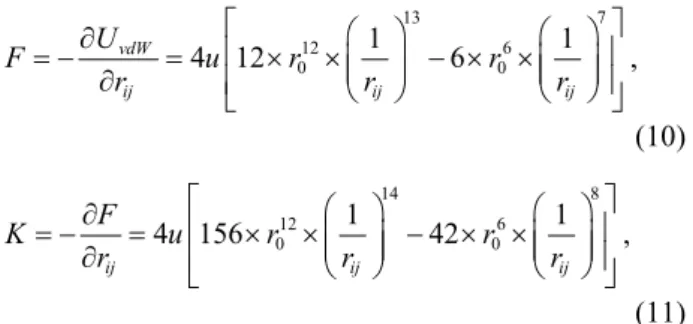Journal of Mechanics
http://journals.cambridge.org/JOMAdditional services for
Journal of Mechanics:
Email alerts: Click here Subscriptions: Click here Commercial reprints: Click here Terms of use : Click here
Characterizing the Mechanical Properties of Graphene and Single Walled
Carbon Nanotubes
S.-H. Tzeng and J.-L. Tsai
Journal of Mechanics / Volume 27 / Issue 04 / December 2011, pp 461 - 467 DOI: 10.1017/jmech.2011.49, Published online: 07 December 2011
Link to this article: http://journals.cambridge.org/abstract_S1727719111001006
How to cite this article:
S.-H. Tzeng and J.-L. Tsai (2011). Characterizing the Mechanical Properties of Graphene and Single Walled Carbon Nanotubes. Journal of Mechanics, 27, pp 461-467 doi:10.1017/jmech.2011.49
Request Permissions : Click here
Journal of Mechanics, Vol. 27, No. 4, December 2011 461
CHARACTERIZING THE MECHANICAL PROPERTIES OF GRAPHENE
AND SINGLE WALLED CARBON NANOTUBES
S.-H. Tzeng * J.-L. Tsai **
Department of Mechanical Engineering National Chiao Tung University Hsinchu, Taiwan 30010, R.O.C.
ABSTRACT
In this study, the mechanical properties of graphene and single walled carbon nanotubes (SWCNTs) were investigated based on molecular dynamics (MD) simulation. During the characterization of the mechanical properties, the atomistic interactions of the carbon atoms were described using the bonded and non-bonded energies. The bonded energy consists of four different interactions: Bond stretching, bond angle bending, dihedral angle torsion, and inversion. On the other hand, the non-bonded interac-tion between the carbon atoms within the cut-off ranges was regarded as the van der Waals (vdW) force. The effect of vdW force on the mechanical properties of graphene and SWCNTs would be mainly of concern. Simulation results indicated that the Young’s modulus of the graphene with vdW force in-cluded is 15 higher than that without considering any vdW interaction. The same tendency also was observed in the armchair and zig-zag SWCNTs. Furthermore, it was revealed that the increment of moduli caused by the vdW force could be primarily attributed to the 1-4 vdW interaction. The influence of the vdW interactions on the mechanical properties of graphene and SWCNTs was then elucidated us-ing the parallel sprus-ing concept.
Keywords: Graphene sheet, SWCNTs, Van der Waals interaction, Mechanical properties, Molecular
dynamics simulation.
1. INTRODUCTION
With characteristics of high strength and stiffness, the graphene and single walled carbon nanotubes (SWCNTs) have been used as reinforcements in com-posite materials [1-3]. In the past decade, the me-chanical properties of graphene and SWCNTs were characterized using molecular dynamics [4-7], struc-tural mechanics [8-10], and finite element analysis [11]. No matter which analysis was performed, the basic process was to specify the atomistic interaction of the atoms and then implement the corresponding interac-tions into the designed model. In fact, for the SWCNTs and graphene with the regular and periodic microstructures, the mechanical properties can be de-termined analytically associated with the atomistic in-teraction [12,13]. In other words, the mechanical properties of graphene and SWCNTs can be exactly characterized once the corresponding atomistic interac-tions of the carbon atoms are appropriately defined. Therefore, the proper description of atomic interactions becomes the key issue in evaluating the mechanical properties of graphene and SWCNTs.
In the characterization of graphite sheet and SWCNTs, the atomistic interactions of the carbon
at-oms are generally characterized by tight-binding (TB) [14-17], Tersoff-Brenner approach [18-21] and the pair- wise potential [22]. It is noted that the pair-wise en-ergy with the advantage of simplicity [23] was adopted in this study to describe the bonded interaction of car-bon atoms. In addition, the non-car-bonded interaction between the carbon atoms within the cut-off ranges was modeled using the van der Waals (vdW) force. In general, the non-bonded interaction was considered to have less contribution on the mechanical behavior and, sometimes, was neglected in atomistic modeling. However, the vdW force may actually have an influence on the mechanical properties of the nano-materi-als/nano-structures [24]. In molecular mechanics, all atoms except those that are bounded to each other or connected to a common atom have to be included in the vdW interaction [25]. It means that the 1-4 interac-tions as well as the higher order terms need to be taken into account in the vdW potentials. Nevertheless, in some molecular dynamics software, such as DL-POLY [26], the selection of the 1-4 vdW interaction as well as its extent is optional. Most studies that focus on the elastic properties of the SWCNTs and graphene rarely explored the effect of vdW force on the mechanical properties of the atomistic structures. In light of the
* Graduate student ** Professor, corresponding author
462 Journal of Mechanics, Vol. 27, No. 4, December 2011
light of the forgoing, it is required to conduct a system-atic molecular simulation in order to clarify the vdW interaction effect, especially the 1-4 vdW force, on the mechanical responses of the atomistic structures.
In this study, the mechanical properties of the SWCNTs and the graphene were constructed, and the corresponding properties were characterized using MD simulation with various degrees of vdW forces. Be-cause SWCNTs and graphene possess simple micro-structures, the influence of vdW interaction on the me-chanical responses can be simply investigated from the molecular simulation. By applying uniaxial tensile loading, the Young’s modulus of the atomistic struc-tures was measured in terms of the corresponding strain field in the deformed configuration. The moduli ob-tained based on the molecular structures with various vdW interactions were compared to each other, and the results were discussed accordingly based on the parallel spring concept.
2. POTENTIAL ENERGY FOR THE ATOMISTIC STRUCTURES
2.1 Graphene
A graphene sheet basically contains a layer of carbon where the carbon atoms are arranged in a hexagonal pattern. The interatomistic distance between the ad-jacent carbon atoms is 1.42Å, and the associated
atom-istic interaction is covalently bonded by SP2 hybridized
electrons, the bond angle of which is 120 degree to each other [8]. In order to investigate the mechanical prop-erties of the graphene, the atomistic structures have to be constructed in conjunction with the appropriately specified atomistic interaction. In the description of graphene, two kinds of atomistic interactions are nor-mally taken in account: One is bonded interaction, such as the covalent bond, and the other is the non-bonded interaction, i.e., vdW forces and electrostatic forces. Among the atomistic interactions, the covalent bond between two neighboring carbon atoms that provides the building block of the primary structure of the gra-phene may play an essential role in the mechanical re-sponses. Such bonded interaction can be described using the potential energy that consists of bond stretch-ing, bond angle bendstretch-ing, torsion, and inversion as illus-trated in Fig. 1 [23]. Therefore, the total potential energy of the graphite contributed from the covalent bond can be written explicitly as
graphite r ,
U
U
U
U
U (1)where Ur is a bond stretching potential; U is a bond
angle bending potential; U is a dihedral angle torsional
potential; and U is an inversion potential. For
gra-phene structure under in-plane deformation, the atom-istic interaction is mainly governed by the bond stretching and bond angle bending; therefore, the dihe-dral torsion and inversion potentials that are related to the out-of-plane deformation were disregarded in the modeling. The explicit form for the bond stretching and bond angle bending can be approximated in terms of elastic springs as [10] i j i n k j (a) (b) (c) (d) i n k j j i k i j i j i n k j i n k j (a) (b) (c) (d) i n k j i n k j j i k j i k
Fig. 1 A schematic of the inter-atomic potential (a) bond stretch, (b) bond angle bending, (c) di-hedral angle torsion, (d) inversion
2 0 1 ( ) , 2 r r U k r r (2) 2 0 1 ( ) , 2 U k (3)
where kr and k are the bond stretching force constant
and angle bending force constant, respectively. The
constants 93800 kcal 2 mole nm r k and k126 2 kcal
mole rad selected from AMBER force field for
carbon–carbon atomic-interaction were employed in our
molecular simulation [27]. The parameters r0 and 0
represent bond length and bond angle in equilibrium position, which are assumed to be 1.42 Å and 120 de-gree respectively, in the graphite atomistic structures.
In addition to the bonded interaction, the non-bonded interaction between the carbon atoms was regarded as the vdW force, which can be characterized using the Lennard-Jones (L-J) potential as 12 6 0 0 vdW 4 , ij ij r r U u r r (4)
where rij is the distance between the non-bonded pair of
atoms. For the hexagonal graphite, the parameters u
0.0556 kcal/mole and r0 3.40 Å suggested for the
carbon–carbon interaction were adopted in the model-ing [28]. Moreover, the cutoff distance for the vdW
force is assigned to be 3r0, which means that beyond
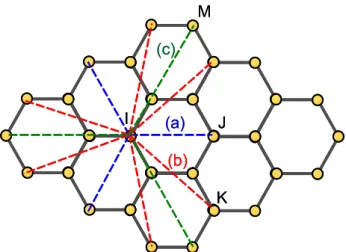
this distance, there are no more vdW interactions taking place. In molecular mechanics, all atoms except those that are bound to each other or connected to a common atom have to be accounted for in the calculation of vdW interactions [25]. When we look closely at the atomic structure of graphene shown in Fig. 2, the possible shortest distance between two carbon atoms needed to be considered for the vdW interaction is 2.84 Å (line (a) in Fig. 2 if it is measured from “atom I”). Thus, the carbon atoms with inter-atomic distance greater than 2.84 Å have to be accounted for in the vdW calculation.
(a) (b) (c) I J K M (a) (b) (c) I J K M
Fig. 2 Interatomistic distance in graphene structure ((a) 2.84Å, (b) 3.757Å, (c) 4.26Å) and J and between atoms I and K are usually denoted as 1-4 vdW interaction since the multiple interactions take place between number 1 atom and number 4 atom. The remaining vdW interactions within the cut-off range are indicated as the higher order vdW interaction. The 1-4 vdW interaction together with the higher order vdW is regarded as the vdW interaction in the molecu-lar mechanics.
2.2 Single Walled Carbon Nanotubes
When modeling the interatomistic behaviors of SWCNTs, in addition to the bond stretching potential and the bond angle bending potential given in Eqs. (2) and (3), the dihedral torsional potential and inversion potential which characterize the out of plane deforma-tion have to be included in molecular simuladeforma-tion. For the dihedral torsional potential and inversion potential, Li and Chou adopted the simplest harmonic form to incorporate the two interactions together into a single equivalent term as [10] 2 0 1 ( ) , 2 U UU k (5)
where k is the torsional constant and equal to
2
40 kcal
mole rad . The parameter 0 represents dihedral torsional angle in equilibrium position, which is equal
to 180 in the SWCNTs atomistic structures. It should
be noted that in the study, the MD simulation was con-ducted using the DL-POLY package where Dreiding potential was utilized to model the inter-atomic poten-tial of carbon–carbon bonding [29]. For the bond stretching and angle changing behaviors, the mathe-matical forms of Dreiding potential are exactly the same as those given in Eqs. (2) and (3). Nevertheless, for the dihedral torsion and inversion, the Dreiding tor-sional potential that accounts for the two portions to-gether is expressed explicitly as
1 cos ( o) .
UUU A m (6)
Because the carbon–carbon bonding in the hexagonal graphite is in resonance, the parameter “m” in Eq. (6) should be equal to 2 [29]. Moreover, the parameter A is decided according to the assumption that Dreiding torsional potential should correspond to the dihedral torsional potential given in Eq. (5). It has been found that when A is equal to 10.02kcal/mole, the Dreiding torsional potential coincides with the dihedral torsional potential well. As a result, the Dreiding torsional
po-tential with m 2 and A 10.02 kcal/mole were
em-ployed in our atomistic simulation. In addition to the bonded interaction, the vdW interaction containing 1-4 vdW force and high order interaction was utilized to characterize the non-bonded interaction of SWCNTs. The influences of 1-4 vdW interaction as well as the high order vdW interaction on the mechanical proper-ties of SWCNTs were evaluated from simple tensile tests.
3. CHARACTERIZATION OF MECHANICAL PROPERTIES
In order to evaluate the material properties of gra-phene sheets and SWCNTs, the simulation box suitable for representing the atomistic structures has to be estab-lished. Figure 3 shows the schematic of the simulation box for the graphene sheet. A periodic boundary con-dition was implemented on all surfaces to demonstrate the infinite graphite structures. It is noted in the gra-phene sheet that the dimension of the simulation box in the thickness direction is set to be large enough that the vdW interaction between the neighboring layers can not be attained. This special design of the simulation box is intended to simulate the single layer of graphene sheets. The atomistic structure with stress-free con-figuration was obtained by performing an NPT ensem-ble with time increments at 1fs for 100ps (the total it-eration steps are 100000) until the potential energy ac-complished a stable value. The NPT ensemble repre-sents that the pressure and temperature of the system may approach the specified values during the MD simulation. This process is accomplished through the Berendsen thermostat by scaling the velocities and po-sitions of atoms at each step to push the temperature
and pressure toward the desired value (P 0 and T =
0K) [30,31]. In the same manner, the simulation box as shown in Fig. 4 was generated to simulate the SWCNTs. The lateral directions of the simulation were designed to be large enough that the neighboring SWCNTs would not be interacted mutually. The MD simulation was carried out under the DL-POLY package originally developed by Daresbury Laboratory [26] in conjunction with the homemade subroutine for post- processing.
Subsequently, the mechanical properties of the at-omistic structures with specified intermolecular poten-tials were determined from the simple tension tests. The methodology developed to evaluate the mechanical properties of the atomistic structures was motivated
464 Journal of Mechanics, Vol. 27, No. 4, December 2011
Fig. 3 Axial stress applied in graphene sheet
(W 46.86 Å, L051.65Å, h 34.0 Å)
Fig. 4 Axial stress applied in SWCNTs
(W 30Å, L0 85.2Å)
solid. The same concept was extended and applied to the atomistic structures by means of a modified NPT ensemble in MD simulation with the characteristics of varying a simulation box in shape and size [32]. In other words, axial stresses were implemented on both sides of the simulation box with other faces being trac-tion free as shown in Figs. 3 and 4 for graphene and SWCNTs, respectively. After the energy minimization process, the equilibrated graphene atomistic structure under axial loading was obtained, and the Young’s modulus was defined in the continuum manner as
,
E
(7)
where is the strain component measured in the loading direction. In Eq. (7), should be the stress directly acting on the atomistic structure. However, for the case of graphene sheets as shown in Fig. 3, because the dimension of the graphene sheet in thickness direction is not compatible to the size of the simulation box, the stress in the graphene sheet has to be converted from
the stress acting on the simulation box, box, in terms of
the geometric parameters as
box , h t (8)
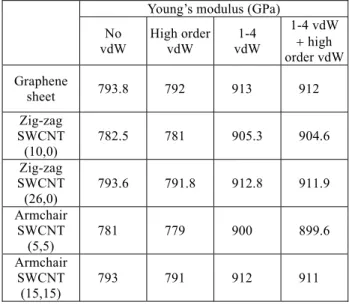
where h is the height of the simulation box, and t is the thickness of the graphene sheet, which is equal to 3.4Å. The Young’s moduli associated with various vdW in-teractions obtained from MD simulation for graphene sheets are presented in Table 1.
Table 1 Mechanical properties of graphene and
SWCNTs obtained based on different degrees of vdW interaction
Young’s modulus (GPa) No vdW High order vdW 1-4 vdW 1-4 vdW high order vdW Graphene sheet 793.8 792 913 912 Zig-zag SWCNT (10,0) 782.5 781 905.3 904.6 Zig-zag SWCNT (26,0) 793.6 791.8 912.8 911.9 Armchair SWCNT (5,5) 781 779 900 899.6 Armchair SWCNT (15,15) 793 791 912 911
It is noted that in the above simulation, the NPT en-semble as well as the modified NPT enen-semble was conducted to characterize the modulus of the nanos-tructures, and thus only active atoms (the one follows the Newtonian second law) were accounted for in the atomistic model. In addition, because the simulation was performed based on load control, the configura-tions of the nanostructures before/after the loading were measured for the evaluation of strain variation. Each time when the loading (0.01GPa) was applied, it was found that the deformed atomistic structure with energy minimization could be achieved after 100ps simulation time. As a result, the strain rate of the deformed process was measured as the strain increment associ-ated with the simulation time. In this study, the strain rate is around 1 106s1.
In the same manner, the mechanical properties of SWCNTs were determined by a modified NPT ensem-ble such that uniaxial stress can be independently ap-plied on the SWCNTs with stress free in the lateral di-rection. Again, after the energy minimization process, the equilibrated SWCNTs atomistic structure was ob-tained, and the modulus was evaluated by measuring the axial strain associated with the applied axial stress. It is noted that the stress implemented on the SWCNTs was calculated from the stress acting on the simulation
box, box, associated with the cross section as
2
boxW ,
A
(9)
where A Dot. The Young’s modulus of SWCNTs
with various radii obtained based on modified NPT ensemble in terms of different vdW interactions are presented in Table 1. Both zig-zag and armchair SWCNTs with different radii were considered in the atomistic simulation.
4. RESULTS AND DISCUSSIONS
Table 1 illustrates the Young’s modulus of graphene sheets and SWCNTs with different degrees of vdW in-teractions. The graphene sheet with 1-4 vdW interac-tion demonstrates the highest Young’s modulus among all cases, and the increment relative to the graphene without any vdW interaction is 15. On the contrary, the graphene sheet with only the high order vdW inter-action included exhibits the lowest value of Young’s modulus. It seems that the higher order vdW interac-tion has a negative effect on the Young’s modulus al-though the effect is not as significant as that of the 1-4 vdW interaction. These phenomena can be explained from the Lennard-Jones potential given in Eq. (4). By taking the derivative of the L-J potential function, the L-J force as well as the corresponding “spring” con-stants in terms of the interatomistic distance is derived as 13 7 12 6 0 0 1 1 4 12 6 , vdW ij ij ij U F u r r r r r (10) 14 8 12 6 0 0 1 1 4 156 42 , ij ij ij F K u r r r r r (11) and the corresponding results were plotted in Fig. 5. It
can be seen that the spring constant at the vdW distance less than 4.2311Å is positive and becomes negative when the distance is greater than 4.2311Å. Based on the parallel spring concept, it is apparent that the posi-tive spring constant may increase the Young’s modulus of the materials; in contrast, the negative spring con-stant may reduce the Young’s modulus. When the atomic interactions are treated as an arrangement of parallel springs, the modulus of the atomistic structures is dependent on the spring constants. Again, when we examine the atomic distances of 1-4 vdW interactions shown in Fig. 2, they are 2.84Å and 3.757Å as denoted by line (a) and (b), and the corresponding “spring” con-stants are positive. Thus, including the 1-4 vdW en-ergy into the interatomistic potential may enhance the modulus of the graphene and the SWCNTs as well. However, for the higher order vdW interaction, the at-omistic distances are no less than 4.26Å, which are greater than critical value of 4.2311Å. Thus, the asso-ciated spring constants are negative, resulting in the negative contribution of the moduli. Moreover, since the negative spring constants of higher order vdW in-teraction usually are much less than the positive spring constants provided by the 1-4 vdW interaction, the higher order vdW interaction plays a minor role in the properties of the graphene sheet. The same tendency is also observed in the zig-zag and armchair SWCNTs with different radii. Agrawal et al. [6] indicated that the moduli of the SWCNTs are not influence by whether the vdW interaction was taken into account or
not.
Infact,ifonly the high order vdW interaction
Distance (Å) S p ri ng cons ta nt (N /m ) Fo rc e (N) 4 6 8 -0.05 -0.025 0 0.025 0.05 0.075 0.1 -1E-11 -5E-12 0 5E-12 1E-11 1.5E-11 2E-11 Spring constant Force 4.2311Å Distance (Å) S p ri ng cons ta nt (N /m ) Fo rc e (N) 4 6 8 -0.05 -0.025 0 0.025 0.05 0.075 0.1 -1E-11 -5E-12 0 5E-12 1E-11 1.5E-11 2E-11 Spring constant Force 4.2311Å
Fig. 5 Interatomistic force and spring constant calcu-lated based on L-J potential
was considered as the vdW interaction in their model-ing, the results presented are quite promising. On the other hand, the literature [24] addressed that the moduli of the SWCNTs can be enhanced by the vdW interac-tion up to 10. According to our earlier examinainterac-tions, it is probable to have the improved properties when 1-4 vdW interaction is accounted for in the atomistic simu-lation. Thus, whether the 1-4 vdW interaction is em-ployed in the molecular simulation would be the crucial issue in the characterization of mechanical properties of graphene and SWCNTs, which should be treated with caution when we model the properties of the atomistic structures.
In addition, the moduli of the SWCNTs derived based on pair-wise potential as well as the 1-4 vdW interaction are around 0.9TPa which is in agreement with the results obtained from the tight-binding poten-tial [15,16] and the Tersoff-Brenner potenpoten-tial [4,18,19, 21] as well as the experimental data [33,34]. Hernández et al. [15] and Cai et al. [16] calculated the Young’s modulus of SWCNTs by second derivative of the strain energy with tight-binding method and Young’s modulus of SWCNTs is equal to 1.2TPa and 0.95TPa, respectively. Bao et al. [4] evaluated the Young’s modulus of graphene and SWCNTs as 1.2TPa and 0.93TPa respectively by conducting MD simulation with Brenner potential. The moduli of 0.7TPa for SWCNTs was obtained by Sammalkorpi et al. [18]. In addition, Reddy et al. [19] modeled the Young’s modulus of finite graphene structure using continuum mechanics approach based on the Brenner potential and the computed Young’s modulus of graphene is equal to 0.7TPa. Jeng et al. [21] investigated the mechanical properties of typical zigzag and armchair SWCNTs through the MD simulation with Tersoff potential. Result indicated that the Young’s modulus of SWCNTs is determined to be 1.06TPa. In light of the published data, it seems that the moduli of SWCNTs are varying from 0.7TPa to 1.2TPa, and our current data are within the range. If we checked the experimental data [33,34], the corresponding value is also fluctuating be-tween 0.9TPa to 1TPa. It is worthy to mention that because the effect of the 1-4 vdW interaction is around
466 Journal of Mechanics, Vol. 27, No. 4, December 2011
15 in terms of the modulus of the SWCNTs, it is really difficult to characterize the inference from the experimental observation. Furthermore, based on our simulation, it is found that the influence of charity on the modulus of SWCNTs is not appreciable, which agrees well with the results addressed in the literature [4,5,12,18].
5. CONCLUSIONS
The mechanical properties of graphene and SWCNTs were investigated according to different extents of the vdW interactions. Both 1-4 vdW interaction and high order vdW interaction are of concern. By applying tensile loading, the deformed configuration of the at-omistic structures was calculated through the molecular simulation, and then the mechanical properties were determined accordingly. Results indicate that the 1-4 vdW interaction can increase the modulus of the atom-istic structures up to 15, and the enhancement is at-tributed to the positive spring constant caused by such atomistic interactions. In contrast, the high order vdW interaction can lessen the modulus, but the decrease is not appreciable as compared to the 1-4 vdW interaction. Thus, combining the high order vdW interaction with the 1-4 vdW potentials in the molecular simulation still can improve the moduli of the graphene and SWCNTs as well.
REFERENCES
1. Lau, K. T., Gu, C. and Hui, D., “A Critical Review on Nanotube and Nanotube/Nanoclay Related Polymer Composite Materials,” Composites Part B Engineering, 37, pp. 425436 (2006).
2. Thostenson, E. T., Li, C. and Chou, T. W., “Nanocomposites in Context,” Composites Science and Technology, 65, pp. 491516 (2005).
3. Fukushima, H. and Drzal, L. T., “Graphite Nano-platelets as Reinforcements for Polymers: Struc-tural and Electrical Properties,” The 17th Technical Conference American Society for Composites, Purdue University, U.S.A. (2002).
4. Bao, W. X., Zhu, C. C. and Cui, W. Z., “Simulation of Young’s Modulus of Single-Walled Carbon Nanotubes by Molecular Dynamics,” Physica B,
352, pp. 156163 (2004).
5. Zhang, C. L. and Shen, H. S., “Temperature- Dependent Elastic Properties of Single-walled Carbon Nanotubes: Prediction from Molecular Dynamics Simulation,” Applied Physics Letters, 89, p. 081904 (2006).
6. Agrawal, P. M., Sudalayandi, B. S., Raff, L. M. and Komanduri, R., “A Comparison of Different Methods of Young’s Modulus Determination for Single-Wall Carbon Nanotubes (SWCNT) Using Molecular Dynamics (MD) Simulations,” Compu-tation Materials Science, 38, pp. 271281 (2006). 7. Tsai, J. L. and Tu, J. F., “Characterizing
Mechani-cal Properties of Graphite Using Molecular Dy-namics Simulation,” Materials and Design, 31, pp. 194199 (2010).
8. Cho, J., Luo, J. J. and Daniel, I. M., “Mechanical Characterization of Graphite/Epoxy Nanocompo-sites by Multi-Scale Analysis,” CompoNanocompo-sites Science and Technology, 67, pp. 23992407 (2007). 9. Scarpa, F., Adhikari, S. and Phani, A. S.,
“Effec-tive Elastic Mechanical Properties of Single Layer Graphene Sheets,” Nanotechnology, 20, p. 065709 (2009).
10. Li, C. and Chou, T. W., “A Structural Mechanics Approach for The Analysis of Carbon Nanotubes,” International Journal of Solids and Structures, 40, pp. 24872499 (2003).
11. Tserpes, K. I. and Papanikos, P., “Finite Element Modeling of Single-Walled Carbon Nanotubes,” Composites Part B Engineering, 36, pp. 468477 (2005).
12. Natsuki, T., Tantrakarn, K. and Endo, M., “Predic-tion of Elastic Properties for Single-walled Carbon Nanotubes,” Carbon, 42, pp. 3945 (2004).
13. Xiao, J. R., Gama, B. A. and Gillespie, Jr. J. W., “An Analytical Molecular Structural Mechanics Model for the Mechanical Properties of Carbon Nanotube,” International Journal of Solids and Structures, 42, pp. 30753092 (2005).
14. Jin, C., Lan, H., Peng, L., Suenaga, K. and Iijima, S., “Deriving Carbon Atomic Chains from
Gra-phene,” Physical Review Letters, 102, p. 205501
(2009).
15. Hernández, E., Goze, C., Bernier, P. and Rubio, A., “Elastic Properties of C and BxCyNz Composite Nanotubes,” Physical Review Letters, 80, pp. 45024505 (1998).
16. Cai, J., Bie, R. F., Tan, X. M. and Lu, C., “Appli-cation of the Tight-Binding Method to the Elastic Modulus of C60 and Carbon Nanotube,” Physica B,
344, pp. 99102 (2004).
17. Molina, J. M., Savinsky, S. S. and Khokhriakov, N. V., “A Tight-Binding Model for Calculations of Structures and Properties of Graphitic Nanotubes,” Journal of Chemical Physics, 104, pp. 46524656 (1996).
18. Sammalkorpi, M., Krasheninnikov, A., Kuronen, A., Nordlund, K. and Kaski, K., “Mechanical Properties of Carbon Nanotubes with Vacancies and Related Defects,” Physical Review B, 70, p. 245416 (2004).
19. Reddy, C. D., Rajendran, S. and Liew, K. M., “Equilibrium Configuration and Continuum Elastic Properties of Finite Sized Graphene,” Nanotechnol-ogy, 17, pp. 864870 (2006).
20. Nardelli, M. B., Yakobson, B. I. and Bernholc, J., “Brittle and Ductile Behavior in Carbon Nano-tubes,” Physical Review Letters, 81, pp. 46564659 (1998).
21. Jeng, Y. R., Tsai, P. C., Huang, G. Z. and Chang, I. L., “An Investigation into the Mechanical Behavior of Single-Walled Carbon Nanotubes Under
Uniax-ial Tension Using Molecular Statics and Molecular Dynamics Simulations,” Computation Materials & Continua, 348, pp. 117 (2009).
22. Jin, Y. and Yuan, F. G., “Simulation of Elastic Properties of Single-Walled Carbon Nanotubes,” Composites Science and Technology, 63, pp. 15071515 (2003).
23. Rappe, A. K. and Casewit, C. J., Molecular Me-chanics Across Chemistry, Sausalito California: University Science Books (1997).
24. Chen, W. H., Cheng, H. C. and Hsu, Y. C., “Me-chanical Properties of Carbon Nanotubes Using Molecular Dynamics Simulations with the Inlayer
Van Der Waals Interactions,” Computer Modeling
in Engineering and Sciences, 20, pp. 123145 (2007).
25. Burkert, U. and Allinger, N. L., Molecular Me-chanics, American Chemical Society (1982). 26. Smith, W. and Forester, T. R., “The DL_POLY
User Manual,” version 2.13, Daresbury Laboratory, (2001).
27. Cornell, W. D., Cieplak, P., Bayly, C. I., Gould, I. R., Merz, Jr. K. M., Ferguson, D. M., Spellmeyer, D. C., Fox, T., Caldwell, J. W. and Kollman P. A., “A Second Generation Force Field for The Simula-tion of Proteins, Nucleic Acids, and Organic Molecules,” Journal of the American Chemical So-ciety, 117, pp. 51795197 (1995).
28. Battezzatti, L., Pisani, C. and Ricca, F., “Equilib-rium Conformation and Surface Motion of Hydro-carbon Molecules Physisorbed on Graphit,” Jour-nal of the Chemical Society, 71, pp. 16291639 (1975).
29. Mayo, S. L., Olafson, B. D. and Goddard III W. A., “Dreiding: A Generic Force Field for Molecular Simulations,” Journal of Physical Chemistry, 94, pp. 88978909 (1990).
30. Berendsen, H. J. C., Postma, J. P. M., van Gun-steren, W. F., DiNola, A. and Haak, J. R., “Mo-lecular Dynamics with Coupling to an External Bath,” Journal of Chemical Physics, 81, pp. 36843690 (1984).
31. Allen, M. P. and Tildesley, D. J., Computer Simu-lation of Liquids, Oxford, Clarendon Press (1987). 32. Melchionna, S., Ciccotti, G. and Holian, B. L.,
“Hoover NPT Dynamics for Systems Varying in Shape and Size,” Molecular Physics, 78, pp. 533544 (1993).
33. Salvetat, J. P., Bonard, J. M., Thomson, N. H., Ku-lik, A. J., Forró, L., Benoit, W. and Zuppiroli, L., “Mechanical Properties of Carbon Nanotubes,” Applied Physics A, 69, pp. 255260 (1999).
34. Yu, M. F., Files, B. S., Arepalli, S. and Ruoff, R. S., “Tensile Loading of Ropes of Single Wall Carbon Nanotubes and Their Mechanical Properties,” Physical Review Letters, 84, pp. 55525555 (2000).
(Manuscript received April 16, 2010, accepted for publication November 22, 2010.)