Electron Transfer-Induced Hydrogenation of Anthracene Catalyzed by Gold and Silver
Nanoparticles
Jin-Pei Deng,†,‡Wen-Chen Shih,† and Chung-Yuan Mou*,†
Department of Chemistry, National Taiwan UniVersity, Taipei, Taiwan 106, and Department of Chemistry, Tamkang UniVersity, Taipei, Taiwan
ReceiVed: December 29, 2006; In Final Form: May 2, 2007
Anthracene hydrogenation in aqueous micellar solutions is catalyzed at room temperature by ionic-surfactant-protected gold and silver nanoparticles (NPs) with well-controlled particle sizes. When sodium borohydride was used as the hydrogen source in the presence of these NPs, the only product obtained in the reaction is 9,10-dihydroanthracene. During the course of the reaction, a blue shift in the surface plasmon resonance band of silver NPs was more prominent and lasted longer than that of gold NPs. The difference in the optical property of metal NPs was related to the charging of their surfaces, indicating that the metal NPs play a role as the nanoelectrode storing electrons from hydrides. At the same time, a remarkable size-dependence of catalytic activity was found for both gold and silver NPs. Mechanism about the electron transfer-relaying effects of metal NPs is proposed for the hydrogenation reaction.
Introduction
Noble metal nanoparticles (NPs), such as gold and silver, are interesting catalytic nanomaterials because the peculiar activities are strongly related to their size, shape and surface charge.1-3Many applications of the metallic NPs in catalysis4,5
and electrocatalysis6-8 have been found. The metal NPs with
high surface-to-volume ratio may be considered as one of the nanoelectrodes which carry and transfer electrons in the elec-trocatalysis. For supported metal NPs, the catalytic activity at metal surfaces is related to both the electronic structure of the metallic surface and the interaction between metal NPs and the support.4 For example, size-sensitive catalysis by gold NPs
deposited on metal oxides has been widely studied for the CO oxidation.5The catalytic activity depends not only on the particle
size of gold NPs, but also on the nature of the support and the preparation method of the catalyst. Because the catalytic activity is related to both the electronic structure of the metallic surface and the interaction between gold NPs and the support, it is difficult to probe the influence of their intrinsic and size-dependent electronic effect on the catalytic activity for supported catalysts.
On the other hand, water-dispersed “naked” gold NP exhibits surprising catalytic activities. Recently, we reported our pre-liminary results on the hydrogenation of anthracene catalyzed by the gold NPs in aqueous micellar solutions at room temperature.9A remarkable non-monotonic size dependence of
catalytic activity was found. The particle sizes of those gold NPs may be controlled by the surfactants used. Both hexade-cyltrimethylammonium bromide (CTAB, a cationic surfactant) and sodium dodecyl sulfate (SDS, an anionic surfactant) were employed to prepare gold NPs with different sizes. In such colloid system, the gold NPs are free of electronic interaction with supports. Therefore, a surfactant-protected metallic colloid system with a solubilized reactant such as anthracene seems to
be an ideal model system to probe the influence of its intrinsic size-dependent property on metal-catalyzed solution reactions due to the absence of any support interaction. Some works on reduction of organic dyes and aromatic nitro compounds catalyzed by metal NPs have been reported recently.10However,
our choice of the anthracene and noble metals offer the advantage of simplicity such that detailed understanding of mechanism may be attempted.
In our preliminary communication on gold NP system,9the
catalysis of gold NPs on the hydrogenation showed high activities at particle sizes below 10 nm. However, the size for optimum activity was not determined. In this paper, the size-dependent catalytic activity of gold NPs of 3-5 nm will be studied in details. The origin of the unique size-dependence of activity was not understood on gold NP system in our previous work.9Partially, this is because the reaction rate in gold NP
system was too rapid to be studied closely. So we would also like to study the catalytic reaction with silver colloids which allows better characterization due to its slower rate. Thus, we also report the hydrogenation of anthracene catalyzed by size-controlled silver NPs in aqueous SDS micellar solution in this paper and compare their activities with gold NPs at the same time. To match the opposite charges of silver ion (Ag+from AgNO3) and surfactant ions, SDS was used to prepare silver
NPs in smaller size. In addition to UV/vis spectroscopy, we employed micellar electrokinetic capillary chromatography (MEKC), a powerful hybrid technique of electrophoresis and chromatography,11to monitor the hydrogenation reaction. The
size-dependent hydrogenation activity on silver NPs was investigated. The differences in catalytic activities between silver NPs and gold NPs were observed. A hydrogenation mechanism is proposed and the role of the metal NPs as nanoelectrodes for electron transfer is discussed.
Experimental Methods
Materials. Sodium borohydride and sodium tetraborate was obtained from Alfa Aesar and Sigma, respectively. All other reagents were purchased from Acros and used as supplied.
* Corresponding author. E-mail: [email protected].
†National Taiwan University. ‡Tamkang University.
10.1021/jp0690042 CCC: $37.00 © 2007 American Chemical Society Published on Web 06/19/2007
Downloaded by NATIONAL TAIWAN UNIV on August 11, 2009
Gold and Silver NPs. The concentrations of HAuCl4 and
AgNO3solutions were used at 25 mM. For SDS-protected gold
NPs, 0.02 mL HAuCl4 was added to 5 mL of 20 mM SDS
solution. Then 0.02 mL of 90 mM NaBH4solution was added.
After the addition of NaBH4, the solution turned pink. The
measurement of gold NPs diameter was performed with the transmission electron microscopy (TEM). The average diameter of gold NPs was 4.1 ( 1 nm. Similar procedures (0.02 mL HAuCl4and 0.03 mL, 45 mM NaBH4) were prepared for the
other gold NPs sample (4.8 ( 1 nm). For SDS-protected silver NPs, 0.09 mL AgNO3in a 5 mL of 10 mM SDS was reduced
with 0.18 mL NaBH4(11 mM). After the addition of NaBH4,
the solution turned yellow. The average diameter of silver NPs was 3.2 ( 0.8 nm. The preparation of another two samples was the same as mentioned above: 0.27 and 0.54 mL AgNO3were
added to 0.54 and 1.08 mL NaBH4(11 mM), respectively. The
average diameters of the two silver NPs were 3.5 ( 1 and 4.4 ( 0.7 nm. Then, all gold and silver NPs-containing solutions were separately mixed with 2.5 mL anthracene (0.1 mM) -containing SDS solution (0.1 M). After stirring, 0.05 mL of 22.5 mM NaBH4solution was separately added to the above
solutions and their absorption spectra were recorded. For all Ag NP systems, the final reaction solutions were further analyzed by MEKC.
For CTAB-protected gold NPs, 0.2 mL HAuCl4solution was
added to 10 mL of 0.1 M CTAB. Then, 0.9 mL of an aqueous 11 mM NaBH4solution was added. After the addition of NaBH4,
the solution turned brown. The average diameter of gold NPs was 3.3 ( 0.8 nm by using TEM. Then, the solution was mixed with 0.15 mL anthracene (0.3 mM)-containing CTAB solution (0.1M). After stirring, 0.01 mL of 11 mM NaBH4solution was
added to the above solutions and their absorption spectra were recorded. For the preparation of the grown gold NPs in the CTAB solution, 9 mL HAuCl4was added to 300 mL of 5 mM
CTAB and followed by 27 mL of 11 mM NaBH4. Then, the
resulting solution was heated at 80°C for 24 h.
Instruments and Methods. The absorption spectra were recorded on the Hitachi U-3310 spectrophotometer. A drop of the NPs solution was placed on a carbon-coated copper grid for TEM experiments. Specimens were examined on a Jeol JSM-1200EX II and JEM-2010 TEM operating at 80 and 200kV. All the capillary electrophoresis experiments were carried out on the P/ACE-MDQ Capillary Electrophoresis System (Beck-man Coulter Inc., Fullerton, CA) with photodiode-array detector. The 100-µm-i.d. and 365-µm-o.d. fused-silica capillary (Polymi-cro Technologies, Phoenix, AZ) with 10-cm effective length was rinsed with H2O and 1M NaOH, and finally filled with the
separation buffer. The separation buffer is 20 mM SDS solution containing 10 mM sodium tetraborate and 10 mM NaH2PO4.
The sample was hydrodynamically injected into the anodic end of the capillary for 5 s at 0.5 psi. Separations were performed by applying an electric field strength of 300 V/cm, the capillary was maintained at 25°C. The turnover frequency (TOF) data were calculated on the basis of metal atoms exposed on the surface, which were obtained from the average particle sizes by assuming metal particles of spherical shape.
Results and Discussion
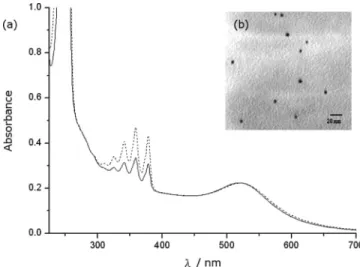
First, we demonstrate that gold NP has a high catalytic activity toward the hydrogenation of anthracene. Figure 1a shows the UV/vis spectrum of the reaction solution containing anthracene and the gold NPs prepared in the SDS solution (dash line). The surface plasmon resonance (SPR) band of gold NPs was observed at 520 nm wavelength. The average diameter of gold
NPs is determined by TEM to be 4.1 ( 1 nm (Figure 1b). To study the catalytic hydrogenation by gold NPs, NaBH4 was
added to the above solution. Figure 1a shows the UV/vis spectrum of the reaction solution 3 min after the addition of NaBH4(solid line). A conspicuous decrease in the intensity of
absorption peaks of anthracene at 359 nm was observed. In the control experiment without gold NPs, the absorption of an-thracene remained unchanged when NaBH4was added to the
anthracene-containing SDS solution. On the other hand, it is necessary to consider the effect of H2on the reaction because
it could be produced when NaBH4was added to water. When
the SDS reaction solution containing anthracene and gold NPs was stirred under an atmosphere of H2, the decrease in the
intensity of absorption of anthracene was much slower (several days). Because of the substantial difference in the reaction rate between the two reductants, it suggests that the influence of H2, produced from NaBH4, on the reaction was insignificant.
The above results indicate that both gold NPs and NaBH4play
important roles on the catalytic reaction. In additional to anthracene, similar reactions catalyzed by gold NPs have been observed for anthracene derivatives such as 9-phenylanthracene and 1,2-benzanthracene.
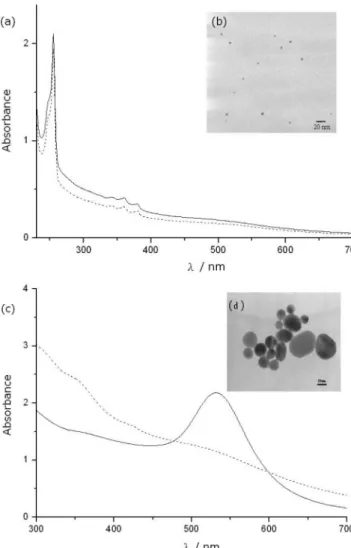
Figure 2a shows the UV/vis spectrum of the reaction solution containing anthracene and the gold NPs prepared in the CTAB solution (dash line). The SPR band of gold NPs is much broader, indicating a smaller mean particle size. The average diameter of gold NPs in Figure 2a is 3.3 ( 0.8 nm, as shown in the TEM micrograph of Figure 2b. When NaBH4was introduced
to the above-described solution, no apparent decrease in the absorbance of anthracene was observed (solid line in Figure 2a). The result indicates that the catalytic activity of those gold NPs prepared in the CTAB solution was much lower than that of the gold NPs in SDS solutions. Further gentle heating was employed to enhance the growth of the gold NP prepared in the CTAB solution. Figure 2c shows that the SPR band of gold NPs (dash line) becomes sharper (solid line) after heating at 80 °C for 24 h, indicating the formation of bigger gold NPs. The size was also confirmed by their TEM images shown in Figure 2d. When the above grown gold NPs were mixed with anthracene followed by the addition of NaBH4, the decrease in
the absorption of anthracene was observed easily. Because the total surface area of the smaller gold NPs originally prepared in the CTAB solution is larger than that of the grown gold NPs Figure 1. (a) Absorption spectra of the gold NPs-containing SDS solution with solubilized anthracene before (dash line) and after (solid line) the addition of NaBH4. (b) TEM image of gold NPs in (a) obtained
through NaBH4reduction of HAuCl4in the SDS solution.
Downloaded by NATIONAL TAIWAN UNIV on August 11, 2009
after heating, it suggests that the size of gold NP, not surface area, is an important factor in its catalytic activity.
Figure 3 is the 1H NMR spectrum of the product in the
CTAB-D2O reaction solution where an excess of NaBH4was
added to the solution of anthracene and gold NPs. The two peaks at 7.4 and 7.3 ppm are assigned to aromatic protons and another singlet peak is observed at 4.0 ppm. The integral ratio of the three peaks is 1:1:1. Thus, 9,10-dihydroanthracene is the sole product of anthracene hydrogenation catalyzed by gold NPs. Anthracene conversion could be estimated by measuring the absorbance of the reaction at 359 nm. Further, it was shown that the hydrogenation reaction was completed in 10 s. Figure
4 gives the TOFs of the hydrogenation catalyzed by the gold NPs of various sizes (TOFs for 5.4, 10.8 and 24.7 nm were obtained by recalculating the data in the previous work9). The
gold NP with the smallest mean size of 3.3 nm has the lowest activity among them. A dramatic change in catalytic activity appears for gold NPs with the size for optimum activity between 3 and 4 nm. This optimum size of 3-4 nm appears to be quite close to that of supported gold NP for CO oxidation.5On the
other hand, little changes in the activity of gold NPs were observed after several runs of reduction. It indicates that the colloidal gold NPs preserved their catalytic activity and were re-usable.9
Next, we turn our attention to silver NP. We prepare SDS-protected silver NPs with the same procedure used for gold NPs. Figure 5a shows the UV/vis spectrum of the reaction solution containing silver NPs and anthracene (solid line). The resulting yellow solution with SPR band at 397 nm indicates the formation of silver NPs. The average diameter of these silver NPs in Figure 5a was 3.2 ( 0.8 nm (Figure 5b). When NaBH4
was added to the above solution, a blue shift of the SPR band with enhanced absorbance and narrowed peak width was observed immediately (Figure 5a, dash line). The shifted peak Figure 2. (a) Absorption spectra of the gold NPs-containing CTAB
solution with solubilized anthracene before (dash line) and after (solid line) the addition of NaBH4. (b) TEM image of gold NPs in panel a
obtained through NaBH4reduction of HAuCl4in the CTAB solution.
(c) Absorption spectra of gold NPs prepared in the CTAB solution before (dash line) and after (solid line) heating at 80°C for 24 h. (d) TEM image of gold NPs in panel c after heating.
Figure 3. 1H NMR spectrum of the product-containing CTAB solution
after reduction with an excess of NaBH4.
Figure 4. Anthracene hydrogenation rates over gold NPs with different sizes.
Figure 5. (a) Successive absorption spectra of the silver NPs-containing solution with solubilized anthracene before (solid line) and after (dash line, 10 min; dot line, 1 h; dash-dot line, 4 h; dash-dot-dot line, 16 h) the addition of NaBH4. (b) TEM image of silver NPs in panel a obtained
through NaBH4reduction of AgNO3in the aqueous SDS solution.
Downloaded by NATIONAL TAIWAN UNIV on August 11, 2009
gradually relaxed toward the original position and intensity over a period about 16 h (Figure 5a, dashed-dotted-dotted line).
1H NMR shows conversion to the product of
9,10-dihroan-thracene. The result showed that anthracene hydrogenation can also be catalyzed by silver NPs.
Because the blue-shifted SPR band of silver NPs overlapped with the absorption peak of anthracene at 359 nm, anthracene conversion could not be directly estimated by the absorbance of its characteristic peak as in the case of gold NPs. Moreover, it was difficult to judge whether the hydrogenation reaction was completed or not. To resolve the problem, we employed MEKC to separate silver NPs from anthracene in the reaction solution. Figure 6 shows the electropherograms, detected at three wavelengths (210, 254, and 405 nm), of the reaction solution 10 min after adding NaBH4. Because the SPR absorption of
silver NPs is near 405 nm, the peak at 12.3 min is assigned to the SDS-protected silver NPs. The absorption of 9,10-dihy-droanthracene is much stronger at 210 nm than that at 254 nm, so the two peaks at 19.8 and 19.2 min are assigned to anthracene and 9,10-dihydroanthracene solubilized in the SDS micelle, respectively. The result shows that silver NPs and anthracene could be successfully separated by MEKC. Anthracene conver-sion could then be estimated by the integrated peak areas at 254 nm because the absorption of anthracene is very high. In the case of Figure 5a, anthracene conversion stops at 42.2%, 10 min after adding NaBH4. Moreover, the progress (or
conversion) of the reaction could be monitored by MEKC. It is interesting to observe the residual amount of anthracene remained unchanged at three different times (10 min, 1 and 4 h later) after adding NaBH4. The observation indicates that
anthracene hydrogenation catalyzed by silver NPs are completed quickly, although the blue shift of the SPR band of silver NPs could last for several hours. Furthermore, two systems of silver NPs with mean diameters of 3.5 ( 1 and 4.4 ( 0.7 nm were prepared with the same procedure to study the size effect of silver NP on catalytic hydrogenation. Figure 7 displays the TOFs of the hydrogenation catalyzed by the silver NPs with different sizes. In the calculation of TOFs for silver NP, we use the same periods of reaction time as gold. The particle with the smallest mean size has the highest activity among them. On the other hand, when the amount of silver NPs in the solution was reduced to∼20% of the original amount of the catalyst, the blue shift of spectrum is still observable but it is surprising that the anthracene conversion suddenly dropped to nearly zero when adding the same amount of NaBH4. The conversion depends
not only strongly on the size of metal NPs, but also the amounts of both the catalyst and the reducing reagent.
It is known that the optical property of silver NPs is influenced by the degree of charging of their surfaces.12-14
Henglein et al. used silver NPs to store electrons from radicals and a blue shift of the SPR band was observed.12 Further,
Mulvaney et al. transferred electrons to silver NPs from the electrode to prevent the ionic adsorption on the surface of silver NPs.13The blue shift of SPR band was found when the electrode
potential was maintained at a value more negative than the open circuit potential. The shift of SPR band depended on the applied potential and was reversible. At the same time, addition of NaBH4resulted in an increase of the electrophoretic mobility
of the negatively charged silver NP. Early on, the same phenomenon of blue shift had been also observed in Mulvaney’s work.14In a control MEKC experiment, NaBH
4was added to
the SDS solution that contained silver NPs only. The change in the electrophoretic mobility of silver NPs was observed after adding NaBH4. These results suggest that the blue shift of the
SPR band and the change in the electrophoretic mobility of silver NPs in our system is due to the electron transfer from hydride to the surface of silver NPs. Going back to the case of gold NPs, we found a small blue shift of the SPR band of gold NPs could be observed only when a very high concentration of NaBH4 was added to the solution and the relaxation back to
original peak was rapid (∼1 s). However, the enhanced absorbance and narrowed peak for the SPR band of gold NPs were much less prominent than that of silver NPs.
On the basis of the above observations, we can draw a picture to account for the hydrogenation of anthracene catalyzed by gold and silver NPs. When NaBH4was added to the reaction
solution, the hydride from NaBH4 could be trapped by the
surfactant at the surface of metal NPs. In fact, gold and silver hydride anions have been previously observed in the gas phase15,16 and in the solid matrix17 of laser-ablated gold and
silver. In this experiment, the hydride could be adsorbed on the surface of metal NPs. It then transfers its electron to the metal NPs. This gives rise to the blue shift in the SPR band of metal NPs. The hydrogen atom, formed from the hydride after electron transfer (ET) to the metal NPs, subsequently attacked an anthracene molecule nearby. ET-induced hydrogenation of anthracene occurred then spontaneously. The metal NPs thus assume a role of storing electrons after ET from the hydride. A negatively charged metal NP may be regarded as a nanoelectrode Figure 6. Electropherograms of the silver NPs-containing solution with
solubilized anthracene and product.
Figure 7. Anthracene hydrogenation rates over silver NPs with different sizes.
Downloaded by NATIONAL TAIWAN UNIV on August 11, 2009
at a negative potential; the electron on the metal NP may be finally released to a suitable acceptor, such as water, to produce H2.18On the other hand, it is possible that other competing
reaction also took place when hydrogen radicals were formed after ET. For example, combination of the hydrogen radical with another radical nearby, may result in the formation of H2which
is highly exothermic. The formation of H2 would be more
favored when the local concentration of hydrogen radicals is high. That seems the case of much reduced amount of silver NP catalyst; little dihydroanthracene could be detected because the concentration of the hydrogen radicals was very high on the surface of the metal NPs and led to hydrogen production instead of hydrogenation. The catalytic hydrogen productions from organic free radicals in water by gold and silver NPs have been studied by Henglein and other researchers.18-22
Thus, the metal NP may be regarded as an electron relay before transferring electron to water. And the size effect of those metal NPs for ET may be explained by their size-dependent redox properties. As the particle size increases, it has less electron affinity.19-23Because the blue shifts of the SPR band
for all the silver NPs were observed, it suggests that all the sizes examined here for the silver NPs exceed the threshold size for the ET to occur. Unfortunately, we could not prepare stable Ag colloid of sizes less than 3 nm to examine the possibility of decreased ET at the very small size region. Meanwhile, our results (shown in Figure 5a) indicate the surface charges of the Ag NPs could be maintained for a long time. Once ET process for the part of NaBH4took place first, the
accumulated electrons on the metal NP could prevent more electrons transferring from the residual hydrides, and thus inhibit its catalytic activity. The smaller NP (3.2 nm) with higher charge density should have higher rate in releasing electrons to water. So the smaller NP in Figure 7 displays higher activity. On the other hand, for the larger sized NP the rate would drop because of higher accumulation of electron on the nanoparticles. An analogue of Coulombic “blockade” may have created a bottle-neck for electron storage.
Finally, we also like to include an indication of how the activities of gold and silver colloidal nanoparticles compare with catalysts where the metals are supported on oxides. We note that micellar catalysts are in general superior for reactions involving two phases which is the case in our investigation. We have tried the same catalytic reduction of anthracene using Au NP supported on mesoporous silica(Au@MCM-41).24We
found no activity under the same condition. This is because that anthracene neither dissolves well in water and nor adsorbes well on silica. Comparing with a literature report on 1,3-butadiene hydrogenation as catalyzed by oxides-supported gold NPs at 423 K,25our unsupported gold NPs show a much higher
TOF(more than 100 times higher) in the active size-range(Figure 4). As we have shown in this paper, we could easily optimize activities to the right particle size while in heterogeneous catalysis, the sizes of gold NP are much more inhomogeneous that a lot of nanoparticles are inefficient in catalysis. In fact, colloidal gold NP would function even better if they are “naked”, e.g., without surfactants. In a recent report of glucose oxidation catalyzed by naked gold NP,26the activity is even higher than
our anthracene reduction. Comparing activities between different reactions may not be so meaningful. We only want to point out here that well-controlled colloidal metal NP could be as active as the best in gas reactions catalyzed by supported metal NP when the size of NP is well-controlled in the right range.
For the gold NP in anthracene hydrogenation, we did observe a sharp decrease of reaction rate at the very small size regime.
It is possible that the semiconducting property of the very small-size gold NPs hindered ET and the electron relay.27Goodman
and co-workers had invoked the metal-semiconductor transition to explain the drop of CO oxidation activity at particle size less than 3 nm.27Previously, Comotti et al.,26when investigating
glucose oxidation catalyzed by gold NP, had observed a rapid drop of activity for sizes greater than 6 nm, but they offered no explanation. At present, the size effect of catalysis of metallic colloidal nanoparticles is not clear, especially in the low size range where we have great difficulties in obtaining stable metal NPs, although small (<3 nm) and stable gold NP can be easily obtained with thiol encapsulation. However, its catalytic activity is very low because the reactants are excluded from the tightly protected surface of Au. For ionic surfactant-protected metal NP, good synthesis methods are urgently needed for the very small (<3 nm) and stable NP in order its catalysis can be explored.
The difference between Au and Ag in catalysis is interesting. It seems to lie in the different capacity for electron storage. The blue shifts of the SPR band for the larger gold NP were hardly observed in our system. The result indicated that the efficiency in ET for the larger gold NP was very low and its lower catalytic activity could be expected. On the other hand, it has been reported that the electron affinity of gold clusters is about 1 eV higher than that of silver clusters.23It is expected
that the ET process for gold NPs is much more efficient than that of silver NPs. Further, surface electron on the gold NP can apparently be quenched by water more easily than that of silver NPs in the aqueous micellar solutions. It is thus reasonable that the catalytic activity of the gold NP is higher than that of the silver NP. Therefore, the prominent difference between gold and silver NPs in activities could be explained by differences in their electron affinities and in the discharge of the ac-cumulated electrons to water.
Conclusions
We have demonstrated that in metal (Au and Ag) NP-catalyzed hydrogenation of anthracene with sodium borohydride, the catalysts gold and silver NPs play a role as the nanoelectrode storing electrons from hydride and the resulting hydrogen radical reacts with anthracene to produce 9,10-dihydroanthracene. The metal NPs at a high negative redox potential give a blue shift of their electronic absorption spectra after an electron-transfer processes. The size-dependent activities of the metal NPs are related to their ability to relay and transfer electrons to water. The function of metal NP as a nanoelectrode makes it possible to catalyze other redox reactions via an electron transfer-storage process.
Acknowledgment. This work was supported by a grant from the National Science Council of Taiwan through the National Nano-Initiative program (NSC-94-2120-M-002-003).
References and Notes
(1) Feldheim, D. L.; Foss, C. A., Jr. Metal Nanoparticles: Synthesis, Characterization and Applications; Marcel Dekker, New York 2002.
(2) Kim, F.; Song, J. H.; Yang, P. J. Am. Chem. Soc. 2002, 124, 14316. (3) Blatchford, C. G.; Siiman, O.; Kerker, M. J. Phys. Chem. 1983, 87, 2503.
(4) Bond, G. C.; Thompson, D. T. Catal. ReV. Sci. Eng. 1999, 41, 319. (5) Haruta, M. Chem. Rec. 2003, 3, 75.
(6) Xiao, Y.; Patolsky, F.; Katz, E.; Hainfeld, J. F.; Willner, I. Science
2003, 299, 1877.
(7) Fan, F. R. F.; Bard, A. J. Science 1997, 277, 1791.
(8) Mallick, K.; Witcomb, M.; Scurrell, M. Mater. Chem. Phys. 2006, 97, 283.
Downloaded by NATIONAL TAIWAN UNIV on August 11, 2009
(9) Deng, J. P.; Shih, W. C.; Mou, C. Y. Chem. Phys. Chem. 2005, 6, 2021.
(10) (a) Jana, N. R.; Pal, T. Langmuir 1999, 15, 3458. (b) Pradhan, N.; Pal, A.; Pal, T Langmuir 2001, 17, 1800. (c) Hayakawa, K.; Yoshimura, T.; Esumi, K. Langmuir 2003, 19, 5517. (d) Esumi, K.; Isono, R.; Yoshimura, T. Langmuir 2004, 20, 237.
(11) Quirino, J. P.; Terabe, S. Science 1998, 282, 465.
(12) Henglein, A.; Mulvaney, P.; Linnert, T. Faraday Discuss. 1991, 92, 31.
(13) Ung, T.; Gierserg, M.; Dunstan, D.; Mulvaney, P. Langmuir 1997, 13, 1773.
(14) Ung, T.; Liz-Marza´n, L. M.; Mulvaney, P. J. Phys. Chem. B 1999, 103, 6770.
(15) Zhai, H. J.; Kiran, B.; Wang, L. S. J. Chem. Phys. 2004, 121, 8231. (16) Buckart, S.; Gantefo¨r, G.; Kim, Y. D.; Jena, P. J. Am. Chem. Soc.
2003, 125, 14205.
(17) Wang, X.; Andrew, L.; Manceron, L.; Marsden, C. J. Phys. Chem. A 2003, 107, 8492.
(18) Henglein, A. J. Phys. Chem. 1979, 83, 2209.
(19) Chen, S.; Murray, R. W.; Feldberg, S. W. J. Phys. Chem. B 1998, 102, 9898.
(20) Henglein, A. Chem. ReV. 1989, 89, 1861.
(21) Miller, D. S.; Bard, A. J.; McLendon, G.; Ferguson, J. J. Am. Chem. Soc. 1981, 103, 5336.
(22) Freund, P. L.; Spiro, M. J. Phys. Chem. 1985, 89, 1074. (23) Taylor, K. J.; Pettiette-Hall, C. L.; Cheshnovsky, O.; Smalley, R. E. J. Chem. Phys. 1992, 96, 3319.
(24) Chi, Y. S.; Lin, H. P.; Mou, C. Y. Appl. Catal. A: Gen. 2005, 284, 199.
(25) Okumura, O.; Akita, T.; Haruta, M. Catal. Today 2002, 74, 265. (26) Comotti, M.; Della Pina, C.; Matarrese, R.; Rossi, M. Angew. Chem., Int. Ed. 2004, 43, 5812.
(27) Valden, M.; Lai, X.; Goodman, D. W. Science 1998, 281, 1647.
Downloaded by NATIONAL TAIWAN UNIV on August 11, 2009