++can happen for a laser
intensity of I≈ 1015W/cm2.
4. Theoretical Treatments of Dissociation of Molecular Ions
It is commonly believed that dissociation of a molecule under the influence of a high-power laser occurs via the initial ionization of the molecule in the intense laser field, followed by the fragmentation of molecular ions.
The dissociation of molecular ions induced by an intense femtosecond laser can proceed in two ways. Within the laser-pulse temporal duration and spatial distribution, the potential surfaces can be modified by the laser electric field, and if the laser intensity is strong enough, it can induce the ion dissociation during the laser duration. This type of dissociation is usually referred to as FAD19-21and occurs in the femtosecond range.
The second type of ion dissociation occurs outside the temporal and spatial influence of the femtosecond laser pulse. In this case, the ions are hot and unstable; this type of dissociation for reasonably large-sized ions can be treated by a statistical theorystypically, the RRKM theory (or QET,
quasi-equilib-F B(t) cos(ωt) ) FB 0exp
[
-(4 ln 2)t2 (∆tF) 2]
cos(ωt) A(t) ) A0exp[-k1(t - t0)] (3-14) Ai+(t) ) A0(∏
j)1 i kj)∑
j)1 i+1exp[-k j(t - t0)]∏
l)1(*j) i+1 (kl- kj) (i ) 1, 2, ..., n - 1) (3-15) An+(t) ) A0(∏
j)1 n kj){
1∏
j)1 n kj -∑
j)1 n exp[-k j(t - t0)] kj∏
l)1(*j) n (kl- kj)}
(3-16) I(R) ) I0exp(
- 8R 2 ∆R2)
Ahi+(t) ) 4π∫
0∞dR R2Ai+(t,R) (3-17)Figure 5. Plots of (a) the photoionization rate constants calculated by
the generalized Keldysh theory for the first and second ionizations of CH4, and (b) the ionization yields for CH4+and CH4++(in arbitrary
units).
rium theory). According to the RRKM theory, the unimolecular dissociation rate constant k(E) can be expressed as28-30
where Wq(E - E
0
q) and F(E) denote the total number of states
for the activated complex with activation energy E0q and the
density of states for the reactant with energy E, respectively. Application of the RRKM theory to the dissociation of CH4
ions will be described in the following discussion.
To investigate the dissociation mechanism of methane ions, ab initio quantum chemical calculations of potential energy surfaces were performed for the ground and excited states of CH4+and CH4++ions and some of their fragments. Geometry
optimizations for the ground state were performed using the B3LYP/cc-pVQZ method. After the geometry optimization, the energies of ions were refined by the CCSD(T)/cc-pVQZ ab initio calculations. The zero-point energy (ZPE) corrections to the total CCSD(T)/cc-pVQZ energies were computed using B3LYP/cc-pVQZ frequencies without scaling. Geometries and energies of excited states of CH4and CH4+were calculated at the CASSCF
/6-31+G* level, where nine active orbitals were occupied by eight and seven electrons for the CH4and CH4+ions,
respec-tively. To avoid any restrictions in the geometries of the transition states and intermediates, we did not use symmetry of the molecules in the excited-state calculations. All ab initio
calculations were performed using the GAUSSIAN 0327 and
MOLPRO 200231packages. To compute the dissociation rate
constants of the ions, the RRKM and microcanonical variational transition-state theories28-30 have been applied.
B3LYP/cc-pVQZ frequencies for all intermediates and transition states were used to perform the RRKM calculations.
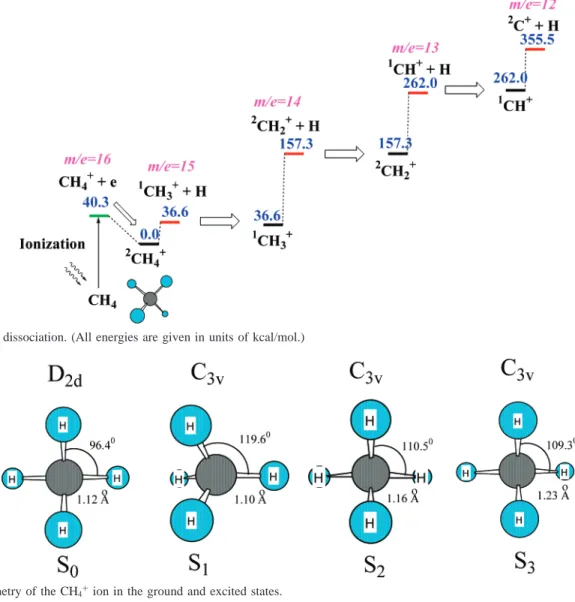
4.1. CH4+Dissociation. The multistep dissociation scheme
for the CH4+ ion is presented in Figure 6. The following
reactions occur without distinct transition states:
In addition, the dissociation rate constant for these reaction channels can be computed by the variational TST theory. Because of the extremely short duration time of the laser pulses, we suppose that the secondary photon absorption by the reaction products is unlikely. Therefore, the appearance of the CH3+,
CH2+, CH+, and C+ions is dependent on the available internal
energy of the initial CH4 ions. Because of the significant
difference between the equilibrium geometries of CH4and CH4+
in the ground electronic state (i.e., the Frank-Condon effect), ionization of the CH4molecule, followed by geometry
relax-Figure 6. CH4+dissociation. (All energies are given in units of kcal/mol.)
Figure 7. Geometry of the CH4+ion in the ground and excited states.
k(E) )1 h
(
Wq(E - E 0q) F(E))
(4-1) CH4+f CH3 + + H (4-2a) CH3+f CH2 + + H (4-2b) CH2+f CH++ H (4-2c) CH+f C++ H (4-2d)ation, can provide sufficient internal energy to the CH4+ion
for its dissociation (see Figure 7). Here, we assume that ionization occurs immediately after excitation of the parent molecule by a high-power laser with energy higher than the ionization threshold, and the ion appears in the ground electronic state. Dissociation of the CH4+ion in the excited2A1state has
been studied by measuring the TREPICO spectra with synchro-tron radiation.32,33It is possible that the excited CH
4+ions might
also be produced in an intense laser. Figure 8 and Table 1 present the geometries of the S1, S2, and S3excited states of
the CH4+ion, excitation energy, and “distortion energy” of these
excited states, in comparison with the equilibrium geometry of
the ground electronic state of CH4+. All these excited states
have C3Vsymmetry. The first and second excited states are the
result of electron transitions from a doubly occupied orbital to the singly occupied HOMO orbital of CH4+. Therefore, the
geometry of these excited states is similar to the tetrahedron shape of the neutral CH4molecule, except that one of the C-H
bonds is elongated in S2and S3, whereas in S1, the HCH′angle
involving the H′ atom on the C3 axis increases to 119.6°.
The higher excited states are the result of electronic transi-tions from doubly occupied orbitals to the lowest unoccupied orbital of CH4+. The geometry of these excited states is similar
to the geometry of the ground triplet state. States S4and S5are
unstable and rapidly decompose to form a H atom and a CH3+ion.
It is necessary to estimate the amount of internal energy of various ions in the reactions shown in eq 4-2 to predict the mass spectra. Dissociation of CH4+occurs if the energy of vibration
along the reaction coordinate exceeds the activation energy, i.e., the barrier height (or the heat of reaction, if the reaction does not have a distinct transition state). The probability of this event can be calculated using the statistical approach of the RRKM theory. For the parent CH4+ ion, the available
internal energy is the part of the excitation energy beyond the ionization threshold, which is converted to vibration energy. After dissociation, the internal energy of secondary fragments is less than the energy of the parent molecule, because of the fact that the excess energy is distributed among the product species and can be approximated as being proportional to the ratio of internal degrees of freedom of the fragment and the parent molecule. The reaction coordinate should be excluded from the total numbers of internal degrees of freedom, because the kinetic energy along the reaction coordinate is supposed to be close to the barrier’s height and is already subtracted from the total internal energy. For the reaction CH4+f CH3+
+ H, the available internal energy of the CH3+fragment is given
as
where E0is the initial photon energy, 6 is the number of internal
degrees of freedom of the CH3+fragment, and 8 is the number
of degrees of freedom of the CH4+ion, excluding the reaction
coordinate (9 - 1 ) 8). According to the scheme presented in Figure 7, the barrier height for the reaction CH3+f CH2++
H is 120.7 kcal/mol, so the minimum energy of the initial photon, which can initiate this secondary reaction, is given as
If the energy of excitation exceeds this value, CH2+fragments
are likely to be observed. For the reaction CH2+f CH++ H, Figure 8. Singly charged ions dissociation rate constants versus
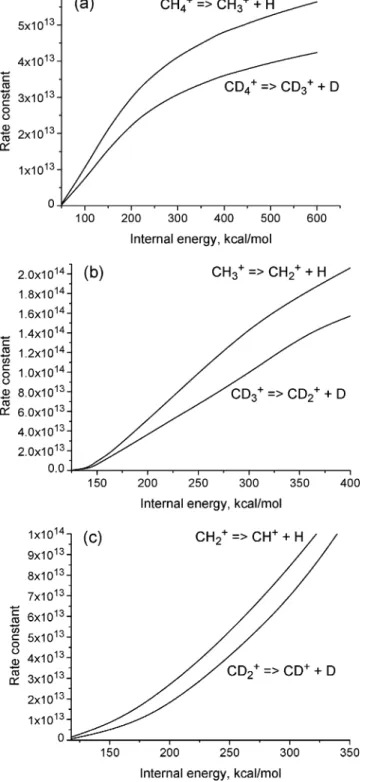
available internal energy of the fragments: (a) CH4+f CH3++ H
and CD4+f CD3++ D, (b) CH3+f CH2++ H and CD3+f CD2+ + D, and (c) CH2+
f CH++ H and CD2+f CD++ D.
TABLE 1: Excitation Energy of the CH4+Ion and the
Internal Energy That Could Be Accumulated by CH4+after
Relaxation from an Excited State, Because of the Difference in the Geometry of the Excited and Ground States
(Distortion Energy) excited state vertical excitation energy (eV) distortion energy (kcal/mol) S1 4.4 19.3 S2 5.3 25.1 S3 10.1 38.6 S4 11.4 S5 13.1 E1) (E0- 36.6) ×6 8 ECH 3) 120.7 ×
(
8 6)
+ 36.6 ) 197.5 kcal/molthe reaction barrier is 104.7 kcal/mol, and the initiation energy is given as
For the reaction CH+f C++ H, the reaction barrier is 93.5 kcal/mol, and the initiation energy is given as
In the last formula, we take into account the fact that the linear CH+fragment has only two rotational degrees of freedom. These results are briefly presented in Table 2. However, note that the decomposition of CH+ cannot be described by a statistical theory such as the RRKM theory.
Excited-states calculations of the neutral CH4molecule reveal
that the first excited state of the CH4molecule, with geometry
close to that of the CH4+ion, is unstable and easily decomposes
to CH2and H2fragments via a barrier of 0.3 kcal/mol. However,
no distinct transition states were observed for the reaction CH4+
f CH2++ H2. The energy of the products is greater than the
energy of the initial CH4+ion by 54.2 kcal/mol.
Figure 8 shows that dissociation rate constants of the parent CH4+ion and its fragments can be higher than 1012s-1if the
internal energy is larger than 100 kcal/mol. To estimate the influence of the isotope effect on the CH4+ dissociation, the
CD4+, CD3+, and CD2+ dissociation rate constants are also
included in Figure 6.
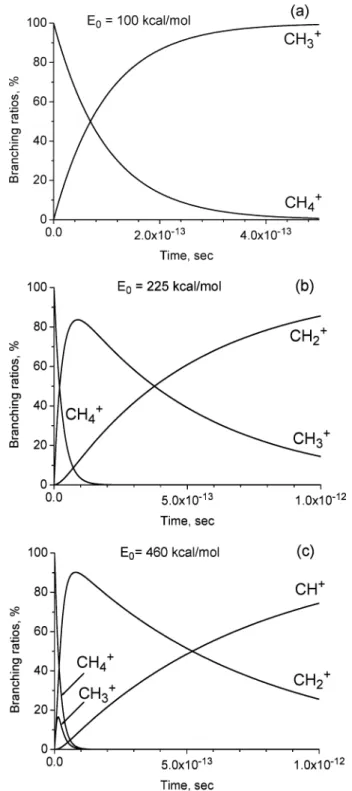
Figure 9 shows the branching ratios of the CH4+dissociation
products at three different values of available internal energies of the initial CH4+ion, up to 1 ps after the ionization. The
internal energy value of E0) 100 kcal/mol is sufficient for the
CH4+ion dissociation, but it is less than that which is necessary
for the CH3+ ion dissociation. Therefore, we have only two
lines: one descending for the CH4+ion concentration, and the
other rising for the CH3+ion concentration. The internal energy
value of E0) 225 kcal/mol (Figure 9b) is sufficient to initiate
the CH3+ion dissociation. In Figure 9b, the concentration of
the CH3+ion initially grows, but later decreases to the zero
value in favor of the CH2+ion production. The internal energy
value of E0) 460 kcal/mol (Figure 9c) is sufficient to initiate
the CH2+ion dissociation. In Figure 9c, the concentration of
the CH3+ion grows at a time interval of ∼0.02 ps and then
decreases fast to the zero value. The concentration of the CH2+
ion also grows at a time interval of 0.1 ps, and then it decreases in favor of the CH+ion production. At this internal energy value, the production of C+ is possible. However, because of the limitations of the RRKM theory, the production of C+is not shown in Figure 9c.
From the aforementioned discussion, we can see that the ionization and dissociation of a molecule are greatly dependent on the intensities of the intense femtosecond laser. This leads to the appearance of the behaviors of mass spectra as a function of laser intensities (see Figure 2).
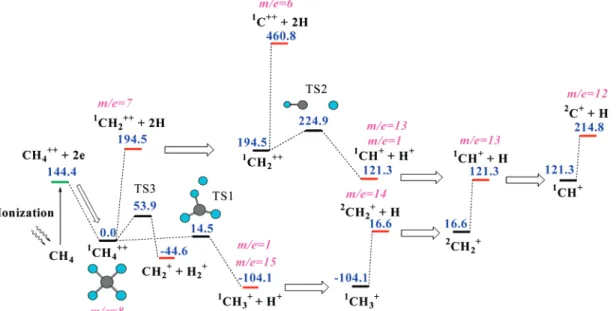
4.2. CH4++Dissociation: Singlet State. CH4++ ion has a
planar square shape (Figure 10). The difference in the geometries of CH4and CH4++provides a large amount of internal energy
for the CH4++ion, which is sufficient for the H+loss reaction
and the H loss reaction to occur. The H+elimination reaction passes through transition state TS1 with a low barrier of 14.5 kcal/mol and produces the CH3+ion. The next dissociation
steps of this fragment have already been described in the previous section.
Because of the high intensity of laser pulses, which is necessary to produce the CH4++ion, we also can suppose that
there are several molecules with an internal energy sufficient for the multistep dissociation of CH3+, to produce the C+ion
TABLE 2: Initiation Energies for the Secondary Dissociation Reactions of the CH4+Ion
Initiation Energy (kcal/mol) fragment CH4+dissociation CH4++dissociation
CH4+(CH4++) 36.6 14.5 CH3+ 197.5 175.4 CH2+ 372.0 349.9 CH+ 559.0 536.9 ECH 2) 104.7 ×
(
5 3)
+ ECH3) 372.0 kcal/mol ECH) 93.5 ×(
2 1)
+ ECH2) 559.0 kcal/molFigure 9. Branching ratios of the CH4+ dissociation fragments at
eventually. Rate constants of the H elimination reaction via TS1 are given in Figure 11.
Because the CH4++ f CH3+ + H+ reaction occurs via
distinct transition state, the initiating energy for this reaction is the barrier’s height at TS1, ECH4) 14.5 kcal/mol. The energy
difference between the barrier’s height and the energy of
products is converted to kinetic energies of the fragments. Using the same argument as for the CH4+ion dissociation, the
available internal energy of the CH3+ fragment is given
as
where E0 is the internal energy of the CH4++ dication. The
initiation energy for the CH3+ion dissociation is given as
For the reaction CH2+ f CH++ H, the initiation energy is given as
For the reaction CH+f C++ H, the initiation energy is given as
Figure 10. CH4++dissociation. Singlet state. (All energies are given in kcal/mol.)
Figure 11. CH4++and CD4++dissociation rate constants versus the
available internal energy.
Figure 12. CH4++dissociation. Triplet state. (All energies are given in kcal/mol.)
E1) (E0- 14.5) ×
(
6 8)
ECH 3) 120.7 ×(
8 6)
+ 14.5 ) 175.4 kcal/mol ECH 2) 104.7 ×(
5 3)
+ ECH3) 349.9 kcal/mol ECH) 93.5 ×(
2 1)
+ ECH2) 536.9 kcal/molBecause the assumption that the energy difference between the barrier’s height and the energy of products is converted to the kinetic energies of the fragments is very crude, and energy difference between the initiation energies of the CH4+ and
CH4++dissociation is small, we do not present kinetic curves
for the CH4++dissociation here.
The CH3++dication is unstable and decomposes to produce
CH2+and H+ions. Similarly, the CH++dication is unstable
and decomposes to the ion pair C+ + H+. We had some difficulties in the calculation of the CH4++ f CH2+ + H2+
reaction pathway, because of the multideterminant character of the wavefunction of the CH4++ ion in the transition-state
Figure 13. CH4mass spectra calculated from the RRKM theory and the generalized Keldysh theory. The peak laser intensities are chosen as (a)
I ) 1.46× 1014W/cm2and (b) I ) 1.1× 1015W/cm2, which correspond to the intensities in Figure 2. In each figure, the upper one is the theoretical result, and the lower one is the experimental result, which is shown in Figure 2. The ion signals are given in arbitrary units, whereas the sum of all mass spectra is normalized to unity.
geometry. To determine the barrier’s height, we assumed that the C-H distances for both eliminating H atoms of the H2+
fragment in the transition-state geometry are the same. There-fore, the barrier’s height at TS3 can be overestimated.
4.3. CH4++ Dissociation: Triplet State. The triplet state
species of the CH4++ion (see Figure 12) easily decomposes to
the CH3++ H+ion pair through a relatively low barrier at TS3.
The subsequent dissociation steps of the CH3+fragment have
been already discussed previously. No pathways leading to the
3CH
2++fragment were found. It could be supposed that if this
fragment can be created, the reaction might occur in an excited state.
5. Discussion and Conclusion
In section 3, we have shown how to calculate the ionization rates and ion yields of CH4as a function of laser intensities. In
section 4, we have applied the RRKM theory to evaluate the rate constants of decomposition for CH4+and CH4++. In this
section, we shall compute the mass spectra and compare them with those experimentally reported in section 2.
From Figure 2, we can see that if the laser intensity is <1015
W/cm2, only CH
4+ion and the ions resulted from the
decom-position are produced; that is, the production of the CH4++ion
and its daughter ions are negligible. On the other hand, when the laser intensity reaches 1015W/cm2, the CH
4++ion and its
daughter ions begin to be observable.
Qualitatively, the observed mass spectra shown in Figure 2 can be understood from Figures 7, 10, and 12. From these
figures, we can see that H+and H2+can only be produced from
CH4++ (in other words, only under the high laser intensity
conditions). Figures 7, 10, and 12 show the mechanisms of the decomposition of CH4+and CH4++(including the singlet and
triplet cases) and the initiation energy required for a particular reaction to occur.
Figure 6 shows the decomposition rate constants of CH4+at
various initiation energies; the process of the decomposition of CH+ is not included, because the RRKM theory cannot be applied in this case. For comparison, the decomposition rate constants for the deuterated species are also shown in Figure 6. For demonstration, some kinetic behaviors of these reactions, under certain particular conditions, are shown in Figure 9, based on the use of the RRKM theory. The initiation energies of CH4+
and CH4++are determined by the laser intensities used in the
production of these ionic species (see section 3) and can be estimated from the generalized Keldysh theory for an applied laser intensity.
From the aforementioned discussion, we can see that, through the use of the theoretical results presented in sections 3 and 4, it is possible to construct mass spectra theoretically and compare the theoretical mass spectra with the experiment mass spectra shown in Figure 2. The results are shown in Figure 13; comparing Figure 13 with Figure 2, we can see that the signals of H2+, C+++(or CH4++++), C++, and CH2++are missing from
the theoretical spectra for I ) 1.9 × 1015 W/cm2 for the
following reasons. The production of C+++ and CH4++++
requires higher ionization energies, such as those observed for
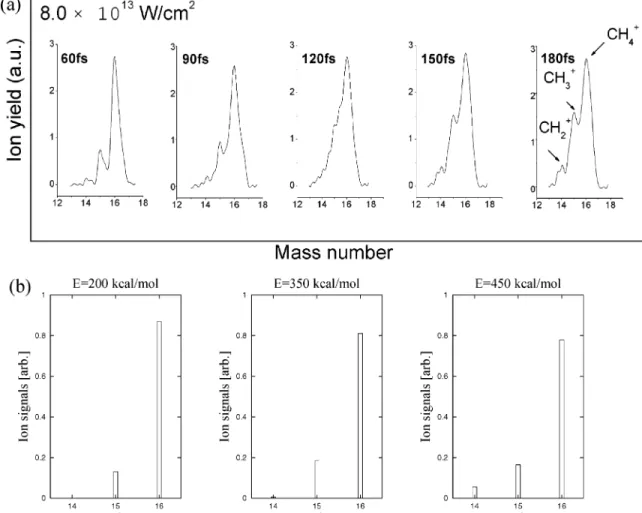
Figure 14. Plotted graphs showing (a) the TOF mass spectra of methane irradiated by an ultrafast laser with different pulse widths of 60, 90, 120,
150, and 180 fs at the same laser peak intensity of 8.0× 1013W/cm2(a CH
4gas pressure of 3.3× 10-3Pa was maintained in the chamber, with
a background pressure of 3.0× 10-4Pa), and (b) the mass spectra of CH4for various initiation energies.
CH4++ f CH4+++and CH4+++f CH4++++, which are not
available to us; the production of H2+and CH2++are predicted
from the theory (see Figure 10), but, because of the fact that their transition state cannot be accurately determined from our level of ab initio calculation, their production is not shown in Figure 13. The products of C++and C+cannot be calculated from the RRKM theory, because of the smallness of their corresponding parent ions. Figure 2 shows that, when the laser intensity increases, the dissociation proceeds more, then the ion signal of C+will be stronger, because C+cannot dissociate any longer.
Recently, Wang et al.19reported the pulse width effect on
the dissociation of CH4+in the intense femtosecond laser field
(see Figure 13). These mass spectra are reproduced in Figure 14; the laser pulse widths vary from 60 fs to 180 fs at the laser peak intensity of 8.0× 1013 W/cm2. We have computed the
mass spectra using the same information discussed previously and shown them in Figure 14 for comparison. As can be seen from Figure 14, the energy dispersion must be included in the calculation to be able to reproduce the experimental spectra completely.
A similar study of high-power laser ionization and dissocia-tion of CH4 has also been reported by Wu et al.20The same
theoretical analysis of mass spectra can be performed but will not be performed in this paper.
In conclusion, in this paper, we have reported the mass spectra of the high-power laser ionization and dissociation of CH4with
intensities varying from 1× 1014W/cm2to 2× 1015W/cm2
and a laser pulse width of 48 fs. In our opinion, the field-assisted dissociation of CH4ions can occur only if the dissociation of
ions happens during the exposure of ions to the laser, because, in this case, the potential surfaces of ions can be greatly modified by the intense laser field for the dissociation to occur. If the dissociation of ions happens outside the influence of the applied laser field, then statistical theory such as the RRKM theory can be applied to calculate the observed mass spectra. This approach has been applied to analyze the experimental mass spectra. The agreement between the experimental spectra and the calculated results seems to be reasonable.
Acknowledgment. This work was supported by Academia
Sinica and NSC (Taiwan).
References and Notes
(1) von Koch, H. Ark. Fys. 1965, 28, 529.
(2) Powis, I. J. Chem. Soc. Faraday Trans. 1979, 75 (2), 1294. (3) Field, T. A.; Eland, J. H. D. J. Electron Spectrosc. Relat. Phenom. 1995, 73, 209.
(4) Stockbauer, R. J. Chem. Phys. 1973, 58, 3800.
(5) Dutuit, O.; Ait-Kaci, M.; Lemaire, J.; Richard-Viard, M. Phys. Scr.,
T 1994, 31, 223.
(6) Furuya, K.; Kimura, K.; Sakai, Y.; Takayanagi, T.; Yonekura, N.
J. Chem. Phys. 1994, 101, 2720.
(7) Chin, S. L. In AdVances in Multiphoton Processes and Spectroscopy, Vol. 16; Lin, S. H., Villaeys, A. A.; Fujimura, Y., Eds.; World Scientific: Singapore, 2004; pp 249-272.
(8) Elshakre, M. E.; Gao, I. R.; Tang, X. P. J. Chem. Phys. 2003, 119, 5397.
(9) Wang, S. F.; Tang, X. P.; Gao, I. R. J. Phys. Chem. A 2003, 107, 6123.
(10) Tang, X. P.; Wang, S. F.; Elshakre, M. E. J. Phys. Chem. A 2003,
107, 13.
(11) Tang, X. P.; Becker, A.; Liu, W.; Sharifi, S. M.; Kosareva, O. G.; Kandidov, V. P.; Agostini, P.; Chin, S. L. Appl. Phys. B 2005, 80, 547-557.
(12) Tang, X. P.; Becker, A.; Liu, W.; Sharifi, M.; Kosareva, O.; Kandidov, V. P.; Agostini, P.; Chin, S. L. Phys. ReV. A 2005, 71, 045401. (13) Assion, A.; Baumert, T.; Bergt, M.; Brixner, T.; Kiefer, B.; Seyfried, V.; Strehle, M.; Gerber, G. Science 1998, 282, 919.
(14) Levis, R. J.; Menkir, G. M.; Rabitz, H. Science 2001, 292, 709. (15) Smith, D. J.; Ledingham, K. W. D.; Kille, H. S. J. Phys. Chem. A 1998, 102, 2519.
(16) Posthumus, J. H.; Frasinski, L. J.; Giles, A. J. J. Phys. B 1995, 28, L349.
(17) Nagaya, K.; Mishima, K.; Lu, H.-F.; Hayashi, M.; Lin, S. H. Chem.
Phys. Lett. 2006, 424, 34.
(18) Nagaya, K.; Mineo, H.; Mishima, K.; Villaeys, A. A.; Hayashi, M.; Lin, S. H. Phys. ReV. A 2007, 75, 013402-1.
(19) Wang, C.; Song, D.; Liu, Y.; Kong, F. Chin. Sci. Bull. 2006, 51, 1269.
(20) Wu, Z.; Wu, C.; Liang, Q.; Wang, S.; Liu, M.; Deng, Y.; Gong, Q. J. Chem. Phys. 2007, 126, 074311.
(21) (a) Wang, S.; Tang, X.; Gao, L.; Elshakre, M. E.; Kong, F. J. Phys.
Chem. A 2003, 107, 6123. (b) Mebel, A. M.; Zyubina, T. S.; Dyakov, Y.
A.; Bandrauk, A. D.; Lin, S. H. Int. J. Quantum Chem. 2005, 105, 506. (22) Mishima, K.; Hayashi, M.; Yi, J.; Chin, S.; Selzle, H. L.; Schlag, E. W. Phys. ReV. A 2002, 66, 033401.
(23) (a) Keldysh, L. V. SoV. Phys.sJETP 1965, 20, 1307. (b) Gribakin, G. F.; Kuchiev, M. Yu. Phys. ReV. A 1997, 55, 3760.
(24) Mineo, H.; Chao, S. D.; Nagaya, K.; Mishima, K.; Hayashi, M.; Lin, S. H. Phys. ReV. A 2007, 75, 027402.
(25) Mineo, H.; Chao, S. D.; Nagaya, K.; Mishima, K.; Hayashi, M.; Lin, S. H. Chem. Phys. Lett. 2007, 439, 224.
(26) Nakashima, N.; Yatsuhashi, T.; Murakami, M.; Mizoguchi, R.; Shimada, Y. In AdVances in Multi-photon Processes and Spectroscopy, Vol. 17; Lin, S. H., Villaeys, A. A., Fujimura, Y., Eds.; World Scientific: Singapore, 2006.
(27) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, Jr., J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R. Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C. and Pople, J. A. Gaussian 03, Revision B.03; Gaussian, Inc.: Pittsburgh, PA, 2003.
(28) Eyring, H.; Lin, S. H.; Lin, S. M. Basic Chemical Kinetics; Wiley: New York, 1980.
(29) Robinson, P. J.; Holbrook, K. A. Unimolecular Reactions; Wiley: New York, 1972.
(30) Steinfeld, J. I.; Francisco, J. S.; Hase, W. L. Chemical Kinetics
and Dynamics; Prentice Hall: Englewood Cliffs, NJ, 1999.
(31) MOLPRO is a package of ab initio programs written by H.-J. Werner and P. J. Knowles, with contributions from J. Almlo¨f, R. D. Amos, M. J. O. Deegan, S. T. Elbert, C. Hampel, W. Meyer, K. Peterson, R. Pitzer, A. J. Stone, P. R. Taylor, and R. Lindh.
(32) Kong, F.; Luo, Q.; Xu, H.; Sharifi, M.; Song, D.; Chin, S. L. J.
Chem. Phys. 2006. 125, 133320.
(33) Gravel, J. F.; Luo, Q.; Boudreau, D.; Tang, X. P.; Chin, S. L. Anal.