行政院國家科學委員會專題研究計畫 期中進度報告
含雙陰離子
咯與酮胺配位基之有機金屬錯合物的合成、鑑
定、反應及應用(1/3)
計畫類別: 個別型計畫 計畫編號: NSC93-2113-M-018-005- 執行期間: 93 年 08 月 01 日至 94 年 07 月 31 日 執行單位: 國立彰化師範大學化學系暨研究所 計畫主持人: 黃瑞賢 報告類型: 精簡報告 報告附件: 出席國際會議研究心得報告及發表論文 處理方式: 本計畫可公開查詢中 華 民 國 94 年 4 月 14 日
含雙陰離子䥇咯與酮胺配位基之有機金屬錯合物的合成、鑑
定、反應及應用(1/3)
計畫編號 93-2113-M-018-005-
國立彰化師範大學化學系暨研究所 黃瑞賢
(含出國報告)
Synthesis, reactivity, and structures of dialuminum complexes containing linked
ketiminate ligands
Jui-Hsien Huang(黃瑞賢)
Department of Chemistry, National Changhua University of Education, Changhua, Taiwan 50058
Abstract
Linked bis(ketimine) (1) can be prepared with the reaction of excess 2,4-pentandione and 4,4’-methylene-bis(2,6-diisopropylaniline) in methanol with small amount of formic acid as catalysts. The dialuminum alkyl complexes containing the linked bis(ketiminate) dianionic ligands, [OC(Me)CHC(Me)N(2,6-iPr2C6H2-4)AlR2]2CH2 (2, R=Me; 3, R=Et), were prepared by a reaction of 2 equiv AlR3 with [OC(Me)CHC(Me)NH(2,6-iPr2C6H2-4)]2CH2 in methylene chloride. Reactions of 2 with 2 and 4 equiv of I2 gave corresponding aluminum iodo complexes 4 and 5, respectively. Treatment of 5 with 2 equiv of
AgBF4, however, gave a diboron complex, [OC(Me)CHC(Me)N(2,6-iPr2C6H2-4)BF2]2CH2 (6) in 18 % isolated yield. All new
complexes have been characterized by 1H and 13C NMR spectroscopy and complexes
1. Introduction
Dinuclear transition metal complexes are rather common and may be useful for acting as catalysts [1-3] or metalloenzymes. [4-6] Among those, dinuclear metal complexes with bis-multidentate ligands, which contain two anionic ancillary ligands linked by a bridge, have been studied [7-14] because of their potential usage as two-center Lewis acid catalysts. Among that, group 13 dimetallic complexes containing bis-bidentate ligands have also been reported by Atwood et al. [15-16] The bis-bidentate ligands used in the dialuminum chemistry are mainly of the Salen classes bridged with backbones such as 1,4-butylene, 1,4-phenylene, and 1,2-cyclohexylene etc. The studies of the aluminum complexes with mono anionic bidentate ketiminate ligands have been reported. [17] Here we report the syntheses and characterization of dialuminum complexes containing linked bis(ketiminate) ligands and their related reactions.
2. Results and Discussion
2.1. Synthesis and characterization
Linked bis(ketimine) (1) can be prepared easily by modifying the published procedure [18] with the reaction of excess (> 2 equiv) 2,4-pentandione and 4,4’-methylene-bis(2,6-diisopropylaniline) in methanol with small amount of formic
acid as catalysts (scheme 1). The excess 2,4-pentandione can be removed under vacuum at elevated temperature to generate a pure bis(ketimine) ligand 1 in high yield. 1H NMR spectrum of 1 reveals that the methylene protons between two phenyl rings
appear at δ 3.97; while the methine proton of the ketiminate backbone appears at
δ 5.18, which serve as an excellent indicator for evaluating the purity of the metal complexes.
Scheme 1 here
Treatment of 1 with two equiv of AlMe3 and AlEt3 in a methylene chloride solution generates the dialuminum complexes 2 and 3, respectively, in high yield (Scheme 2). The reactions were proceeding along with the elimination of two equiv of methane or ethane. The complexes have been characterized by 1H and 13C NMR spectra, and X-ray structure determination. For both complexes 2 and 3, 1H NMR spectra exhibit one methine resonance and two methyl resonances for the isopropyl fragments, which is consistent with fast ring inversion and slow aryl rotation. [17] Moreover, the 1H NMR spectra of the methine protons of the ketiminate backbones of
2 and 3 exhibit one singlet at δ 5.33 and 5.32,respectively. The AlMe2 fragments of
complex 2 appears as a singlet at δ−0.94 and the AlEt2 of complex 3 exhibits one set triplet and multiplet at δ −0.22 and 0.84 for the CH2 and CH3 fragments. These data indicate symmetric structures for both complex 2 and 3.
Scheme 2 here
Reactions of 2 with 2 and 4 equiv of iodine yield the expected aluminum methyl iodo complex 4 and aluminum diiodo complex 5, respectively (Scheme 2). Complexes 4 and 5 have been characterized by elemental analysis and 1H and 13C NMR spectroscopy. The methine proton resonances of ketiminate backbone for complexes 4 and 5 are shown as single resonance at δ 4.00 and 4.01, respectively. Complex 4 remains a C2 symmetry through the bridged CH2 fragment, however, the
isopropyl groups of the phenyl rings exhibit asymmetrical geometry due to the unsymmetrical coordination geometry of the aluminum center. These phenomena can be seen from the 13C NMR spectra showing more complicate phenyl and isopropyl resonances.
Attempts to remove the iodide of complex 5 to yield cationic aluminum center by adding AgBF4 have resulted an unexpected di-borane complex 6 with 18 % isolated yield (Scheme 3). 1H and 13C NMR spectra proved the existence of the linked ketiminate ligands and 11B NMR spectrum also indicate the presence of boron in complex 6. The 11B NMR spectrum exhibits a triplet at δ 0.67 relative to the standard of BF3⋅OEt2 showing the coupling of JBF = 15.4 Hz.
Scheme 3 here
A crystal structure determination of 2 confirms the bimetallic nature of the complex. The colorless crystals of 2 were obtained by cooling a saturated diethyl ether solution at −20 °C. The molecular structure of 2 and selected bond distances and angles are shown in Figure 1 and Table 1, respectively. As expected, both the aluminum atoms in complex 2 are surrounded by two methyl groups and one bidentate ketiminate ligand forming a tetrahedral geometry. The biting angles of ketiminate ligands, N(1)−Al(1)−O(1) (which is 94.58(17)°), is smaller than a regular tetrahedral bond angle of 109.28°. The backbone of ketiminate, O(1)−C(3)−C(4)−C(5)−N(1), forms a plane and the aluminum atom is deviated from the plane at 0.5821 Å. A large dihedral angle (98.5º) is observed between the phenyl ring and the ketiminate back bone plane, suggesting minimal steric interaction exists between the aluminum methyl groups and the phenyl rings in the solid state. In viewing half part of the molecular structure of 2, it is similar to that of (OCMeCHCMeNAr)AlMe2. [17]
Figure 1 and Table 1 here
Crystals of 3 were obtained from a concentrated diethyl ether solution at −20 °C. The molecular structure of 3 is shown in Figure 2 and selected bond distances and angles are listed in Table 1.
Figure 2 here
from sublimation of solids. Due to the small molecule size, some of the high angle reflection data were omitted. The molecular structure of 6 is shown in Figure 3 and selected bond distances and angles are listed in Table 1. Complex 6 is consistent with a C2 symmetry with two boron atoms located on the two ketiminate fragments.
Each boron atom is surrounded by two fluorine atoms and one ketiminate fragments forming a tetrahedral geometry. The two B−F bond distances (ca. 1.36 Å) are similar to reported bidentate ligand chelated BF2 complexes. [19-20] The ketiminate fragment chelates to the boron atom with an angle of 109.8(3), similar to the bond angles of tetrahedral geometry.
Figure 3 here
3. Conclusions
In conclusion, the synthesis of a linked bis(ketimine) ligand is described. Reactions of the bis(ketimine) with AlMe3 yield a new type of dialuminum alkyl complex 2, which can be converted to corresponding aluminum iodo complexes by adding various amount of I2. A diboron compound, [OC(Me)CHC(Me)N(2,6-iPr2C6H2-4)BF2]2CH2 (6), can be obtained from the reaction of 2 with 2 equiv of AgBF4 via metal exchange. A further study of aluminum hydride complexes containing this bis(ketiminate) ligands is currently undergoing.
4. Experimental Section
4.1. General Procedures.
All reactions were performed under a dry nitrogen atmosphere using standard Schlenk techniques or in a glove box. Toluene and diethyl ether were dried by refluxing over sodium benzophenone ketyl. CH2Cl2 was dried over P2O5. All solvents were distilled and stored in solvent reservoirs which contained 4Å molecular sieves and were purged with nitrogen. 1H and 13C NMR spectra were recorded on a Bruker AC 200 or Bruker Avance 300 spectrometer. Chemical shifts for 1H and 13C spectra were recorded in ppm relative to the residual protons and 13C of CDCl3 (δ 7.24, 77.0) and C6D6 (δ 7.15, 128.0). 11B NMR spectra were recorded on a Bruker Avance 300 M Hz spectrometer with BF3•OEt2 as reference at δ 0 ppm. Elemental analyses were performed on a Heraeus CHN-OS Rapid Elemental Analyzer at the Instrument Center, NCHU.
4.2. Synthesis of [OC(Me)CHC(Me)NH(2,6-iPr2C6H2-4)]2CH2 (1)
Excess 2, 4-pentanedione (6.0 g, 60.0 mmol) and 4,4’-methylenebis-(2,6-diisopropylaniline) (10 g, 27.3 mmol) were placed in a flask and dissolved in 50 mL methanol. Small amount of formic acid was added as
catalyst. The mixture was stirred at room temperature for 12 h and volatiles were removed under vacuum to generate 14.4 g pale yellow solids in 99.4% yield. 1H NMR (CDCl3): δ 1.07 (d, 12H, CHMe2), 1.14 (d, 12H, CHMe2), 1.62 (s, 6H, CMe), 2.09 (s, 6H, CMe), 2.95 (m, 4H, CHMe2), 3.97 (s, 2H, CH2), 5.18 (s, 2H,
CMeCHCMe), 6.93 (s, 4H, Ph), 11.92 (s, br, 2H, NH). 13C NMR (CDCl3): δ 19.1 (q, JCH = 128 Hz, CH3), 22.6 (q, JCH = 126 Hz, CH3), 24.4 (q, JCH = 127 Hz, CH3), 28.3 (d, JCH = 128 Hz, CHMe2), 28.6 (q, JCH = 126 Hz, CH3), 41.5 (t, JCH = 127 Hz, CH2), 95.6 (d, JCH = 161 Hz, CMeCHCMe), 124.0 (d, JCH = 155 Hz, phenyl CH), 131.2 (s, phenyl Cipso), 140.5 (s, phenyl Cipso), 145.9 (s, phenyl Cipso), 164.2 (s, CN), 195.8 (s,
CO). Anal. Calcd. for C35H50N2O2: C, 79.20; H, 9.49; N, 5.28. Found: C, 79.07; H,
9.91; N, 5.22.
4.3. Synthesis of [OC(Me)CHC(Me)N(2,6-iPr2C6H2-4)AlMe2]2CH2 (2)
A solution of trimethylaluminum (2M in toluene, 18.87 mL, 37.74 mmol) was added dropwise at 0 °C to a stirred solution of 1 (10.0 g, 18.87 mmol) in methylene chloride (70 mL). The resulting solution was stirred at room temperature for 5 h. Volatiles were removed under vacuum to generate 12.0 g of yellow solids in 98 % yield. Colorless crystals of 2, which are suitable for x-ray structure determination, can be obtained from a saturated diethyl ether solution. 1H NMR (CDCl3): −0.94 (s, 12H, AlMe2), 1.03 (d, 12H, CHMe2), 1.12 (d, 12H, CHMe2), 1.75 (s, 6H, CMe), 2.07 (s, 6H,
CMe), 2.87 (m, 4H, CHMe2), 3.99 (s, 2H, CH2), 5.33 (s, 2H, CMeCHCMe), 6.90 (s,
4H, Ph). 13C NMR (CDCl3): −11.1 (q, JCH = 112 Hz, AlMe2), 23.1 (q, JCH = 129 Hz,
CMe), 24.4 (q, JCH = 129 Hz, CHMe), 24.6 (q, JCH = 129 Hz, CHMe), 25.8 (q, JCH = 128 Hz, CMe), 27.9 (d, JCH = 132 Hz, CHMe2), 41.0 (t, JCH = 130 Hz, CH2), 100.5 (d, JCH = 162 Hz, CMeCHCMe), 124.9 (d, JCH = 154 Hz, phenyl CH), 136.9 (s, phenyl
Cipso), 139.4 (s, phenyl Cipso), 142.7 (s, phenyl Cipso), 176.3 (s, CN), 180.7 (s, CO).
Anal. Calcd. for C39H60N2O2Al2: C, 72.86; H, 9.41; N, 4.36. Found: C, 72.19; H, 9.04; N, 4.27.
4.4. Synthesis of [OC(Me)CHC(Me)N(2,6-iPr2C6H2-4)AlEt2]2CH2 (3)
A solution of triethylaluminum (1.9 M in toluene, 9.93 mL, 18.87 mmol) was added dropwise at 0 °C to a stirred solution of 1 (5.0 g, 9.43 mmol) in methylene chloride (50 mL). The resulting solution was stirred at room temperature for 12 h. Volatiles were removed under vacuum to generate yellow solids; which were recrystallized from a diethyl ether solution to yield 6.45 g pale yellow solids in 98 % yield. 1H NMR (CDCl3): −0.23 (m, 8H, AlCH2CH3), 0.84 (t, 12H, AlCH2CH3 ), 1.05 (d, 12H,
CHMe2), 1.16 (d, 12H, CHMe2), 1.75 (s, 6H, CMe), 2.09 (s, 6H, CMe), 2.89 (m, 4H, CHMe2), 3.99(s, 2H, CH2), 5.31 (s, 2H, CMeCHCMe), 6.93 (s, 4H, Ph). 13C NMR
(CDCl3): δ −1.1 (t, JCH = 111 Hz, AlCH2CH3), 8.9 (qt, JCH = 124 Hz, 2JCH = 5 Hz, AlCH2CH3), 23.0 (q, JCH = 129 Hz, CMe), 24.2 (q, JCH = 126 Hz, CHMe), 24.6 (q, JCH
= 126 Hz, CHMe), 25.7 (q, JCH = 128 Hz, CMe), 27.8 (d, JCH = 129 Hz, CHMe), 41.1 (t, JCH = 127 Hz, CH2), 100.6 (d, JCH = 162 Hz, CMeCHCMe), 124.9 (d, JCH = 154 Hz, phenyl CH), 137.1 (s, phenyl Cipso), 139.5 (s, phenyl Cipso), 142.7 (s, phenyl Cipso),
176.4 (s, CN), 181.2 (s, CO).
4.5. Synthesis of [OC(Me)CHC(Me)N(2,6-iPr2C6H2-4)AlIMe]2CH2 (4)
To a 50 mL Schlenk flask containing 1 (3.6 g, 5.6 mmol) was added 20 mL toluene and iodine (2.9 g, 11.4 mmol) and heated at 100 °C for 48 h. Volatiles were removed under vacuum and the resulting yellow-brown solid was recrystallized from toluene to yield 4.50 g of yellow brown solids (yield 93%). 1H NMR (CDCl3): δ −0.55 (s, 6H, AlMe), 0.97 (d, 6H, CHMe2), 1.07~1.17 (m, 18H, CHMe2), 1.84 (s, 6H, CMe), 2.14 (s, 6H, CMe), 2.66 (m, 2H, CHMe2), 3.25 (m, 2H, CHMe2), 4.00 (s, 2H, CH2), 5.65 (s,
2H, CMeCHCMe), 6.90 (s, 2H, Ph), 6.97 (s, 2H, Ph). 13C NMR (CDCl3): δ –7.5 (q, JCH = 116 Hz, AlMe2), 23.2 (q, JCH = 128 Hz, CHMe2), 23.4 (q, JCH = 128 Hz, CMe),
24.1 (q, JCH = 128 Hz, CHMe2), 24.7 (q, JCH = 126 Hz, CHMe2), 25.7 (q, JCH = 128 Hz,
CMe), 26.8 (q, JCH = 126 Hz, CHMe2), 28.0 (d, JCH = 128 Hz, CHMe2), 28.4 (d, JCH =
128 Hz, CHMe2), 40.9 (t, JCH = 127 Hz, CH2), 102.6 (d, JCH = 163 Hz, CMeCHCMe), 124.9 (d, JCH = 155 Hz, phenyl CH), 125.4 (d, JCH = 155 Hz, phenyl CH), 135.4 (s, phenyl Cipso), 140.0 (s, phenyl Cipso), 142.5 (s, phenyl Cipso), 143.7 (s, phenyl Cipso),
N, 3.23. Found: C, 49.94; H, 6.12; N, 3.42.
4.6. Synthesis of [OC(Me)CHC(Me)N(2,6-iPr2C6H2-4)AlI2]2CH2 (5)
By a similar procedure, the reaction of 2 (3.6 g, 5.6 mmol) with I2 (5.8 g, 22.8 mmol) in toluene gave brown solids 4 in 98 % yield (6.0 g). 1H NMR (CDCl3): δ 1.04 (d, 12H, CHMe2), 1.21 (d, 12H, CHMe2), 1.91 (s, 6H, CMe), 2.20 (s, 6H, CMe), 2.95 (m, 2H, CHMe2), 4.01 (s, 2H, CH2), 5.75 (s, 2H, CMeCHCMe), 6.96 (s, 4H, Ph). 13C
NMR (CDCl3): δ 24.5 (q, JCH = 126 Hz, CMe), 24.6 (q, JCH = 126 Hz, CHMe), 25.4 (q, JCH = 127 Hz, CHMe), 25.8 (q, JCH = 129 Hz, CMe), 28.7 (d, JCH = 128 Hz, CHMe2), 41.1 (t, JCH = 130 Hz, CH2), 103.3 (d, JCH = 162 Hz, CMeCHCMe), 125.5 (d, JCH = 154 Hz, phenyl CH), 135.1 (s, phenyl Cipso), 140.6 (s, phenyl Cipso), 143.3 (s, phenyl
Cipso), 180.3 (s, CN), 182.5 (s, CO). Anal. Calcd. For C35H48N2O2Al2I4: C, 38.56; H,
4.44; N, 2.57. Found: C, 38.10; H, 4.38; N, 2.43.
4.7. Synthesis of [OC(Me)CHC(Me)N(2,6-iPr2C6H2-4)BF2]2CH2 (6)
To a 50 mL Schlenk flask containing 4 (0.5 g, 0.46 mmol) and AgBF4 (0.18 g, 0.92 mmol) was added 20 mL toluene and heated at 90 °C for 8 h. The solution was filtered through Celite and volatiles were removed under vacuum. The resulting solids were recrystallized from a toluene solution to generate 0.050 g of yellow solids in 18% yield. 1H NMR (CDCl3): δ 1.07 (d, 12H, CHMe2), 1.20 (d, 12H, CHMe2), 1.82 (m, THF), 1.88 (s, 6H, CMe), 2.19 (s, 6H, CMe), 2.80 (m, 2H, CHMe2), 3.72 (m,
THF), 3.96 (s, 2H, CH2), 5.56 (s, 2H, CMeCHCMe), 7.02 (s, 4H, Ph). 13C NMR
(CDCl3): δ 21.2 (q, JCH = 129 Hz, CMe), 22.8 (q, JCH = 130 Hz, CMe), 24.2 (q, JCH = 132 Hz, CHMe), 24.5 (q, JCH = 126 Hz, CHMe), 28.3 (d, JCH = 131 Hz, CHMe2), 41.6 (t, JCH = 132 Hz, CH2), 67.9 (t, THF), 98.7 (d, JCH = 168 Hz, CMeCHCMe), 124.9 (d, JCH = 153 Hz, phenyl CH), 132.5 (s, phenyl Cipso), 140.8 (s, phenyl Cipso), 144.6 (s,
phenyl Cipso), 172.9 (s, CN), 176.8 (s, CO). 11B NMR (CDCl3): δ 0.67 (t, JBF = 15.4
Hz). MS (FAB): 607 (M−F), 320 (M−306)
4.8. X-Ray structure determination of complexes 2, 3, and 6.
Crystals of complex 2 were obtained from a concentrated diethyl ether solution of 2 at
−20 °C. Crystals of 6 were obtained directly from the sublimation of viscous complex 6 by storing it in glove box. Crystals of 2 and 6 were mounted on a goniostat and data collections were preceded at 150(2) K and data of crystal of 3 were collected at 298(2) K. Data were collected on a Bruker SMART CCD diffractometer with graphite-monochromated Mo Kα radiation. Structural determinations were
made using the SHELXTL package of programs. All refinements were carried out by full-matrix least squares using anisotropic displacement parameters for all non-hydrogen atoms. All the hydrogen atoms are calculated. The crystal data are summarized in Table 2.
Acknowledgments
We thank the National Science Council of Taiwan for financial support and the National Center for High Performance Computing for databank search.
References
1. C. P. Casey, J. D. Audett, Chem. Rev. 86 (1986) 339−352.
2. J. Holton, M. F. Lappert, R. Pearce, P. I. W. Yarrow, Chem. Rev. 83 (1983) 135−201.
3. D. Zhang, G. –X. Jin, Organometallics 22 (2003) 2851−2854.
4. N. H. Williams, B. Takasaki, M. Wall, J. Chin, Acc. Chem. Res. 32 (1999) 485−493.
5. B. J. Wallar, J. D. Lipscomb, Chem. Rev. 96 (1996) 2625−2657. 6. B. Bosnich, Inorg. Chem. 38 (1999) 2554−2562.
7. S. A. Schuetz, V. W. Day, A. L.Rheingold, J. A.Belot, J. Chem. Soc. Dalton Trans. (2003) 4303−4306.
8. Y. Kasahara, Y. Hoshino, M. Kajitani, K. Shimizu, G. P. Sato, Organometallics 11 (1992) 1968−1971.
9. Z. Li, C. Jablonski, Inorg. Chem. 39 (2000) 2456−2461.
40 (2001) 1386−1390.
11. P. E. Kruger, N. Martin, M.Nieuwenhuyzen, J. Chem. Soc. Dalton Trans. (2001) 1966−1970.
12. N. Yoshida, H. Oshio, T. Ito, Chem. Commun. (1998) 63−64. 13. N. Yoshida, K. Ichikawa, Chem. Commun. (1997) 1091−1092. 14. D. A. Atwood, M. J. Harvey, Chem. Rev. 101 (2001) 37−52. 15. M. S. Hill, P. Wei, D. A. Atwood, Polyhedron 17 (1998) 811−819.
16. M. A. van Aelstyn, T. S. Keizer, D. L. Klopotek, S. Liu, M. –A. Munoz-Hernandez, P. Wei, D. A. Atwood, Organometallics 19 (2000) 1796−1801.
17. R. –C. Yu, C. –H. Hung, J. –H. Huang, H. –Y. Lee, J. –T. Chen, Inorg. Chem. 41 (2002) 6450–6455.
18. J. E. Parks, R. H. Holm, Inorg. Chem. 7 (1968) 1408−1416.
19. J. Chen, A. Burghart, C. –W. Wan, L. Thai, C. Ortiz, J. Reibenspies, K. Burgess, Tetrahedron Lett. 41 (2000) 2303–2307.
20. J. Chen, A. Burghart, A. Derecskei-Kovacs, K. Burgess, J. Org. Chem. 65 (2000) 2900-2906.
Scheme 1. O N O N H H NH2 H2N O O 2 + 1 formic acid
Scheme 2 O N O N Al Al Me Me Me Me O N O N Al Al I I I I O N O N Al Al I Me Me I 2 AlMe3 4 I2 2 I2 1 2 4 5 O N O N Al Al Et Et Et Et 3 2 AlEt3
Scheme 3 O N O N Al Al I I I I O N O N B B F F F F 2 AgBF4 5 6 O N O N Al Al I I [BF4]2 2 AgBF4
Captions to figures
Figure 1. The molecular structure of complex 2. Thermal ellipsoids were drawn at 30 % probability and all hydrogen atoms were omitted for clarity.
Figure 2. The molecular structure of complex 3. Thermal ellipsoids were drawn at 30 % probability and all hydrogen atoms were omitted for clarity.
Figure 3. The molecular structure of complex 6. Thermal ellipsoids were drawn at 30 % probability and all hydrogen atoms were omitted for clarity.
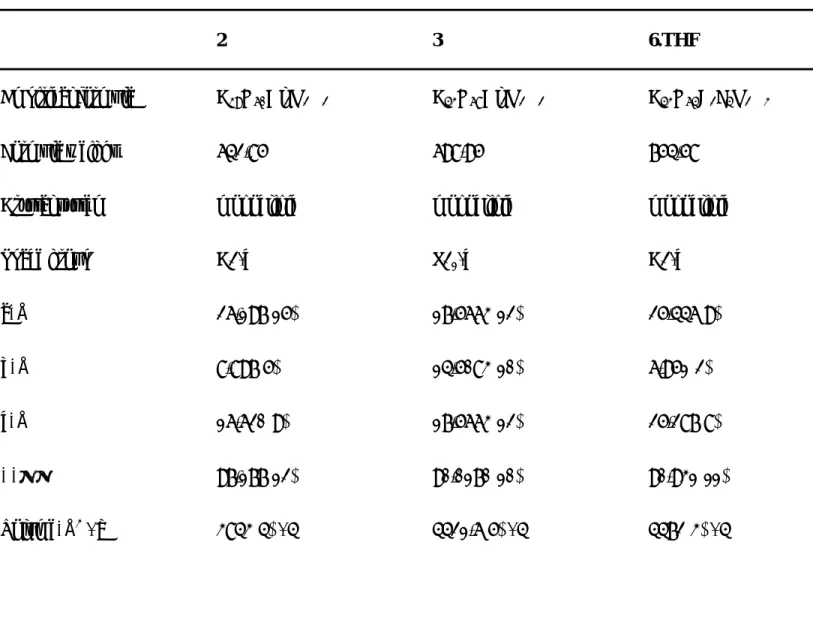
Table 1. Selected bond distances and angles of complexes 2, 3, and 6
2
Al(1)−O(1) 1.784(4) Al(1)−N(1) 1.934(4) Al(1)−C(1) 1.972(6) Al(1)−C(2) 1.937(5) O(1)−C(4) 1.305(5) N(1)−C(6) 1.334(5) C(3)−C(4) 1.511(6) C(4)−C(5) 1.355(6) O(1)−Al(1)−N(1) 94.58(17) O(1)−Al(1)−C(2) 111.8(2) N(1)−Al(1)−C(2) 111.6(2) O(1)−Al(1)−C(1) 107.9(2) N(1)−Al(1)−C(1) 111.3(2) C(2)−Al(1)−C(1) 117.3(2)
3
Al(1)−O(1) 1.7990(18) Al(1)−N(1) 1.9356(19) Al(1)−C(8) 1.955(3) Al(1)−C(6) 1.962(3) Al(2)−O(2) 1.7957(18) Al(2)−N(2) 1.9316(19) Al(2)−C(40) 1.948(3) Al(2)−C(42) 1.964(3) N(1)−C(4) 1.317(3) N(1)−C(10) 1.463(3) N(2)−C(36) 1.316(3) N(2)−C(26) 1.453(2) O(1)−C(2) 1.303(3) O(2)−C(38) 1.303(3) C(2)−C(3) 1.354(3) C(3)−C(4) 1.427(3) C(37)−C(38) 1.361(3) C(36)−C(37) 1.423(3)
O(1)−Al(1)−N(1) 93.26(8) O(1)−Al(1)−C(8) 111.53(10) N(1)−Al(1)−C(8) 113.03(11) O(1)−Al(1)−C(6) 107.63(11) N(1)−Al(1)−C(6) 112.21(10) C(8)−Al(1)−C(6) 116.62(13) O(2)−Al(2)−N(2) 94.26(8) O(2)−Al(2)−C(40) 109.20(13) N(2)−Al(2)−C(40) 114.77(11) O(2)−Al(2)−C(42) 118.39(15) N(2)−Al(2)−C(42) 108.83(11) C(40)−Al(2)−C(42) 118.39(15) 6 B−O(1) 1.462(5) B−N(1) 1.572(5) B−F(1) 1.367(5) B−F(2) 1.365(5) N(1)−C(4) 1.313(4) O(1)−C(2) 1.311(5) C(2)−C(3) 1.344(6) C(3)−C(4) 1.412(5) O(1)−B−N(1) 109.8(3) F(2)−B−O(1) 110.0(3) F(2)−B−N(1) 109.6(3) F(1)−B−O(1) 108.6(3) F(1)−B−N(1) 110.0(3) F(2)−B−F(1) 108.9(3)
Table 2. The summary of crystallographic data for complexes 2, 3, and 6.THF
2 3 6.THF
Empirical formula C39H60Al2N2O2 C43H68Al2N2O2 C43H64B2F4N2O3
Formula weight 642.85 698.95 754.58
Crystal system monoclinic monoclinic monoclinic
Space group C2/c P21/c C2/c a, Å 26.197(15) 17.5663(12) 25.446(9) b, Å 8.897(5) 14.5083(10) 6.951(2) c, Å 16.620(9) 17.5663(12) 25.287(8) β, ° 97.177(12) 90.0170(10) 90.931(11) Volume, Å3/ Z 3843(4) / 4 4421.6(5) / 4 4472(3) / 4
Density (cald.), Mg/m3 1.111 1.050 1.121
Absorption coefficient 0.109 mm-1 0.099 0.080 mm-1
F(000) 1400 1528 1624
Crystal size 0.33 x 0.29 x 0.26 mm 0.65 x 0.53 x 0.45 mm 0.31 x 0.77 x 0.26 mm
θ range for data collection 2.42 to 27.65° 1.17 to 27.54° 2.25 to 24.00°
Reflections collected 11858 27426 11394
Independent reflections 4398 (Rint = 0.1669) 11095 (Rint = 0.0673) 3490 (Rint = 0.0952)
Max. and min. transmission 0.9486 and 0.5667
Data / restraints / parameters 4398 / 01/ 208 10095 / 01/ 458 3490 / 0 / 249
Goodness-of-fit on F2 0.675 0.729 1.009
R indices (all data) R1=0.2637, wR2=0.1933 R1=0.1304, wR2=0.1359 R1=0.1168, wR2=0.2626
出席第 228 次美國化學會全國大會會議報告 報告人:彰化師大化學系黃瑞賢
第228 次美國化學會全國大會於 2004 年八月 22-26 日於美國東岸的費城舉 行。本人於台北時間八月20 日星期五從台北出發,並於八月 21 日星期六到達費 城。本次所發表之論文“Zirconium and Hafnium Complexes Bearing Substituted Pyrrolyl Ligands. Synthesis, Characterization, and Ring-Opening Polymerization of Lactide and ε-Caprolactone”是以壁報方式呈現。本論文於會議中共報告兩次,第 一次是在八月 22 日(星期日)晚上七至十時的無機組論文發表會,第二次是在八 月23 日晚上七至十時(星期一)的 SCI-MIX 論文發表會。在會議期間除論文發表 外,亦利用有限的時間盡量聽取其他頂尖科學家的最新結果。 會議於八月 26 日星期四結束,本人於八月 27 日星期五啟程返台並於八月 28 日星期六深夜到達。綜觀本次會議,除成功報告所發表之論文,亦獲得許多 新的知識及靈感,對於研究有莫大的幫助。