行政院國家科學委員會專題研究計畫成果報告
帶有顯著日間差異的遺傳性進行性肌肉張力不全症之研究
Molecular Study of Her editar y Pr ogr essive Dystonia with Mar k
Diur nal Fluctuation in Chinese
計畫編號:NSC 87-2314-B-002-140
執行期限:86 年 8 月 1 日至 87 年 7 月 31 日
主持人:王本榮 國立台灣大學醫學院小兒科
中文摘要 帶有顯著日間差異的遺傳性進行性肌肉張 力不全症(以下簡稱 HPD)是一個顯性遺傳 的臨床疾病。它具有明顯日間症狀變動, 兒童期發病,並且對L-dopa具有完全療效 之特徵。我們自從1988年陸續有6名HPD的 病例被診斷治療。最近,Ichinose等首先 發現了日本HPD病人肇因於GTP cyclohydrolase I(GTPCH)基因的突變,我 們在本研究中分析了患者的基因變化。 GTPCH 缺 乏 會 引 起 tetrahydro-biopterine (BH4)不足,所以同時也是引 起苯酮尿症的原因之一。然而 GTPCH 缺乏 所引起的苯酮尿症卻是隱性遺傳的。 在本研究 中我們 發現了一 個病人 有 R249S (747CàG)突變。病人血球經 PHA 刺 激後可以測到很低的 GTPCH 活性,但是 mRNA 的表現卻是正常的。突變株如果在大 腸菌中表現其活性正常,但是在細胞中表 現的活性及蛋白質卻是偏低的。所以 R249S 致病的原因是因為其穩定度不足。本研究 指出突變的機轉才是決定疾病表現(HPD 或 苯酮尿症)的主要因子。疾病表現和遺傳型 式並沒有絕對的關係。 關鍵詞:帶有顯著日間差異的遺傳性進行 性肌肉張力不全症,Segawa, GTP cyclohydrolase I Abstr act GTP cyclohydrolase I (GTPCH), which catalyzes the rate limiting step of tetrahydrobiopterin (BH4) biosynthesis, isassociated with two clinically distinctive
human diseases. Hyperphenylalaninemia is a severe disease induced by recessive GTPCH mutations, whereas dopa-responsive dystonia (DRD) is a benign disease in which only dominant mutations have been found. We found a homologous (recessive) GTPCH mutation (R249S, 747CàG) in a DRD patient who had low but measurable GTPCH activity in her PHA-stimulated mononuclear blood cells. Arginine 249 is located at the C-terminus of GTPCH, outside the enzyme catalytic site. The expression of GTPCH mRNA in the patient's mononuclear blood cells was normal. Enzyme specific activity and kinetics of R249S mutant protein expressed in E. coli and eukaryotic cells were
also normal. However, the amount of R249S mutant protein expressed in transfected cells was lower than the wild-type protein. In pulse-chase experiment, the synthesis of mutant protein was normal, but its stability was decreased. This is the first destabilizing mutation found in the GTPCH gene. Our data raise the possibility that DRD could be either dominantly or recessively inherited. The phenotype of GTPCH deficiency seems to be determined by residual activity, while the inheritance is more likely determined by the mechanism of mutation.
Keywor ds: Hereditary progressive dystonia
with marked diurnal fluctuation, HDP, Segawa, GTP cyclohydrolase I
Intr oduction
Fib GTP cyclohydrolase I (EC 3.5.4.16) (GTPCH) is responsible for the rate-limiting step of tetrahydrobiopterin (BH4)
important human enzymes including phenylalanine hydroxylase, tyrosine hydroxylase, tryptophan hydroxylase and nitric oxide synthase. The latter three are involved in the production of bioactive humoral factor dopamine, serotonin and nitric oxide.
GTPCH deficiency is first described in a girl with hyperphenylalaninemia in 1984 (Niederwieser et al. 1984). Neopterin, biopterin, serotonin and dopamine are all very low in her body fluids, and GTPCH activity is not detectable in her liver.
Hyperphenylalaninemia induced by GTPCH deficiency is a rare disease, and only 12 cases have been reported from a recent
international database (Blau et al. 1996). These patients have progressive mental retardation and seizure. Variable tone and posture ranging from marked hypotonia to opisthotonos are characteristic. Treatment with L-dopa relieves their abnormal tone and seizure effectively, while diet control itself does not.
Dopa-responsive dystonia (DRD), also called hereditary progressive dystonia with marked diurnal fluctuation (HPD) or Segawa disease, is a disease that also shows dramatic response to L-dopa (Nygaard et al. 1988; Segawa et al. 1971). Compared to
hyperphenylalaninemia, dystonia is the major symptom in DRD, and patients become symptom-free when they are treated with low dose L-dopa. HPD/DRD was recently
mapped to chromosome 14q22.1-q22.2 where human GTPCH was located (Nygaard et al. 1993; Tanaka et al. 1995). GTPCH was then proved to be the gene responsible for DRD (Ichinose et al. 1994).
Hyperphenylalaninemia and DRD differ not only in clinically manifestations but also in inheritance. Hyperphenylalaninemia is a recessive disease, and affected siblings have been found in families. After the cloning of GTPCH gene, two homologous mutations have been found in hyperphenylalaninemia patients (Blau et al. 1995; Ichinose et al. 1995). On the contrary, DRD was believed to be an autosomal dominant disease with low
penetrance and female predominance (Nygaard 1995). All GTPCH mutations previously found in DRD patients showed dominant transmission (Ichinose et al. 1995; Furukawa et al. 1996; Bandmann et al. 1996).
Hyperphenylalaninemia patients (with two mutated GTPCH alleles) have very low GTPCH activities (Niederwieser et al. 1984; Blau et al. 1996). Theoretically, DRD patients (with one mutated and one normal GTPCH alleles) should have 50% GTPCH activity. However, DRD patients usually have below 20% GTPCH activity (Segawa 1996; Ichinose and Nagatsu 1997). DRD mutation may exert dominant-negative effects since GTPCH is a homodecamer (Nar et al. 1995), but some DRD mutations
produce truncated proteins that may be unable to join the complex (Hirano 1995). The mechanism of DRD mutation is still not solved.
In this paper, we described a DRD mutation with unusual inheritance and mechanism. The characterization of this mutation by eukaryotic expression system has increased our understanding in the molecular basis of GTPCH
deficiencies.roblast growth factors (FGFs) approach to the treatment of genetic disease involving FGFRs.
Mater ials and Methods:
Description of the patient
The 12-year-old girl with hereditary progressive dystonia with marked diurnal fluctuation (HPD, Segawa syndrome) has been described previously (Hwu et al. 1989; Wang et al. 1994). It was later known that HPD is equal to Dopa-responsive dystonia (DRD). Her symptoms developed when she was 2 years and 8 months old. She had posture dystonia, posture tremor, posture rigidity and neck tilting that were more severe toward the end of a day. She was free of symptoms under the treatment of L-dopa 20 mg/kg/day since the age of 3 years. She
has no family history and both her parents are normal. The parents are not consanguineous. Informed consents were obtained before blood sampling.
Biochemical analyses
Plasma amino acids analyses were performed on a Biotronic LC 3000 amino acids analyzer (Biotronic, Germany). Urinary pterin derivatives were analyzed by HPLC after oxidation (Antonozzi et al. 1988). GTPCH activities of patients were measured in PHA-stimulated mononuclear blood cells, and analyzed by reversed phase HPLC after oxidation and phosphatase treatment (Blau et al. 1985; Werner et al. 1996).
Formate release assay was performed according to the method of Burg and Brown with modifications (Burg and Brown 1968).
3
H-GTP (5-20 Ci/mmol) was obtained from Amersham (Amersham, USA), and cold GTP was from Borghinger (Borghinger Manham, USA). A 50 µl reaction mixture containing 45 µl cell extract, 0.1 M Tris-HCl pH 8.0, 2.5 mM EDTA, 1 µCi 3H-GTP and 200 µM GTP was incubated at 37oC for 30 min with prokaryotic protein or for 1 hr with eukaryotic protein. Eukaryotic protein is stable for at least 3 hours in this assay.
Reaction was stopped by 25 µl 4 M HCOOH. After adjusting the volume to 500 µl with water, 0.1 ml (0.2 g/ml) activated charcoal was added to the mixture. After extraction, 0.3 ml of supernatant was subjected to scintillation counting. Protein concentration was determined by Bradford dye reagent kit (Bio-Rad, USA). GTPCH activity was expressed as pmol/mg/hr or pmol/µg/hr. Our method was different from what described by Burg and Brown that cold GTP was added to the reaction mixture. The total concentration of GTP was thus brought closer to the Km of the enzyme to increase the sensitivity of the assay (manuscript in preparation).
PCR and sequencing
Amplification of all six exons of the
GTPCH gene was performed as described by Ichinose et al. (Ichinose et al. 1995). PCR products were purified by a QIAEX II gel extraction kit (QIAGEN, Germany). Sequencing was performed by a CirmVent Thermal Cycle Dideoxy DNA sequencing kit (BioLabs, USA). PCR products of exon 6 were cloned to confirm the sequence change. Fnu4HI (BioLab, USA) digestion was performed following the recommendation of the manufacturer. DNA samples from normal, unrelated individuals were used as controls. GTPCH mRNA expression
Total cellular RNA was extracted from PHA-stimulated whole blood. First strand synthesis was performed by AMV reverse transcriptase and oligo dT (Promega, USA). PCR was performed with primer F2 (5’-GCCCCGCAGCGAGGAGGATAAC) and R3 (5’-GACAGACAATGCTACTGGCAGT) as described by Hirano et al. except that 0.5
µCi of α-32P-dCTP was added to the PCR mixture, and the cycle number was decreased to 20 (Hirano et al. 1995). PCR products were digested with Fnu4HI and were separated by a 5% sequencing gel. After electrophoresis, the gel was dried and exposed to a phosphoscreen. Images were analyzed by a Phosphoimager and quantified by the ImageQuant program (Molecular Dynamics, USA). As an internal control, the GAPDH gene product was amplified with primer GAPDH-upper
(5'-CCATGGAGAAGGCTGGGG) and GAPDH-lower
(5'-CAAAGTTGTCATGGATGACC). Mutagenesis and construction of plasmids
GTPCH cDNA was kindly provided by Dr. Nagatsu. This cDNA produces functional protein although it lacks a short 5’ piece (Ichinos et al. 1995). Mutagenesis was performed by a PCR method with the wild-type cDNA as a template (Vallejo et al. 1994). Two separated PCR reactions were
(5’-CTCTCATTAGCAGCTGAGCTT, the mutated nucleotide is underlined) and GTP-m747F
(5’-AAGCTCAGCTGCTAATGAGAG) / R3. PCR products were then mixed and amplified with F3/R3. The final product was digested with BsrGI, and was used to replace the corresponding fragment in wild-type cDNA. The replaced region was confirmed by sequencing.
Both the wild-type and mutant cDNAs were cloned into pGEX vector (Pharmacia, Sweden) to produce glutathione fusion proteins in E.coli JM109. Similar amounts of
the wild-type and mutant fusion proteins, determined by both Commasie blue staining and western blot analysis, were subjected to GTP cyclohydrolase I activity measurement.
Eukaryotic expression plasmids CMV-A16-GTPCH, driven by CMV promoter, were constructed by inserting GTPCH cDNAs (NarI/EcoRI fragment) into pCMV-AGP/EBP (replacing its BstBI/ EcoRI fragment) (Lee et al. 1996). This generated an N-terminal AGP/EBP epitope (called A16) to the expressed GTPCH protein. To
construct a full length GTPCH cDNA, the 5’ end of the cDNA (starting from ATG) was added by PCR. Expression plasmid CMV-GTPCH contains this full-length cDNA. Production of GTPCH antiserum and western blot analysis
GTPCH cDNA was cloned into pRSET vector, expressed in E.coli DE3, and purified
by a nickel column. The purified protein was used to immunize rabbits to generate
antiserum. This antibody was used in both western blot analysis and
immunoprecipitation. Western blot was performed using GTPCH antibody at 1: 1600 dilution or monoclonal antibody A16 (Lee et al. 1996) at 1: 2500 dilution. Secondary antibodies used were HRP-coupled sheep anti-mouse Ig or sheep anti-rabbit Ig
(Amersham, USA) at 1: 1600 dilution. Blots were developed using the ECL detection system (Amersham, USA).
Cells and Transfections
Baby hamster kidney cells (BHK) were cultured in Dulbecco’s MEM with 10% FCS. Calcium phosphate precipitation method was used for transfection (Graham and van der Erb, 1973). BHK or HeLa cells in 10 cm or 6 cm dishes were transfected with 10 or 5 µg of the GTPCH plasmid and 1 µg of pRSV-gal. Forty hours after transfection, cells were harvested for GTPCH assay, western blot analysis and β-galactosidase assay. β
-galactosidase assay was performed according to the instructions provided by Pharmacia (Pharmacia, Sweden). It was used as a transfection efficiency control.
Pulse-chase experiment
4µg GTPCH-wt or CMV-GTPCH-R249S was transfected into BHK cells grown in 6 cm culture dishes. Forty hours after transfection, cells were washed twice with PBS, incubated in DMEM-10% FCS without methionine and cysteine for 1 hr, and pulsed with Pro-mix (Amersham, USA) for 30 min. After chasing for 1, 2, 4 or 8 hours, cells were harvested and lysed by RIPA buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 2.5 mM EDTA, 1 mM PMSF, and 1 µg/ml leupeptin), and were subjected to
immunoprecipitation with GTPCH antibody and protein A-sepharose. After washing for 4 to 5 times, samples were separated by SDS-PAGE. Exposure and quantification were done by Image Analyzer (BAS 1500, FUJI).
Results
The patient had GTPCH deficiency GTPCH activities were measured in PHA-stimulated mononuclear blood cells of
a DRD patient and her parents by HPLC. The patient’s cells had low, but measurable GTPCH activity (4.2 pmol/mg/hr, normal 38.4-102.6 pmol/mg/hr) which is compatible with DRD (with residual activity up to 20% of normal) (Segawa 1996, Ichinose and Nagatsu, 1997). GTPCH activity was 7.8 pmol/mg/hr for her mother and 32.4 pmol/mg/hr for her father. Although the reason for her mother’s low GTPCH activity is not clear, DRD does affect females more than males. Both the patient and her parents had normal plasma phenylalanine levels (0.018 µmol/ml for patient, 0.020 µmol/ml for her mother, and 0.021 µmol/ml for father, normal 0.01-0.03 µmol/ml). The patient’s urinary neopterin level was 0.143 mmol/mol CRE (average of two analyses) (normal 0.1-0.5 mmol/mol), and her urinary neoptein to biopterin ratio was 69.4% (normal 49-85%). Therefore, she had partial GTPCH deficiency with borderline low urinary neopterin level. Homologous R249S mutation
Genomic DNA was extracted from peripheral blood of the family. Direct sequencing after amplification of all six exons of the GTPCH gene of the patient showed a 747CàG change in exon 6 which changes amino acid residue 249 from arginine to serine (R249S). This is a
homologous mutation that happens on both her GTPCH alleles (Figure 2A). Both of her parents were heterozygous for this mutation (Figure 2A), so this is a recessive mutation. After cloning of the PCR products, ten independent clones were analyzed for each member of the family. Only mutant clones could be found in the patient, but both mutant and wild-type clones were found in parents (Figure 2A). This mutation was searched in normal population by Fnu4HI digestion which specifically cleaves the mutant allele. In 100 unrelated normal controls (200 alleles), no R249S mutation was found (data not shown). No other mutation could be found in the coding region and exon-intron borders of the GTPCH genes in this family.
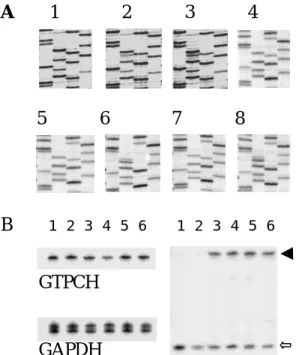
A 1 2 3 4
5 6 7 8
B 1 2 3 4 5 6 1 2 3 4 5 6
Figure 2. Detection of GTPCH R249S mutation. (A) GTPCH exon 6 PCR products were sequenced either directly (panels 1-3) or after cloning (panels 4-8). The pictures are direct sequencing for the patient, her father and mother (panel 1-3), sequencing of a representative clone for the patient (panel 4), mutant and wild-type clones for her mother (panels 5-6), and father (panels 7-8). (B) Quantification of mononuclear cell GTPCH mRNA for the family by RT-PCR. Upper panel shows the PCR products of GTPCH, and lower panel the products of GAPDH. All tests were duplicated. Lanes 1-2, 3-4 and 5-6 represent the patient, her father and mother respectively. (C) Fnu4HI cleavage of RT-PCR products. Lanes 1-2, 3-4 and 5-6 are the results for the patient, her father and mother respectively. The uncut (wild type) products were marked by an arrow, and the cut (mutant) products by an open arrow. R249S mRNA was expressed normally in peripheral blood
RT-PCR was performed to measure the expression of GTPCH mRNA in PHA-stimulated blood of the family. For better quantification, radioisotope labeling with fewer amplification cycles prevented the
GTPCH
6
saturation of PCR reactions. The results showed that the expressions of GTPCH mRNA were equal in this family (Figure 2B). GAPDH mRNA expression was used as an internal control. RT-PCR products were futher digested by Fnu4HI which specifically cleaves the mutant allele. As shown in Figure 2C, the amounts of uncut (wild type) and cut (mutant) DNA products were equal for her heterozygous parents. Therefore, GTPCH mRNA expression is not affected by the R249S mutation.
R249S mutant protein expressed in E.coli has
normal activity
To further characterize this mutation, expression of R249S and wild-type
glutathione fusion proteins in E. coli was
established. The expression levels of recombinant proteins were determined by commasie blue staining and western blot analysis. Enzyme activities in crude bacterial lysates analyzed by HPLC and formate release assay showed that recombinant R249S protein had normal GTPCH specific activity. In kinetic study it also showed normal Vmax and apparent Km. R249S mutant protein expressed in transfected cells showed normal specific activity but decreased expression levels
Transient expression in cultured cells was also used to characterize the mutant protein because the processing and
degradation of over-expressed recombinant proteins in E. coli may be abnormal (Laiho et
al. 1997). To facilitate the detection of the mutant protein, we added an N-terminal tag (A16 epitope of AGP/EBP) to the cDNAs. This tag does not affect GTPCH function as cells transfected with the wild-type construct showed good GTPCH activities. In the
experiments, either CMV-A16-GTPCH-wt or CMV-A16-GTPCH-249, together with
pRSV-gal, was transfected into HeLa (Fig 4A, upper panel) and BHK cells (Fig. 4A, lower
panel). Transfection efficiencies monitored by β-galactosidase assay were similar in these experiments. Western blot analysis with A16 monoclonal antibody showed that
R249S protein was expressed at a lower level (23% in HeLa, 28% in BHK cells) as
compared to the wild-type protein (Figure 4A). GTPCH activity in cells transfected with R249S cDNA was also low. After normalization for protein expression, specific activity of R249S mutant protein was normal. Data from a typical kinetic study was plotted on Fig. 4B.
R249S showed decreased stability
To explore the mechanism of decreased mutant protein expression, a pulse-chase experiment was designed. Full-length GTPCH cDNAs (starting from ATG) were used in this study. As shown in Figure 5A, after a 30-min pulse, wild-type and mutant GTPCH proteins were equally labeled.
Control transfection without GTPCH plasmid produced no GTPCH protein (picture not shown). Therefore, the synthetic rate of R249S mutant protein was normal. These newly synthesized GTPCH proteins were chased for up to 8 hr. The amount R249S protein decreased more rapidly than the wild-type protein during the chase period. The quantitative result was shown in Fig. 5B. Half-lifes calculated from Fig. 5B were 8.94 hr for the wild-type and 2.39 hr for the mutant protein. A cross-reacting protein (open arrow, Fig. 5A) with a size slight larger than GTPCH was pretty stable during the experiment. These results indicate that R249S mutation decreases the stability of GTPCH protein.
A
B
1 2 3 4 5 6 WT R249S HeLa BHK 800 1000 1200 Ac tiv ity (pm ol/ mg r)Figure 4. Transient expression of wild-type and R249S GTPCH proteins. (A) Western blot analysis of BHK and HeLa cells transfected with either wild-type or R249S GTPCH cDNAs. 6, 12 or 25 µg of total cell extracts was loaded onto adjacent lanes of a polyacrylamide gel. Western blot analysis was performed using a mouse monoclonal antibody against A16 (AGP/EBP) epitope. Film exposure time was 1 min for BHK protein and 5 min for HeLa protein. (B) A kinetic study of GTPCH activities in transfected BHK cells. Equal amounts of crude lysates were assayed under different substrate concentrations.
Discussion
In this paper, we describe an HPD patient who has a homologous GTPCH mutation. Both her parents are carriers of this mutation, so DRD is recessively inherited in this family. This inheritance pattern is unusual because it was known that dominant GTPCH mutations induced DRD, while recessive mutations induced
hyperphenylalaninemia (Nygaard 1995). GTPCH monomer is composed of a helical N-terminal part, a compact C-terminal domain and a C terminus (Nar et al. 1995). The C-terminal domain contains the enzyme catalytic site. Although R249S is an
unconserved change, it is located at the C-terminus of GTPCH. This could explain why R249S mutant protein possesses normal specific enzyme activity. However, R249S decreases the stability of GTPCH. This is the first destabilizing mutation described in the GTPCH gene. Previously, only
loss-of-function mutations or possibly dominant-negative mutations have been found. Supposedly, complete GTPCH deficiency produces hyperphenylalaninemia, while partial deficiency produces DRD. Because R249S mutation results in higher residual enzyme activity than other known mutations, homologous R249S mutation retains enough activity to prevent the development of hyperphenylalaninemia. The inheritance of GTPCH deficiency is more likely determined by the mechanism of mutation.
Recently, mutations affecting protein processing or stability are more recognized as etiologies of genetic diseases. Mutant
proteins could be degraded by different mechanisms before they are processed to a mature form. For example, the cystic fibrosis transmembrane conductance regulator (CFTR) △F508 mutant protein could act as normal chloride channels, however, it fails to exit the ER and is degraded by the
ubiquitin/proteosome pathway (Cheng et al. 1990; Ashkenas and Byers, 1997). We do not know how R249S mutant protein is degraded. The interaction between the wild-type and mutant proteins in GTPCH homodecamer is another issue that may be concerned for other GTPCH mutations.
Currently, more than 14 GTPCH mutations have been found in DRD, and three mutations have been found in hyperphenylalaninemia (Thony and Blau, 1997). In the studies of Ichinose et al,
recombinant expression of GTPCH in E. coli
could not tell the difference between DRD and hyperphenylalaninemia because no mutation gave any activity (Ichinose et al. 1994, 1995). We suggest that these mutations should be tested in eukaryotic expression system. Just before we submit this manuscript, there is a report describing dystonia with motor delay in compound heterozygotes for GTPCH gene mutations (Furukawa et al. 1998). Therefore, GTPCH activities should be measured in dopa-responsive dystonia with recessive inheritance (Segawa and Nomura, 1993; Gorke and Bartholome, 1990) unless the
deficiency of tyrosine hydroxylase could be proved (Knappskog et al. 1995).
計畫成果自評 本這個計畫開創了一個新的研研究領 域,也有了新的發現,目前我們正在擴展 這方面的研究。經由這個計畫的經驗,我 們對於蛋白質分析的知識和能力都增強 了。由於下一個世紀將是蛋白質的世紀, 這個計畫的執行也將有助於我們日後的研 究工作。 Refer ences
Antonozzi I, Carducci C, Vestri L,
Pontecorvi A, Moretti F (1988) Rapid and sensitive method for high-performance liquid chromatographic analysis of pterins in biological fluids. J Chromatography 459:319-324
Blau N, Joller P, Atarés M, Cardesa-Garcia J, Niederwieser A (1985) Increase of GTP cyclohydrolase I activity in mononuclear blood cells by stimulation: detection of heterozygotes of GTP cyclohydrolase I deficiency. Clinica Chimica Acta 148:47-52
Blau N, Ichinose H, Nagatsu T, Heizmann CW, Zacchello F, Burlina AB (1995) A missense mutation in a patient with guanosine triphosphate cyclohydrolase I deficiency missed in the newborn
screening program. J Pediatr 126:401-405 Blau N, Barnes I, Dhondt JL (1996)
International database of
tetrahydrobiopterin deficiencies. J Inher Metab Dis 19:8-14
Burg AW, Brown GM (1968) The
biosynthesis of folic acid. J Biol Chem 243:2349-2358
Cheng S, Gregory R, Marshall J, Paul S, Souza D, White G, O'Riordan C, et al (1990) Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63:827-834
Furukawa Y, Kish SJ, Bebin EM, Jacobson
RD, Fryburg JS, Wilson WG, Shimadzu M, et al (1998) Dystonia with motor delay in compound heterozygotes for GTP-cyclohydrolase I gene mutations. Ann Neurol 44:10-16
Gorke W, Bartholome K (1990) Biochemical and neurophysiological investigation in two forms of Segawa's syndrome. Neuropediatrics 21:3-8
Graham F, van der Erb A (1973) A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 52:456-457
Hirano M, Tamaru Y, Nagai Y, Ito H, Imai T, Ueno S (1995) Exon skipping caused by a base substitution at a splice site in the GTP cyclohydrolase I gene in a Japanese family with hereditary progressive dystonia / dopa responsive dystonia. Biochem Biophys Res Commun 213:645-651
Hwu WL, Wang PJ, Shen YZ (1989) Hereditary progressive dystonia with marked diurnal fluctuation: report of a case. Acta Paediatr Sin 30:46-51 Ichinose H, Ohye T, Takahashi E, Seki N,
Hori A, Segawa M, Nomura Y, et al (1994) Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat Genet 8:236-242
Ichinose H, Ohye T, Matsuda Y, Hori T, Blau N, Burlina A, Rouse B, Matalon R, Fujita K, Nagatsu T (1995) Characterization of mouse and human GTP cyclohydrolase I genes. Mutations in patients with GTP cyclohydrolase I deficiency. J Biol Chem 270:10062-10071
Ichinose H, Nagatsu T (1997) Molecular genetics of hereditary dystonia – mutations in the GTP cyclohydrolase I gene. Brain Res Bulletin 43:35-38 Knappskog PM, Flatmark T, Mallet J,
Lüdecke B, Bartholomé K (1995)
Recessively inherited L-dopa-responsive dystonia caused by a point mutation (Q381K) in the tyrosine hydroxylase gene. Hum Mol Genet 4:1209-1212
Teller DC, Niemi K-M, Saarialho-Kere U, et al (1997) Transglutaminase 1 mutations in autosomal recessive congenital
ichthyosis: private and recurrent mutations in an isolated population. Am J Hum Genet 61:529-538
Lee YM, Miau LW, Chang CJ, and Lee SC (1996) Transcriptional induction of alpha-1 acid glycoprotein gene by synergistic interaction of two alternative activator forms of AGP/EBP (C/EBPβ) and NF-kB or Nopp140. Mol Cell Biol 16:4257-4263 Nar H, Huber R, Meining W, Schmid C,
Weinkauf S, Bacher A (1995) Atomic structure of GTP cyclohydrolase I. Structure 3:459-466
Niederwieser A, Blau N, Wang M, Joller P, Atarés M, Cardesa-Garcia J (1984) GTP cyclohydrolase I deficiency, a new enzyme defect causing hyperphenylalaninemia with neopterin, biopterin, dopamine, and serotonin deficiencies and muscular hypotonia. Eur J Pediatr 141:208-214 Nygaard TG, Marsden CD, Duvoisin RC
(1988) Dopa-responsive dystonia, In Fahn S, Marsden CD, Calne DB (Ed) Advances in Neurology, Vol 50, Raven Press, New York, 1988, pp.377-384
Nygaard TG, Wilhelmsen KC, Risch NJ, Brown DL, Trugman JM, Gilliam TC, Fahn S, et al (1993) Linkage mapping of dopa-responsive dystonia (DRD) to chromosome 14q. Nat Genet 5:386-391 Nygaard TG (1995) Dopa-responsive
dystonia. Curr Opin Neurol 8:310-313 Segawa M, Ohmi K, Itoh S, Aoyama M,
Hayakawa H (1971) Childhood basal ganglia disease with remarkable response to L-Dopa, hereditary basal ganglia disease with marked diurnal fluctuation. Shinryo (Tokyo) 24:667-672
Segawa M, Nomura Y (1993) Hereditary progressive dystonia with marked diurnal fluctuation, pathophysiological
importance of the age of onset. Adv Neurol 60:568-576
Segawa M (1996) Segawa disease (hereditary progressive dystonia with marked diurnal fluctuation-HPD) and abnormalities in
pteridin metabolism. Rhinsho Shinkeigaku 36:1322-1323
Tanaka H, Endo K, Tsuji S, Nygaard TG, Weeks DE, Nomura Y, Segawa M (1995) The gene for hereditary progressive dystonia with marked diurnal fluctuation maps to chromosome 14q. Ann Neurol 37:405-408
Thony B, Blau N (1997) Mutations in the GTP cyclohydrolase I and 6-pyruvoyl-tetrahydropterin synthase genes. Human mutation 10:11-20
Vallejo, A. N., Pogulis, R. J. and Pease, L. R. (1994) PCR Methods and Applications 4, S123-S130
Wang PJ, Ko YM, Young C, Hwu WL, Shen YZ (1994) Hereditary progressive
dystonia with marked diurnal fluctuation (Segawa syndrome) in Taiwan. Brain Develop 16:126-131
Werner ER, Werner-Felmayer G, Wachter H (1996) High-performance liquid
chromatographic methods for the quantification of tetrahydrobiopterin biosynthetic enzymes. J Chromatography B, 684:51-58