國 立 交 通 大 學
生物資訊及系統生物研究所
碩士論文
改進 RNA 三級結構的多重比對
Improving Multiple Alignment of
RNA Tertiary Structures
研 究 生:程美齡
指導教授:林苕吟 博士
盧錦隆 博士
改進 RNA 三級結構的多重比對
Improving Multiple Alignment of
RNA Tertiary Structures
研 究 生:程美齡
Student:Mei-Ling Cheng
指導教授:林苕吟 博士 Advisor:Dr. Tiao-Yin Lin
盧錦隆 博士 Dr. Chin Lung Lu
國 立 交 通 大 學
生 物 資 訊 及 系 統 生 物 研 究 所
碩 士 論 文
A Thesis Submitted to Institute of Bioinformatics
College of Biological Science and Technology
National Chiao Tung University in partial Fulfillment of the
Requirements for the Degree of Master in
Biological Science and Technology
July 2012, Hsinchu, Taiwan
I
中文摘要
中文摘要
中文摘要
中文摘要
近年來人們對非編碼 RNA (ncRNAs) 的興趣正快速地成長,儘 管這些 ncRNAs 不會被轉譯成蛋白質,但它們在細胞內卻扮演著許多 重要的角色。事實上,大多數已有的 ncRNAs 的功能仍是未知並且是 需要被預測的。從演化的角度而言,分子的結構往往會比它的序列來 得保守些,因此,偵測出 RNA 三級結構之間的相似程度,將更能洞 察出 RNA 分子的功能以及它們的演化關係,而這些功能與演化關係 是無法單從 RNA 序列的資訊中給偵查出來的。因此,本研究的目的 是設計出一個軟體工具可以有效率且正確地計算出多個 RNA 三級結 構的相似程度。我們的方法是利用一個新的結構字元集將 RNA 三級 結構轉成一級的結構字元式序列,然後再利用傳統的多重字元序列比 對工具 CLUSTAL W,以及新的類 BLOSUM 置換分數矩陣來比對出 多重 RNA 結構字元式序列,進而推算出原 RNA 三級結構之間的相似 程度。接著,我們利用上述的方法實做出一個軟體工具,稱之為 iMARTS。最後我們利用一些 RNA 三級結構來測試 iMARTS,並將其 實驗結果與我們先前開發出來的軟體工具 MARTS 做比較,實驗的結II
果顯示出 iMARTS 確實比 MARTS 有更好的表現。因此,我們相信 iMARTS 在結構生物學的研究上可以做為一個有用的生物資訊工具。
III
Abstract
Recently, there is a fast growing interest in noncoding RNAs (ncRNAs) because they play a lot of essential roles in many cellular processes, even though the transcripts of these ncRNAs are not translated into proteins. Actually, the function of most available ncRNAs is still unknown and needs to be determined. Since molecular structures are typically more evolutionarily conserved than sequences, detecting structural similarities among RNA three-dimensional (3D) structures can bring more significant insights into their functional and even evolutionary relationships that would not be detected by sequence information alone. Therefore, the purpose of this study is to design a software tool that can efficiently and accurately compute the structural similarity of multiple RNA 3D structures. Our method first uses a new structure alphabet to transform RNA 3D structures into 1D SA-encoded sequences and then uses a traditional multiple sequence alignment tool CLUSTAL W and a new BLOSUM-like scoring matrix to align the SA-encoded sequences of several RNAs for detecting the structural similarity of these RNAs. Next, we have implemented the above method into a software tool called iMARTS. Finally, we have tested iMARTS on some RNA 3D structures and compared its experimental results with those obtained by our previously developed tool MARTS. Consequently, the experimental results show that iMARTS indeed has a better performance when compared with MARTS. Therefore, we believe that iMARTS can serve as a useful tool in the
IV study of structural biology.
V
Acknowledgement
還清晰地記得自己第一次踏進老師辦公室和實驗室的模樣,有 點雀躍,還有點緊張。也永遠記得為了 R3D-BLAST 第一次和大夥兒 一起奮鬥到天亮的情景,為了趕進度總是 coding 到半夜,一個人走 在那忽然覺得好漫長的紅磚步道,還有口試結束那如釋重負的滋 味。兩年來,在研究和生活上都經歷了不少事情,也學會了很多, 感謝我的指導老師盧錦隆教授,嚴謹又不失親切的教導,總是能用 很生活化的比喻,讓我們更深刻地感受邏輯的重要,在您的身上真 的有好多值得我們學習的地方。此外還要感謝李家同教授每個禮拜 給我們在研究上以及英文寫作和聽力的指導,也要感謝林苕吟教授 擔任我的另一位指導老師。 謝謝昆澤學長總是耐心地與我討論研究上的問題,在研究和生 活上都給了我很多的幫助。謝謝忠翰學長在我碰到問題時總能告訴 我可以嘗試的辦法,快分我四分之一滔滔不絕的能力吧!感謝亙亘學 長總是大方地分享自己所知道的一切,讓我省了很多力氣呢!謝謝昱VI 全學長的時時關心與經驗分享,瑪莉歐理論在好多地方都好受用呢! 謝謝仁駿學長之前常陪我聊聊抒解我的壞心情,在今年我生日一過 十二點時就送上最棒的祝福。謝謝芸蓁學姊和彥菱學姊總是記得有 我這個小學妹,給了我很多溫暖和關心。感謝晟宸學長提供我許多 找工作上的建議和提醒,讓我的面試加分不少,謝謝慶恩學長精闢 地解說了台灣軟體產業的結構,還提供我找工作幾個要注意的事 項,也謝謝演富學長和志偉學長提供我不同的意見。謝謝瑋芸學妹 和熾隆學弟平常的幫忙,有了你們實驗室變得更可愛些了。 另外,還要感謝呂威甫教授,從大學到研究所這幾年來給我的 指導與關心,總在我需要的時候拉我一把、給我信心。最後,要感 謝我的男朋友惠弘,這兩年來不論晴雨的接送,一路的陪伴與支持, 並且給我最大的體諒和包容。感謝親愛的家人永遠無條件的支持, 讓我順利的完成學位。謝謝那些曾經幫助過我,給我關心和鼓勵的 朋友們。
VII
Contents
中文摘要 ... I Abstract ... III Acknowledgement... V Contents ... VII List of tables ... VIII List of figures ... IXChapter 1 Introduction ... 1
Chapter 2 Materials and Methods ... 8
2.1 假扭轉角與類 Ramachandran 的 η - θ 平面圖 ... 9
2.2 以結構字元式方法將三級結構編碼成一級結構序列 ... 13
2.3 類 BLOSUM 之結構字元置換分數矩陣 ... 18
Chapter 3 Implementation of Software Tool ... 26
3.1 iMARTS 之輸入(Input) ... 26
3.2 iMARTS 之輸出(Output) ... 29
Chapter 4 Results and Discussions ... 31
4.1 Pseudoknot 多重結構比對 ... 32 4.2 tRNA 多重結構比對 ... 34 4.3 Ribozyme 多重結構比對 ... 36 4.4 5S Ribosomal RNA 多重結構比對 ... 38 4.5 16S Ribosomal RNA 多重結構比對 ... 40 Chapter 5 Conclusions ... 43 References ... 44
VIII
List of tables
Table 2-1. 由 23 個字母組成的結構字元集與其 η 與 θ 角度。 ... 16 Table 4-1. Pseudoknot 多重結構比對的 RMSD 比較。 ... 32 Table 4-2. tRNA 多重結構比對的 RMSD 比較。 ... 34 Table 4-3. Ribozyme 多重結構比對的 RMSD 比較。 ... 36Table 4-4. 5S Ribosomal RNA 多重結構比對的 RMSD 比較。 ... 38
IX
List of figures
Figure 2-1. (a) 六個標準扭轉角,α、β、γ、δ、ε 與 ζ。(b) 以兩個假 扭轉角 η 與 θ 表示一個核苷酸。 ... 10 Figure 2-2. 所有在 PDB 結構資料集內不重複且非末端的核苷酸η - θ 圖。每個點代表一個核苷酸。 ... 12 Figure 2-3. 以 AP 演算法所分出來的 23 個群組。 ... 14 Figure 2-4. 23 個分群中心點核酸的三級結構圖。 ... 15 Figure 2-5. 分群數目與其總平均誤差值關係。 ... 16 Figure 2-6. 經由序列比對後產生的 4 個 blocks。 ... 18Figure 2-7. iPARTS 所使用的類 BLOSUM 置換分數矩陣。 ... 23
Figure 2-8. 第五次 iteration 所產生的類 BLOSUM 置換分數矩陣。 . 24 Figure 2-9. 第六次 iteration 所產生的類 BLOSUM 置換分數矩陣。 . 25 Figure 3-1. iMARTS 之操作介面。 ... 27 Figure 3-2. iMARTS 之輸出介面。 ... 29 Figure 3-3. iMARTS 之輸出介面,視覺化呈現結構的多重比對。 . 30 Figure 4-1. 對五個 Pseudoknots 的三級結構進行多重比對。 ... 33 Figure 4-2. 對六個 tRNAs 的三級結構進行多重比對。 ... 35 Figure 4-3. 對三個 Ribozyme 的結構進行多重比對。 ... 37
Figure 4-4. 對三個 5S Ribosomal RNA 的結構進行多重比對。 ... 39
1
Chapter 1
Introduction
近年來,RNA 分子在生物學上受到了特別的重視,尤其是那些 不會轉譯(translated)成蛋白質的非編碼 RNA(non-coding RNAs, 簡稱 ncRNAs)[1]。它們並非是過去所認為的遺傳訊息的攜帶者, 而是在許多的生理機制調控過程中扮演了相當重要的角色。例如在 轉錄轉譯的基因調節、核醣體移碼、化學修飾等調節功能、染色體 複製以及協助穩定 mRNA 的結構等等[2, 3, 4, 5]。事實上,大多數目 前已有的 ncRNAs 其功能仍然是未知的。RNA 分子可以分成三種不 同的層級來看:(1)由 A、U、C、G 所組成的核苷酸序列,為 RNA 的一級結構(primary structure)。(2)根據 Watson-Crick 鹼基對關係 (A/U 與 C/G)與 Wobble 鹼基對關係(U/G)形成的鍵結,使得 RNA 產生折疊,即為二級結構(secondary structure)。(3)除了上述的鹼 基對關係,也可能因為在立體空間上距離很近而產生額外的鍵結, 或者彼此有相互作用,相連而形成立體結構,級所謂的三級結構
2
(tertiary structure)。目前 RNA 的三級結構有些可以利用 X-ray 與
NMR 等實驗方法來得知。如同蛋白質,若想預測一個 RNA 其可能
存在的功能時,最常見也最為有效的方法即是去搜尋資料庫並找出 相似且功能已知的 RNA,所以目前已有許多資料庫被建構出來,如
NOCODE [6]、RNAdb [7]、miRBase [8]、fRNAdb [9]和 ncRNAdb [10]。但是相較於蛋白質 20 個字母的字元集(alphabet),RNA 則只 有 4 個字母的字元集(A、U、C、G)就顯得特別地小,使得利用
RNA 序列的比對(sequence alignment)去找出相似的 RNA 分子則
無法像在搜尋相似蛋白質般正確與有效。事實上,RNA 分子的功能 也是取決於它的三級結構,因為在演化過程中,RNA 二級與三級結 構會比其一級序列來得更為保守(conservative)而不易改變,過去 許多研究也顯示出比對 RNA 三級結構之間的相似程度會比只比較 一級序列獲得更多的資訊[11]。因此我們可以透過分析 RNA 三級結 構間的相似程度,從已知功能的 RNA 推估出未知的 RNA 可能存在 的功能,進而幫助生物學家深入了解 RNA 的功能,甚至是演化上的 關係。
然而,現今存入 PDB(Protein Data Bank)資料庫[12]中的 RNA 三級結構,不論數量還是大小都是逐年快速的增加,使得我們若是
3 用人工手動的方式去比較和分析這些 RNA 結構則越顯困難並且耗 費時間,所以發展出一個精確、快速且自動化分析比對 RNA 三級結 構工具的需求就變得越來越高了。但理論上,在三級結構的層級上 去計算兩個蛋白質或 RNA 之間的相似程度仍然是一件困難的工 作。因為對於計算出兩個結構中最大的相似子結構的問題要求得一 個常數倍的近似解,已經被證明是一個 NP-hard 的問題[13]。基於這 個理由,近來用於比對 RNA 三級結構的工具都是建構在啟發式的方 法上,例如 ARTS [14, 15]、DIAL [16]、SARSA [17] 及 iPARTS [18]。
ARTS 是 一 個 用 來 偵 測 兩 個 RNA 結 構 間 最 大 相 似 子 結 構
(substructure)的軟體工具,它使用了一個時間複雜度為三次方的 演 算 法 。 首 先 在 RNA 結 構 中 找 出 任 兩 對 連 續 的 鹼 基 配 對 (base-pairs),由這兩對鹼基配對上的四個磷原子構成一個稱之為
seed 的單位。接著藉由比較兩個 RNA 結構中的這些 seed,利用貪婪
(greedy)的方式從 seed 往結構的兩邊延伸並計算出相似程度最大 的區域,也就是局部相似的結構片斷(作者 Doro 等人稱之為 seed
matches),進而達到兩個 RNA 結構的比對。而這樣的做法有一個限
制,兩個要進行比對的 RNA 結構都必須要有二級結構的存在才能完 成。在偵測 RNA 結構模體(RNA structural motifs),ARTS 算是一個
4 還不錯的軟體工具,但是對於兩個大的 RNA 分子結構 (如 ribosomal RNA) 間的比較,對 ARTS 而言則是一件極為耗時的工作,另有文 獻指出 ARTS 的結構比對有時是不正確的[16]。 因此 Ferrè 等人提出另一種方法,改進了 ARTS 太花時間與準確 度不夠的不足之處,進而開發出可比對兩個 RNA 結構的軟體工具 DIAL。DIAL 則是利用一個時間複雜度為二次方的動態規劃演算法
(dynamic programming algorithm),藉由計算兩個 RNA 分子間的一 級序列、鹼基配對、扭轉角(torsion angles)與假扭轉角(pseudo-torsion
angles)來計算兩個 RNA 三級結構之間的相似程度。DIAL 提供三種
不同類型的比對方式: (1) global alignment,可比對出整個 RNA 結構 的相似程度。 (2) local alignment, 可找出結構上局部相似的地方。
(3) semi-global alignment,一種對 end gap 不計分的 global alignment。
雖然 Ferre 等人在文獻上[16]表示 DIAL 不論是執行的速度還是預測 的準確度,都比 ARTS 來得更好,但是就我們實際的操作與觀察 [17],發現在某些情況下 DIAL 所產生的 RNA 結構比對仍然有誤, 並且 DIAL 在計算鹼基配對相似度時所花費的時間也是相當地多。 我們實驗室在先前的研究提出了一個結構字元式(structural alphabet-based)的演算法,發展出一個可進行兩個或多個 RNA 三級
5
結構比對的網頁工具,SARSA(Structural Alignment of RNA Using a
Structural Alphabet )。 首 先 我 們 利 用 所 謂 的 向 量 量 化 ( vector quantization)的方式,根據四個扭轉角(α、γ、δ 與 ζ)將 RNA 殘
基主幹(backbone)在三維空間的構形(conformation)劃分成 23 類, 每一類各由一個英文字母來表示,我們將此 23 個字母視為一個 RNA 三級結構的字元集(structural alphabet; 簡稱 SA)。接著,再透過結 構字元集把每個 RNA 三級結構重新編碼成由這 23 個結構字母所構 成的一級序列(SA-encoded sequence)後,則可以使用傳統的序列 比對(sequence alignment)演算法並搭配 BLOSUM-like 的置換分數 矩陣,以此決定出兩個結構之間的相似程度。我們的 SARSA 提供了 兩種比對工具,一為 PARTS(Pairwise Alignment of RNA Tertiary
Structures)可進行兩個 RNA 三級結構的比對,另一為 MARTS
(Multiple Alignment of RNA Tertiary Structures)可進行多個 RNA 三 級結構之間的比對。在 PARTS 裡面,我們提供了四種不同的兩個
RNA 結構比對方式: (1) global alignment,供使用者比較與分析兩
個 RNA 三級結構整體的相似程度。 (2) semi-global alignment,即對
end gap 不計分的 global alignment。 (3) local alignment,供使用者找
出 兩 個 RNA 三 級 結 構 局 部 相 似的 區 塊 。 (4) normalized local
6
對兩兩 RNA 結構的比對而言,我們先前的實驗結果顯示出我們的
PARTS 在執行速度上通常都是比 DIAL 和 ARTS 要來得快,並且 PARTS 和 DIAL 的比對結果是差不多。
之後,我們實驗室又發展出一個新的結構字元式的演算法,用 來比對兩個 RNA 三級結構,並將 PARTS 改版成 iPARTS(improved
PARTS)。我們蒐集了一些具有代表性的 RNA 三級結構,改採用兩
個假扭轉角 η 與 θ 來表示一個 RNA 三級結構。然後我們將這些 η 與 θ 角繪製在一張 η - θ 平面圖上,並且我們利用近來發表的親和性 互動式分群演算法(Affinity Propagation clustering algorithm)[19], 分類出 23 個結構字元(SA letters),建構新的結構字元集。隨後以 BLOSUM-like 的方式重新計算置換分數矩陣。經過實驗分析後,我 們發現 iPARTS 確實能比 PARTS 來得準確。 在本研究中,我們首先利用 iPARTS 的結構字元集合將 RNA 三 級結構編碼成一級結構字元式序列,然後再利用傳統的比對工具 CLUSTAL W 以進行多重結構字元式序列的比對。為了要使結構字元 能夠更精確地比對,我們利用 Henikoff 與 Henikoff [20]提出的統計方 法,改進並建立出新的屬於 RNA 三級結構字元 23×23 的置換分數 表。基於上述的精神,我們發展出一個新的 RNA 三級結構多重比對
7
的軟體工具,稱之為 iMARTS(improved MARTS)。最後,實驗結 果也顯示我們新的 iMARTS 確實有比先前的 MARTS 有更好的表現。
8
Chapter 2
Materials and Methods
本研究方法首先我們利用 RNA 核苷酸的兩個假扭轉角 η 與 θ 角,畫出一張類似於 Ramachandran 的平面圖。接著我們利用親合性 互動式(Affinity Propagation)分群演算法來對在 η - θ 平面圖上的 RNA 核苷酸進行分群,並且得到一組含有 23 個核苷酸結構的字元 集。最後我們便可將 RNA 三級結構個別編碼成由結構字元所組成的 一級結構序列,然後再利用傳統的多重序列比對工具 CLUSTAL W 去分析比對這些結構字元式的一級序列,以決定出原 RNA 三級結構 之間的相似程度。在這個章節我們將會進一步的介紹關於 (1) 假扭 轉角以及類 Ramachandran 平面圖,(2) 如何產生結構式字元集,並 將 RNA 3D 結構轉為 1D 序列,(3) 如何從 1D 結構字元式序列的 比對產生類似於 BLOSUM 的 RNA 結構字元式置換分數矩陣。
9
2.1 假假假假扭轉角扭轉角扭轉角扭轉角與與與與類類 Ramachandran 的類類 的的 η - θ 平面的 平面平面圖平面圖圖 圖
以蛋白質而言,對於每一個胺基酸只需要用兩個標準扭轉角 (torsion angle) φ 和 ψ 便能描述其構形。而每個 RNA 結構上的核苷 酸則是具有六個標準的扭轉角(α、β、γ、δ、ε 與 ζ)(Figure 2-1a)。 過去我們在 SARSA 上是使用其中四個扭轉角(α、γ、δ 與 ζ)來表 示一核苷酸構形,因為已有文獻[21]表示利用 α、γ、δ 與 ζ 角便足 夠來區分不同核苷酸的構形,加入 β 與 ε 角對於核苷酸構形的分 析並無太大差異。但是要同時考慮六個或是四個標準扭轉角來,分 析和分類出這些核苷酸構型將會是一個高維度的問題,在計算上是 件不容易解決的問題,而且也不容易將分析的結果給予視覺化,以 方便用目視的方式去評估其結果。為了要解決考慮多個標準扭轉角 的上述缺點,Duarte 與 Pyle 等人[22]提出以兩個假扭轉角 η 與 θ 角 來表示核苷酸的骨架(Figure 2-1b),他們認為有些情況下使用假扭 轉角來代表核苷酸構形會比用標準扭轉角來得更好。
10 Figure 2-1. (a) 核苷酸的六個標準扭轉角,α、β、γ、δ、ε 與 ζ。 (b) 以兩個假扭轉角 η 與 θ 表示一個核苷酸(標示為 n),η 是由 C4′n-1, Pn, C4′n 與 Pn+1四個原子所形成的假扭轉角度,θ 則是由另四 個原子 Pn, C4′n, Pn+1 與 C4′n+1所形成的假扭轉角度。 雖然利用 η 與 θ 兩個假扭轉角來表示核苷酸的骨架,可能不 如使用六個標準扭轉角來得精準,但如此一來可以把高維度的核苷 酸分析降為二維的計算,同時也可以把核苷酸與其分析的結果視覺 化地表示在一個二維的平面上,此舉有助於進一步地把核苷酸的骨 架構型進行分類[22, 23]。利用此精神的 Duarte 與 Pyle 等人也發展了
11 PRIMOS [24]與 COMPADRES [25]兩個工具,分析結構之間的相似程 度,以搜尋出在 PDB 資料庫中特定的 RNA 結構模體。 我們準備了一組不重複(non-redundant)且解構解析度最低到 3.0Å 的 PDB 裡的 RNA 資料集來繪製 η - θ 圖,總共有 117 個 RNA 結構,包含 9,527 個核苷酸。接著我們使用由 Duarte 與 Pyle 發展的 AMIGOS 工具[26],將這些 RNA 三級結構中各個核苷酸的 η 與 θ 角度計算出來(不包含每個結構的開頭與結尾的核苷酸,一共 9267 個)。然後將這些計算出來角度繪製在以 η 角度為 X 軸、以 θ 角 度為 Y 軸的 η - θ 圖上(Figure 2-2)。如前段所敘述,二維的 η - θ 圖可以幫助我們以圖形表示法的方式量化不同結構之間的差異性, 以利於進行 RNA 三級結構的分析。
12
Figure 2-2. 所有在 PDB 結構資料集內不重複且非末端的核苷酸 η - θ
圖。每個點代表一個核苷酸。中間兩灰底且垂直重疊的區域(150° ≤ η ≤ 190° 與 190° ≤ θ ≤ 260°)為 RNA 結構中的螺旋結構區域(helical
13 2.2 以結構字元式方法將三級結構編碼成一以結構字元式方法將三級結構編碼成一以結構字元式方法將三級結構編碼成一以結構字元式方法將三級結構編碼成一級級級結構序列級結構序列結構序列 結構序列 接下來我們將先前得到的 η - θ 圖,利用親和性互動式分群演算 法將圖上的所有的點分成 23 群(Figure 2-3),並且將此 23 群各個中 心點(center)視為該群的代表結構如 Figure 2-4 所示,而且分別以 一個字母來表示一個代表性結構,最後這 23 個字母便稱為 RNA 的 結構字元集(Table 2-1)。之所以決定分成 23 群,是因為當初我們有 統計所謂的平均誤差值(average error),將一群內所有的點到中心點 的距離加總起來除以群內的個數即為平均誤差值,假設分成 23 群, 則會有 23 個平均誤差值,把這 23 個值加總起得到總平均誤差值, 我們希望不論分多少群,總平均誤差值是越小越好。利用親和性互 動式分群演算法我們一共產生了 3 到 60 不同的分群數(如 Figure 2-5 所示),雖然 23 群的總平均誤差值並非最小的,但我們仍然選擇 23 為我們最後的分群數,除了群數 23 在 Figure 2-5 的曲線上尚為趨緩, 主要原因為過多的群數會使得 η - θ 圖上中間螺旋結構區域過度分 群(overpartition),另外 23 群便可以讓我們直接套用在 BLAST 上去 做快速的資料庫搜尋。
14
Figure 2-3. 以 AP 演算法所分出來的 23 個群組。每群分別以不同顏
15
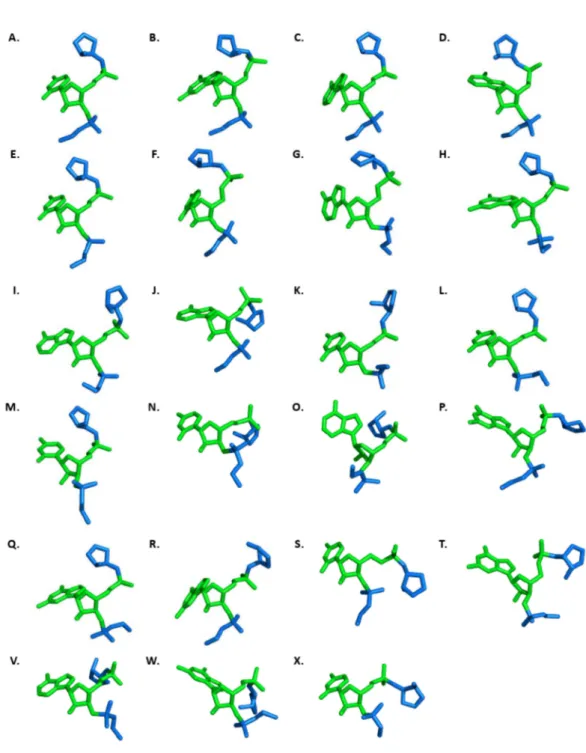
Figure 2-4. 23 個分群中心點核酸的三級結構圖。中心點核酸以綠色
表示;中心點前後各一個會影響中心點核酸假扭轉角度的核酸以藍 色表示。
16
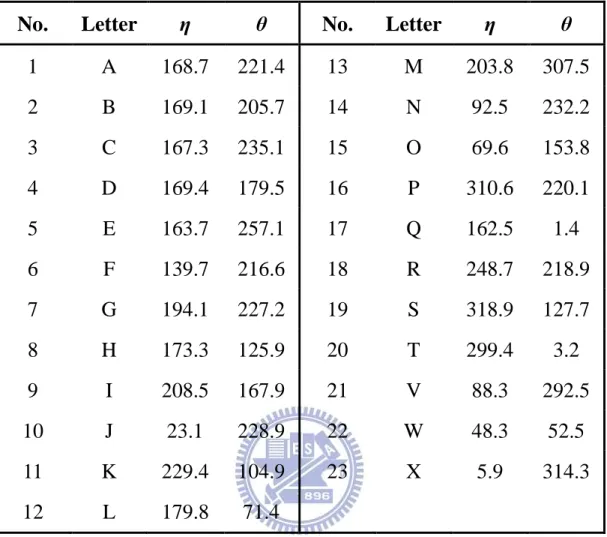
Table 2-1. 由 23 個字母組成的結構字元集與其 η 與 θ 角度。
No. Letter η θ No. Letter η θ
1 A 168.7 221.4 13 M 203.8 307.5 2 B 169.1 205.7 14 N 92.5 232.2 3 C 167.3 235.1 15 O 69.6 153.8 4 D 169.4 179.5 16 P 310.6 220.1 5 E 163.7 257.1 17 Q 162.5 1.4 6 F 139.7 216.6 18 R 248.7 218.9 7 G 194.1 227.2 19 S 318.9 127.7 8 H 173.3 125.9 20 T 299.4 3.2 9 I 208.5 167.9 21 V 88.3 292.5 10 J 23.1 228.9 22 W 48.3 52.5 11 K 229.4 104.9 23 X 5.9 314.3 12 L 179.8 71.4 Figure 2-5. 分群數目與其總平均誤差值關係。 0 100 200 300 400 500 600 700 800 900 1000 1100 1200 1300 1400 1500 1600 1700 1800 1900 2000 2100 2200 2300 0 5 10 15 20 25 30 35 40 45 50 55 60 總 平 均 誤 差 值 總 平 均 誤 差 值 總 平 均 誤 差 值 總 平 均 誤 差 值 群數 群數群數 群數
17
結構字元式的演算法其最主要精神就是將三級結構轉換成由結 構字元所組成的一級序列。對於每個 RNA 結構,我們逐一計算每個 核苷酸在 η - θ 圖上的位置與各中心點位置之間的距離,依據最近鄰 居規則(nearest neighbor rule)將此核苷酸的結構逐一編碼成與其距 離最近的中心點之結構字元,這些編碼後的結構字元就組成了所謂 的 RNA 一級結構序列。 雖然將三級結構根據以 η - θ 角為基礎的結構字元集轉成一級 的結構序列,勢必會損失掉一些資訊。但好處是我們可以利用傳統 的字串比對演算法,去比較兩條一級結構序列以快速地決定出兩個 RNA 三級結構的相似程度。
18
2.3 類類類類 BLOSUM 之之之結構字元置換之結構字元置換分結構字元置換結構字元置換分分分數數數數矩陣矩陣矩陣矩陣
在使用傳統序列的比對時,我們需要一個結構字元的計分函式 (scoring function),才能精確地比對出兩條或多條 RNA 結構字元式 的序列。因此我們參考 1992 年 Henikoff 與 Henikoff 提出的統計方法 去建構出一個 23×23 類 BLOSUM(BLOcks Substitution Matrix)的
RNA 結構字元的置換分數矩陣。 當年 Henikoff 與 Henikoff 的做法是從一個公開並且已分類的蛋 白質資料庫得到一個蛋白質序列的集合,然後根據資料庫的分類, 分 別 將 每 一 群 內 的 蛋 白 質 進 行 序 列 的 多 重 比 對 ( multiple alignment),以產生這些胺基酸序列比對中具有高度保留性的區域, 也就是比對後那些沒有插入空白(gap)的區域,Henikoff 與 Henikoff 稱之為 blocks。舉一個例子來說(如 Figure 2-6 所示),我們可以從 一組多重序列比對的結果中得到 4 個 blocks。 Figure 2-6. 經由序列比對後產生的 4 個 blocks。
19
接著 Henikoff 與 Henikoff 就去計算出 blocks 集合裡每一個胺基酸出 現的頻率,以及胺基酸與胺基酸配對(align)在一起的頻率,然後 再利用統計的方法去建構出 BLOSUM 置換分數矩陣。 在本研究中,我們利用 DARTS [23]資料庫裡頭 RNA 三級結構來 產生我們的 RNA 結構字元的置換分數矩陣,方法如下: 首先將 DARTS 裡頭的每一群 RNA 三級結構轉成結構字元式的一級序列, 然後再將每一群的結構字元式序列進行多重序列比對,從比對的結 果中蒐集出一群屬於 RNA 結構字元式的 blocks 集合。接著我們再去 計算出此 blocks 集合裡每一個 RNA 結構字元出現的頻率,以及 RNA 結構字元與 RNA 結構字元配對的頻率,最後再利用 Henikoff 與
Henikoff 所提出的統計方法去建構出 RNA 結構字元的類 BLOSUM
置換分數矩陣。 以下介紹我們用來建構出 23×23 的 RNA 結構字元類 BLOSUM 置換分數矩陣的統計方法: 令 , , ⋯ , 為我們的 RNA 結構 字元集合。 表示在 RNA 結構字元式 blocks 集合中所有 , 的數量(即結構字元 配對到結構字元 的數量)。 表示每一個 結構字元 在 RNA 結構字元式 blocks 集合裡出現的頻率。 為 , 配對頻率的觀察值(observed frequency), 為 , 配
20 對頻率的期望值(expected frequency)。 首先我們先從 RNA 結構字元式 blocks 集合裡計算出每一對 , 的配對數量 ,接著再去計算出所有 , 配對頻率的觀 察值 : = ∑ ∑ 然後從每個結構字元在 blocks 集合裡出現的頻率 ,可以推估出 每一對 , 配對頻率的期望值 : = 2 = ≠ 接著再將 , 配對頻率的觀察值 與期望值 根據下列公式 求得 , 配對的分數 score , :
score , = "# log & ' ± 0.5,
21 當 , 配對頻率的觀察值大於其期望值時, score , 的分數 會大於 0,這就表示 與 這兩個結構字元傾向配對在一起,反之 則表示 與 不傾向配對在一起。 事實上,依照上述方法所產生的置換分數矩陣會有一個缺點, 此缺點發生於當有多條結構相似的 RNAs 被拿來進行多重結構字元 式序列比對以產生一些 blocks。因此在每一個 block 裡會有數條關係 較近的 RNAs,這些數條關係較近的 RNAs 在 block 中的同一欄位 (column)其共有字元出現的頻率會偏高,因而導致在計算各種結 構字元配對頻率時的偏差(bias)。為了解決這個問題,Henikoff 與
Henikoff 還提出了分群(cluster 或 group)的觀念,將每個 block 內
的序列較為相近的分群在一起並將它們視為一條序列。在本研究 中,我們將相似程度大於或等於 85% 的結構字元式序列分成一群。 根據上述的演算法,我們利用 iPARTS 的置換分數矩陣(Figure 2-7)去做結構字元式序列的多重比對以產生 blocks 集合,然後利用 這些 blocks 產生一個新的置換分數矩陣,有了這個新的置換分數矩 陣,我們又可以用相同的方法產生更新的置換分數矩陣,如此步驟 一直重複直到置換分數矩陣呈現收斂為止。
22
在本研究中,我們利用 DARTS 資料庫裡已分類好的 RNA 三級 結構來建構出屬於 RNA 結構字元的類 BLOSUM 置換分數矩陣。
DARTS 利用 RNA 三級結構的相似程度將 1333 個 RNAs 分成 244
群,其中有 89 群皆只包含一個 RNA 結構,故無法被拿來進行多重 的結構字元式序列比對,因此我們只考慮其餘的 155 群,而這 155 群總共包含了 816 個 RNA 結構。接著,我們用至少 85% 的相同字 元比去做為分群的門檻(cut-off)以產生我們所需的 blocks,最後再 用這些 blocks 產生一個類 BLOSUM 的結構字元置換分數矩陣,這個 過程我們重複執行了數次(iteration),發現在第五次(Figure 2-8) 與第六次(Figure 2-9)所產生的分數矩陣有呈現收斂的現象。
23
Figure 2-7. iPARTS 所使用的類 BLOSUM 置換分數矩陣,其中藍色
24
25
26
Chapter 3
Implementation of Software Tool
基於前面章節介紹的結構字元式的演算法,我們發展了一個軟體 工具,稱之為 iMARTS(Improved Multiple Alignment of RNA Tertiary
Structures),可供使用者進行 RNA 三級結構的多重比對。接下來我 們會介紹關於 iMARTS 的操作介面(Figure 3-1),並且逐步地說明如 何使用 iMARTS。 3.1 iMARTS 之之之輸入之輸入輸入輸入((((Input)))) 1. 輸入或是貼上多個(至少兩個)RNA 三級結構,格式為 <pdb|ndb id>:<chain id>:[residue]-[residue],第一項為 PDB 資料庫或者 NDB 資料庫的代碼,第二項為其鏈(chain)的代號,第三項可輸入 欲比對結構起始與結束的殘基位置。舉例來說,“ 1ASZ:R ” 表 示是 PDB 代碼為 1ASZ,chain 編號為 R 的這整個 RNA 結構,
27
而 “ 1ASZ:R:620-660 ” 則表示是 PDB 代碼 1ASZ,chain 編號
R,並且是從第 620 號到 660 號的殘基這個 RNA 子結構。另外, “ 1ASZ:R:-660 ” 則為 PDB 編碼 1ASZ 的 R chain 之子結構,從
第 1 個殘基到第 660 號殘基。
2. 除了上述的輸入方式外,使用者也可隨意的上傳任何 RNA 三級
結構(PDB 檔案),iMARTS 允許使用者最多可上傳 20 個 PDB 檔案。
28
3. 若使用者欲使用預設參數來執行 iMARTS,請直接按下 “ Run iMARTS ” 即可。否則,須按照以下步驟來設定參數。
4. 選用一個 RNA 結構字元置換分數矩陣。iMARTS 提供第五次 iteration 之類 BLOSUM 置換分數矩陣、第六次 iteration 之類 BLOSUM 置換分數矩陣,以及 iPARTS 之類 BLOSUM 置換分數
矩陣。
5. 選用一組空白罰分(gap penalty)。iMARTS 預設的空白罰分為 開 gap(existence)是扣四分,延長 gap(extension)是扣一分。
29 3.2 iMARTS 之輸出之輸出之輸出之輸出((((Output)))) 在 iMARTS 的輸出介面上,首先會顯示所有輸入的 RNA 三級結 構與其相關資訊,以及所使用的參數。接著,iMARTS 會顯示將這 些 RNAs 的一級結構字元式序列進行多重比對的結果,與其對應的 原始核苷酸序列,如 Figure 3-2 所示。 Figure 3-2. iMARTS 之輸出介面。
30
此外,使用者可以從 “ Superposition display ”連結到 Jmol 視窗,以 觀察多重比對後在三維空間中 RNAs 結構的重疊情形,如 Figure 3-3 所示。
31
Chapter 4
Results and Discussions
在這個章節,我們將使用一些例子對 iMARTS、MARTS 和 Modified-MARTS(此為使用 iPARTS 的置換分數矩陣的 MARTS 版 本)來進行測試,並且進一步地說明與討論我們的實驗結果。我們 使用了 Pseudoknots、tRNAs、Ribozymes、5S Ribosomal RNAs 與 16S
Ribosomal RNAs 這五種不同的 RNA 三級結構來進行多重結構比對
的實驗。
在參數的設定上,iMARTS 分別使用了第五次 iteration 與第六次
iteration 之類 BLOSUM 置換分數矩陣,以及六組不同的空白罰分
(-X, -Y),其中 -X 表示開 gap 的扣分而 -Y 表示延長 gap 的扣分, 這六組空白罰分分別為(-4, -1)、(-4, -2)、(-5, -1)、(-5, -2)、(-6, -1) 與(-6, -2)。MARTS 是使用其預設的類 BLOSUM 置換分數矩陣和 預設的空白罰分(-5, -2)。Modified-MARTS 則是使用 iPARTS 的類
32 4.1 Pseudoknot 多重結構比對多重結構比對多重結構比對 多重結構比對
第一組實驗我們以五個 Pseudoknot 的結構(1l2x:A, 2ap0:A,
2ap5:A, 1yg4:A 與 1kpy:A)來進行多重結構比對,結果如下表所示:
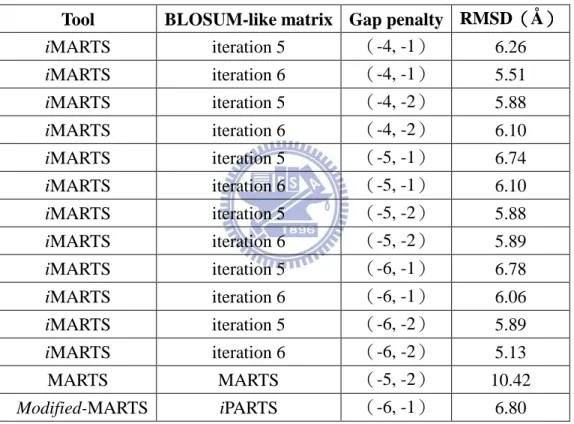
Table 4-1. Pseudoknot 多重結構比對的 RMSD 比較
Tool BLOSUM-like matrix Gap penalty RMSD(((Å)( )))
iMARTS iteration 5 (-4, -1) 6.26 iMARTS iteration 6 (-4, -1) 5.51 iMARTS iteration 5 (-4, -2) 5.88 iMARTS iteration 6 (-4, -2) 6.10 iMARTS iteration 5 (-5, -1) 6.74 iMARTS iteration 6 (-5, -1) 6.10 iMARTS iteration 5 (-5, -2) 5.88 iMARTS iteration 6 (-5, -2) 5.89 iMARTS iteration 5 (-6, -1) 6.78 iMARTS iteration 6 (-6, -1) 6.06 iMARTS iteration 5 (-6, -2) 5.89 iMARTS iteration 6 (-6, -2) 5.13 MARTS MARTS (-5, -2) 10.42 Modified-MARTS iPARTS (-6, -1) 6.80 由 Table 4-1 裡 14 個多重結構比對的結果,我們可以看到 iMARTS 在使用第六個 iteration 的類 BLOSUM 置換分數矩陣和空白罰分(-6, -2)時的 RMSD(5.13 Å)為最小為(其結構的重疊如 Figure 4-1a 所示)。然而,MARTS 的 RMSD 則為 10.42 Å(其重疊的結構如 Figure
33
4-1b)。而 Modified-MARTS 的 RMSD 為 6.8 Å,剛好介於 iMARTS 與 MARTS 之間。由此可見, iMARTS 的三級結構的比對結果確實 比 MARTS 來得好,尤其是 1l2x:A 與 1kpy:A(如 Figure 4-1b 紅色與 藍色的 RNAs)這兩個 RNAs 三級結構,MARTS 明顯沒有把它們與 其它三個 RNAs 對得很好。
(a) (b)
Figure 4-1. 對五個 Pseudoknots 的三級結構進行多重比對。(a)
iMARTS 多重結構的比對,其 RMSD 為 5.13 Å。(b)MARTS 多重結 構的比對,RMSD 為 10.42 Å。
34 4.2 tRNA 多重結構比對多重結構比對多重結構比對多重結構比對
第 二 組 實 驗 我 們使 用 六 個 tRNA 的結構(1H4S:T, 2CSX:C,
1ASZ:R:620-660, 1EVV:A, 1IL2:C, 1J2B:C)來進行多重結構比對,其
實驗結果如下表所示:
Table 4-2. tRNA 多重結構比對的 RMSD 比較。
Tool BLOSUM-like matrix Gap penalty RMSD(((Å)( )) )
iMARTS iteration 5 (-4, -1) 9.36 iMARTS iteration 6 (-4, -1) 10.11 iMARTS iteration 5 (-4, -2) 9.93 iMARTS iteration 6 (-4, -2) 10.10 iMARTS iteration 5 (-5, -1) 9.43 iMARTS iteration 6 (-5, -1) 10.38 iMARTS iteration 5 (-5, -2) 8.83 iMARTS iteration 6 (-5, -2) 8.44 iMARTS iteration 5 (-6, -1) 9.35 iMARTS iteration 6 (-6, -1) 10.10 iMARTS iteration 5 (-6, -2) 8.57 iMARTS iteration 6 (-6, -2) 8.40 MARTS MARTS (-5, -2) 12.48 Modified-MARTS iPARTS (-6, -1) 11.43
在 Table 4-2 中,我們可以發現 iMARTS 在使用第六個 iteration 的類 BLOSUM 置換分數矩陣和空白罰分(-6, -2)時的 RMSD(8.4 Å) 為最小,重疊起來的 tRNA 結構如 Figure 4-2a 所示。MARTS 的多重
35
結構比對結果其 RMSD 為 12.48 Å (重疊的 tRNA 結構參見 Figure
4-2b),造成此結果的主要原因是 MARTS 沒有把而 1IL2:C (如 Figure
4-2b 中 黃 色 的 結 構 ) 與 其 它 的 tRNA 結 構 對 得 很 好 。 對 於
Modified-MARTS,其 RMSD 則為 11.43 Å,結果仍沒有比 iMARTS 好。
(a) (b)
Figure 4-2. 對六個 tRNAs 的三級結構進行多重比對。(a)iMARTS
多重結構的比對,其 RMSD 為 8.40 Å。(b)MARTS 多重結構的比 對,RMSD 為 12.48 Å。
36 4.3 Ribozyme 多重結構比對多重結構比對多重結構比對 多重結構比對 第三組實驗我們使用同一個 Ribozyme(1X8W),但不同 chain 的 RNA 結構(1X8W:A, 1X8W:B 和 1X8W:C)來進行三級結構的多 重比對,實驗結果如 Table 4-3 所示: Table 4-3. Ribozyme 多重結構比對的 RMSD 比較。
Tool BLOSUM-like matrix Gap penalty RMSD((((Å))))
iMARTS iteration 5 (-4, -1) 7.39 iMARTS iteration 6 (-4, -1) 7.35 iMARTS iteration 5 (-4, -2) 7.41 iMARTS iteration 6 (-4, -2) 7.35 iMARTS iteration 5 (-5, -1) 7.39 iMARTS iteration 6 (-5, -1) 7.35 iMARTS iteration 5 (-5, -2) 7.41 iMARTS iteration 6 (-5, -2) 7.35 iMARTS iteration 5 (-6, -1) 7.39 iMARTS iteration 6 (-6, -1) 7.29 iMARTS iteration 5 (-6, -2) 7.41 iMARTS iteration 6 (-6, -2) 7.29 MARTS MARTS (-5, -2) 35.50 Modified-MARTS iPARTS (-6, -1) 7.71 觀察這組實驗結果可以發現,iMARTS 與 Modified-MARTS 的 RMSD 皆在 8 Å 以下,而 MARTS 的 RMSD 卻高達 35.5 Å,其結構 的重疊(Figure 4-3b)相當地不理想。
37
(a) (b)
Figure 4-3. 對三個 Ribozyme 的結構進行多重比對。(a)iMARTS 多
重結構的比對,其 RMSD 為 7.29 Å。(b)MARTS 多重結構的比對,
38 4.4 5S Ribosomal RNA 多重結構比對多重結構比對多重結構比對 多重結構比對
第四組實驗我們使用了三個 5S Ribosomal RNA 的結構(3CC2:9,
1Y69:9 與 2J03:B),來進行 RNA 三級結構的比對,實驗結果如 Table
4-4 所示:
Table 4-4. 5S Ribosomal RNA 多重結構比對的 RMSD 比較。
Tool BLOSUM-like matrix Gap penalty RMSD(((Å)( )) )
iMARTS iteration 5 (-4, -1) 3.68 iMARTS iteration 6 (-4, -1) 3.68 iMARTS iteration 5 (-4, -2) 3.91 iMARTS iteration 6 (-4, -2) 3.91 iMARTS iteration 5 (-5, -1) 3.91 iMARTS iteration 6 (-5, -1) 3.70 iMARTS iteration 5 (-5, -2) 3.70 iMARTS iteration 6 (-5, -2) 3.70 iMARTS iteration 5 (-6, -1) 3.70 iMARTS iteration 6 (-6, -1) 3.91 iMARTS iteration 5 (-6, -2) 3.70 iMARTS iteration 6 (-6, -2) 3.70 MARTS MARTS (-5, -2) 18.73 Modified-MARTS iPARTS (-6, -1) 5.28 由上表得知,iMARTS 在使用第五個與第六個 iteration 的類 BLOSUM 置換分數矩陣和空白罰分(-4, -1)進行多重結結構比對時 的 RMSD(3.68 Å)皆為最小為(其結構的重疊如 Figure 4-5a 所示),
39
而 MARTS 結構比對的 RMSD 為 18.73 Å (見 Figure 4-5b), Modified-MARTS 則為 5.28 Å,其結果比 MARTS 好,但仍不及 iMARTS。
(a) (b)
Figure 4-4. 對三個 5S Ribosomal RNA 的結構進行多重比對。(a)
iMARTS 多重結構的比對,其 RMSD 為 3.68 Å。(b)MARTS 多重結 構的比對,RMSD 為 18.73 Å。
40
這三個結構本身也極為相似,而其中 2J03:B(如 Figure 4-5 黃色 的 RNA)與 1Y69:9 (如 Figure 4-5 橘色的 RNA)這兩個 RNAs 在
MARTS 的結構比對,相較於 iMARTS 的結構比對並無太大的差異,
而 MARTS 整體的比對表現較差,其主要原因則是因為 3CC2:9(如
Figure 4-5 紅色的 RNA)在結構序列比對上為了將特定的幾個字元配
對在一起而開了許多 gap,導致在 MARTS 的結構比對時無法將
3CC2:9 與其餘兩個 RNAs 比對得很好,以 Figure 4-5a 來看,3CC2:9
與其它兩個結構是非常地相似。
4.5 16S Ribosomal RNA 多重結構比對多重結構比對多重結構比對多重結構比對
最後一組實驗我們將五個 16S Ribosomal RNA 的結構(2UU9:A,
2E5L:A, 1FJG:A, 2HGR:A 與 1HNW:A)進行結構的多重比對,實驗
結 果 如 Table 4-5 所 示 。 觀 察 這 組 實 驗 的 結 果 , iMARTS 與 Modified-MARTS 進行多重結構比對的 RMSD 皆不超過 3 Å,而
MARTS 結構比對的 RMSD 卻高達 45.25 Å。Figure 4-6a 為 iMARTS
41
時所得到的五個 Ribosomal RNA 重疊的結構,其 RMSD 為 2.49 Å。
Figure 4-6b 則為 MARTS 的結構比對結果,以此組結果得知, MARTS
的比對結果有誤,其中的 2UU9:A(即 Figure 4-6b 紅色的 RNA)明 顯地旋轉了至少 90 度,因此造成整體比對的結果極差,RMSD 值極 大。
Table 4-5. 16S Ribosomal RNA 多重結構比對的 RMSD 比較。
Tool BLOSUM-like matrix Gap penalty RMSD(((Å)( )) )
iMARTS iteration 5 (-4, -1) 2.60 iMARTS iteration 6 (-4, -1) 2.51 iMARTS iteration 5 (-4, -2) 2.68 iMARTS iteration 6 (-4, -2) 2.50 iMARTS iteration 5 (-5, -1) 2.62 iMARTS iteration 6 (-5, -1) 2.50 iMARTS iteration 5 (-5, -2) 2.68 iMARTS iteration 6 (-5, -2) 2.68 iMARTS iteration 5 (-6, -1) 2.62 iMARTS iteration 6 (-6, -1) 2.62 iMARTS iteration 5 (-6, -2) 2.68 iMARTS iteration 6 (-6, -2) 2.68 MARTS MARTS (-5, -2) 45.25 Modified-MARTS iPARTS (-6, -1) 2.76
42
(a) (b)
Figure 4-5. 對五個 16S Ribosomal RNA 的結構進行多重比對。(a)
iMARTS 多重結構的比對,其 RMSD = 2.49 Å。(b)MARTS 多重結 構的比對,RMSD = 45.25 Å。
43
Chapter 5
Conclusions
在本研究中,我們使用了 iPARTS 的結構字元以及利用分群的概 念改善了 iPARTS 的類 BLOSUM 置換分數矩陣,並且重新建立一個 可供使用者比對多個 RNA 三級結構的軟體工具 iMARTS。除此之 外,我們的實驗結果也顯示出新的 iMARTS 確實比之前的 MARTS 和 Modified-MARTS 有更好的表現。最後,我們相信這個使用結構字 元式的演算法搭配上新的類 BLOSUM 置換分數矩陣的 iMARTS,可 以在結構生物學的研究成為一種有用的工具。目前 iMARTS 是利用 CLUSTAL W 來進行 RNA 結構字元式序列的多重比對,事實上應有 其它更好的軟體工具如 T-Coffee 也可以來進行 RNA 結構字元式序列 的多重比對。因此,如何利用 T-Coffee 來取代 iMARTS 目前所使用 的 CLUSTAL W,將是一件值得研究的課題。44
References
1. Storz, G. (2002) An expanding universe of noncoding RNAs.
Science, 296, 1260−1263.
2. Doudna, J.A. (2000) Structural genomics of RNA. Nature Structural
Biology, 7, 954-956.
3. Eddy, S. R. (2001) Non-coding RNA genes and the modern RNA world. Nature Reviews Genetics, 2, 919–929.
4. Mattick, J.S. and Makunin, I.V. (2006) Non-coding RNA. Human
Molecular Genetics, 15, R17-R29.
5. Storz, G. (2002) An expanding universe of noncoding RNAs.
Science, 296, 1260-1263.
6. He, S.M., Liu, C.N., Skogerbo, G., Zhao, H.T., Wang, J., Liu, T., Bai, B.Y., Zhao, Y. and Chen, R.S. (2008) NONCODE v2.0: decoding the non-coding. Nucleic Acids Research, 36, D170-D172. 7. Pang, K.C., Stephen, S., Dinger, M.E., Engstrom, P.G., Lenhard, B.
and Mattick, J.S. (2007) RNAdb 2.0-an expanded database of mammalian non-coding RNAs. Nucleic Acids Research, 35, D178-D182.
8. Griffiths-Jones, S., Saini, H.K., van Dongen, S. and Enright, A.J. (2008) miRBase: tools for microRNA genomics. Nucleic Acids
Research, 36, D154-D158.
9. Kin, T., Yamada, K., Terai, G., Okida, H., Yoshinari, Y., Ono, Y., Kojima, A., Kimura, Y., Komori, T. and Asai, K. (2007) fRNAdb: a platform for mining/annotating functional RNA candidates from
45
non-coding RNA sequences. Nucleic Acids Research, 35, D145-D148.
10. Szymanski, M., Erdmann, V.A. and Barciszewski, J. (2007) Noncoding RNAs database (ncRNAdb). Nucleic Acids Research, 35, D162-D164.
11. Capriotti, E., Marti-Renom, M.A. (2010) Quantifying the relationship between sequence and three-dimensional structure conservation in RNA. BMC Bioinformatics, 11, 322.
12. Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N., and Bourne, P. E. (2000) The protein data bank. Nucleic Acids Research, 28, 235–242.
13. Kolodny, R. and Linial, N. (2004) Approximate protein structural alignment in polynomial time. Proceedings of the National Academy
of Sciences of the United States of America, 101, 12201-12206.
14. Dror, O., Nussinov, R. and Wolfson, H. (2005) ARTS: alignment of RNA tertiary structures. Bioinformatics, 21, ii47-53.
15. Dror, O., Nussinov, R. and Wolfson, H.J. (2006) The ARTS web server for aligning RNA tertiary structures. Nucleic Acids Research, 34, W412-415.
16. Ferré, F., Ponty, Y., Lorenz, W.A. and Clote, P. (2007) DIAL: a web server for the pairwise alignment of two RNA three-dimensional structures using nucleotide, dihedral angle and base-pairing similarities. Nucleic Acids Research, 35, W659-W668.
17. Chang, Y.F., Huang, Y.L. and Lu, C.L. (2008) SARSA: a web tool for structural alignment of RNA using a structural alphabet. Nucleic
Acids Research, 36, W19-W24.
18. Wang, C.W., Chen, K.T. and Lu, C.L. (2010) iPARTS: an improved tool of pairwise alignment of RNA tertiary structures. Nucleic Acids
46
Research, 38, W340-347.
19. Frey, B.J. and Dueck, D. (2007) Clustering by passing messages between data points. Science, 315, 972-976.
20. Henikoff, S. and Henikoff, J.G. (1992) Amino-acid substitution matrices from protein blocks. Proceedings of the National Academy
of Sciences of the United States of America, 89, 10915-10919.
21. Hershkovitz, E., Sapiro, G., Tannenbaum, A., and Williams, L. D. (2006) Statistical analysis of RNA backbone. IEEE/ACM
Transaction on Computational Biology and Bioinformatics, 3, 33–
46.
22. Duarte, C.M. and Pyle, A.M. (1998) Stepping through an RNA structure: A novel approach to conformational analysis. Journal of
Molecular Biology, 284, 1465-1478.
23. Wadley, L.M., Keating, K.S., Duarte, C.M. and Pyle, A.M. (2007) Evaluating and learning from RNA pseudotorsional space: Quantitative validation of a reduced representation for RNA structure. Journal of Molecular Biology, 372, 942-957.
24. Duarte, C.M., Wadley, L.M. and Pyle, A.M. (2003) RNA structure comparison, motif search and discovery using a reduced representation of RNA conformational space. Nucleic Acids
Research, 31, 4755–4761.
25. Wadley, L.M. and Pyle, A.M. (2004) The identification of novel RNA structural motifs using COMPADRES: an automated approach to structural discovery. Nucleic Acids Research, 32, 6650–6659.