Alumina-Promoted Sulfated Mesoporous Zirconia Catalysts
Chi-Chau Hwang†and Chung-Yuan Mou*,†,‡Department of Chemistry, National Taiwan UniVersity, Taipei 106, Taiwan, and Center of Condensed Matter Science, National Taiwan UniVersity, Taipei 106, Taiwan
ReceiVed: NoVember 28, 2008; ReVised Manuscript ReceiVed: January 23, 2009
Mesoporous zirconia, hydrothermally synthesized from surfactant templating, was directly impregnated with aluminum sulfate to give the acidic Al-promoted sulfated mesoporous zirconia (AS/MP-ZrO2). A series of
AS/MP-ZrO2catalysts were characterized by Brunauer-Emmett-Teller and X-ray diffraction for their texture
properties and crystalline phases. The catalytic behavior for n-butane isomerization was found to be strongly promoted at relatively low temperature by the addition of a proper amount of alumina as a promoter.27Al
S.S. magic-angle spinning nuclear magnetic resonance results indicated that Zr atoms were partially substituted by Al, giving a considerable increased concentration of Brønsted acids. X-ray photoelectron spectroscopy and diffuse-reflectance infrared Fourier-transformed spectra (DRIFT) analysis were then employed to identify and relatively quantify properties of acid sites on catalyst surface. A balanced distribution of acid sites strength was proven to prevent a catalyst from deactivating rapidly due to coke formation on the catalyst surface. A small concentration of olefins formed by oxidation of n-butane and proven to be key intermediates during n-butane isomerization on sulfated zirconia was found by the Baeyer test. Electron paramagnetic resonance and in situ DRIFT results show that this occurs via oxidative dehydrogenation of butane by the sulfate groups to form butene which leads to butyl carbenium species for skeleton isomerization. A modified biomolecular mechanism for the isomerization of butane is examined to explain the catalysis results.
1. Introduction
Since the discovery of its strong acidity, sulfated zirconia (SZ) has attracted much attention as a promising process catalyst. The main interest is associated with increasing need for environmentally benign catalysts and reaction processes. SZ is a solid acid catalyst with great potential application in industrial acid-catalyzed processes, such as the skeleton isomer-ization of alkanes1,2and esterification in biodiesel production.3
It is a good substitute for the conventional liquid-phase or halide-containing solid acid catalysts that have caused much environ-mental concerns.
Initially, Arata and co-workers devoted their attention to the preparation of SZ, its physicochemical properties, and catalytic performance in alkane isomeriation at relatively mild temperatures.1,2
But SZ catalysts suffer from the disadvantages of deactivation and possibly from sulfur loss during reaction and regeneration. Later, SZ catalysts have been successfully incorporated with transition metals (Fe and Mn)4so as to enhance substantially
its catalytic performance in n-alkane isomerization reactions. In spite of the high catalytic activity and product selectivity achieved by Fe/Mn-promoted SZ, serious concerns about the long-term stability still remain and cast doubt on its industrial applications. Thus, the noble metal Pt often has to be used to stabilize the catalytic activity.5 Then, one would have a
drawback in Pt-promoted SZ catalyst that it is sensitive to sulfur in the feed gas.
Lately, the promotion effect of main group metals (alumina or gallium)6,7on SZ in the isomerization reaction of n-alkanes has
received much attention for it stabilized and promoted activity
without the use of Pt.8-13We reported that the addition of an
optimum amount of Al onto the SZ catalyst, in various morphol-ogies, gives rise to a catalyst much more active than the corre-sponding unpromoted one.14,15These promotional effects have been
demonstrated on the isomerization reactions of butane,16pentane,13
and hexane.11The promotion and stabilization effects of Al on SZ
in alkane isomerization without the use of expensive Pt are technologically encouraging. We are thus developing a new generation of catalyst for alkane isomerization.
In the past, the main active site of SZ for the catalytic isomerization of alkane is considered to be the acid sites, although there have been debates about the relative importance of Brønsted vs Lewis acid sites. However, recently there has been a change of our understanding in the catalytic mechanism of SZ. SZ catalyst not only plays the role of solid acid, but also it was shown to initiate the catalytic cycle by dehydrogenation to olefin which then forms carbine for the chain initiation.17,18
The redox property of SZ is responsible for the oxidative dehydrogenation. In our previous work,14we have been focusing
the role of Al on the acidity SZ. So in view of the recent work on redox property of SZ, we should come to understand the role of Al also in the promotion of redox reaction.
In this work, alumina-promoted sulfated mesoporous zirconia catalysts (named as AS/MP-ZrO2) were used to investigate the
catalytic behavior of n-butane isomerization reaction. Sulfated mesoporous zirconia (S/MP-ZrO2) was first successfully
syn-thesized in 199919and was tested for n-alkane isomerization.20
As it has relatively high surface area, one would expect a better catalytic performance over the low surface area SZ catalysts.21
In addition to the information about identifying Brønsted and Lewis acid sites22on the catalyst after chemisorption of probe
molecules, various characterization techniques, such as electron paramagnetic resonance (EPR), in situ diffuse-reflectance infrared Fourier-transformed spectra (DRIFTs), and olefin * To whom correspondence should be addressed. E-mail: cymou@
ntu.edu.tw.
†Department of Chemistry, National Taiwan University.
‡Center of Condensed Matter Science, National Taiwan University.
10.1021/jp810465n CCC: $40.75 2009 American Chemical Society Published on Web 03/11/2009
Downloaded by NATIONAL TAIWAN UNIV on August 11, 2009
titration with aqueous potassium permanganate KMnO4(Baeyer
test), were employed to reveal the role of Al in the promotion of redox reaction. Our primary goal is to understand the origin and function of the catalytic active sites, and this information will in turn help us in optimizing the composition of aluminum and sulfur for a better performing catalyst.
2. Experimental Section
2.1. Catalyst Preparation. The mesoporous ZrO2(MP-ZrO2)
was synthesized by the surfactant-assisted route, as in the previous report of Ciesla et al..19An ordered MP-ZrO
2material
loaded with aluminum sulfate (Hayashi Osaka, denoted as xAS/ MP-ZrO2with x corresponding to the nominal AS concentration
in wt % based on the amount of MP-ZrO2), aluminum nitrate
(Acros, A/MP-ZrO2), and ammonium sulfate (Acros,
S/MP-ZrO2) was prepared via incipient wetness impregnation: the
required quantity of each source dissolved in 75 % (vol %) alcohol solution was impregnated by the as-synthesized meso-phase zirconia-containing surfactant. The slurry was stirred for 5 h and oven-dried at 100°C overnight. All the catalysts were calcined at 400°C for 5 h to eliminate the template and then at 630 °C for 5 h to obtain the tetragonal phase of zirconia (in static air with a heating ramp of 3°C/min).
2.2. Characterization Techniques. Elemental analysis was determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES) using a Jarrel-Ash ICAP 9000 instru-ment. Carbon analysis was done with a Heraeus VarioEL-III instrument.
In situ diffuse-reflectance infrared Fourier-transformed (DRIFT) spectra of ammonia chemisorbed on catalyst were taken with a Thermo Nicolet 380 FTIR spectrometer. The sample was first dried at 400°C for at least 1 h prior to ammonia adsorption. The adsorption of ammonia was done by flowing 5% NH3/N2
mixture through the catalyst at 120°C for 5 min, and the system was then flushed with dry N2 for 15 min. The catalyst with
adsorbed ammonia was then subjected to a desorption process at increasing temperatures. Spectra were recorded after the catalyst’s temperature was held for 15 min at each temperature. The similar methodology was also applied to adsorb butane on the activated sample at 50°C.
X-ray photoelectron spectroscopic (XPS) analysis of pyridine chemisorbed on catalyst was performed using a Thermo VG Scientific ESCALAB 250 instrument with a monochromatic Al KR radiation (1486.8 eV) and under a residual pressure of∼5 × 10-10torr. The pyridine-adsorbed sample was first degassed
ex situ under vacuum at about 120°C for 0.5 h. The pretreated sample was then transferred into the XPS instrument, and spectra were collected in the N 1s region. Charge effects were compensated by electron flux from a flooding gun, and the Zr 3d5/2binding energy was set at 182.2 eV. The spectral profiles
were deconvoluted into peaks by using XPSPEAK software from RCSMS laboratory.
The Baeyer testsa conventional method to check the oxida-tion of alkenes with aqueous potassium permanganate (KMnO4)swas adopted to check certain unsaturated olefin
species produced during the n-butane isomerization. The reaction gases exiting the reactor were passed through a potassium permanganate aqueous solution with 5 mL of 0.5µM, and the time-dependent UV-vis experiment was monitored by a Hitachi U-3310 spectrophotometer.
The solid-state 27Al MAS NMR spectra were taken to
determinate the coordination states of aluminum. The experi-ments were performed at room temperature on a Bruker DSX-400WB NMR spectrometer with a 4 mm-diameter ZrO2rotor
spun at 4.0 kHz at 9.4 T magnetic field.
EPR spectra were obtained at 84 and 300 K with a Bruker EMX spectrometer working in the X-band (9.53 GHz). A weighted catalyst of 0.2 g was placed inside a 4-mm outside diameter quartz tube with stopcocks. Prior to each measurement, the catalysts were evacuated at room temperature below 1× 10-3Torr.
2.3. Catalytic Study. Catalytic reaction was carried out in a fixed bed continuous flow quartz reactor at atmospheric pressure. The catalyst (usually 1.0 g) was first pressed into 10-20 mesh size pellets and mixed with quartz sand. It was then pretreated under an air flow at 450°C for 3 h and then prereduced with H2at 300°C for 1 h. After flushing the system
with N2at the reaction temperature for 0.5 h, the reaction was
started by introducing a mixture of n-butane (gU99.9%, San-fu) and H2(n-C4:H2) 1:10 v/v) at a total inlet flow rate of
15.5 mL min-1. The reaction product was analyzed by an online Agilent 6890N gas chromatograph equipped with a flame ionization detector and a 60-m DB-1 column.
3. Results
3.1. Structure Study. Before we present the catalytic properties of alumina-promoted AS/MP-ZrO2, we would like
to have definite information about their textual properties. In addition, the thermal stability of crystalline phase and surface features such as surface acidity and redox property of these sulfated mesoporous zirconia catalysts promoted by alumina were also studied.
Textural properties of various sulfated mesoporous zirconia catalysts with or without alumina (i.e., AS/MP-ZrO2or
S/MP-ZrO2) were characterized by X-ray diffraction and N2
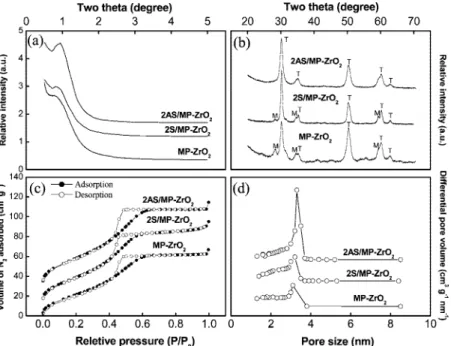
adsorption-desorption isotherm. In Figure 1a, X-ray diffraction patterns for the 2AS/MP-ZrO2and 2S/MP-ZrO2catalysts exhibited a single
broad peak at∼1.0°due to their somewhat disordered meso-structure. From TEM images, we found that its mesostructures are of wormlike nanopores.15For the MP-ZrO
2sample without
alumina or sulfate dopants, the low-angle XRD peak shows a less-ordered mesostructure suggesting collapse of its nanochan-nels. The high angle region of XRD peaks of these three catalysts (Figure 1b) are characteristic of the tetragonal crystal-line phase (demoted as T) of ZrO2which is the active phase
for the catalysis of alkane isomerization.8It is worth noting that
the 2AS/MP-ZrO2catalyst can maintain not only better
meso-structure of zirconia but also retard the crystalline transformation from tetragonal to monoclinic phase (demoted as M in Figure 1a). The Brunauer-Emmett-Teller isotherms of these three samples (Figure 1c) with the hysteresis between adsorption and desorption branches are typical for a mesoporous material. Apparently, considerable loss of the volume of N2adsorbed in
2S/MP-ZrO2and MP-ZrO2catalysts also indicates some collapse
of their mesoporous structure. This observation of structural instability is consistent with the XRD result for samples with calcination temperature at 630°C. Compared to 2S/MP-ZrO2
and MP-ZrO2 catalysts, Figure 1d then depicts that a more
homogeneous pore size distribution with a sharper peak centered at 3.3 nm found on the alumina-promoted catalyst 2AS/MP-ZrO2.
3.2. Catalytic Reaction. Butane isomerization reaction is a valuable model reaction for understanding the skeleton isomer-ization of alkanes catalyzed by strong solid acid such as SZ.23
It is interestingly complex enough to show both the roles of acidity and redox properties of the catalyst. To understand the individual role played by Al and S in the catalytic reaction, we tried to fine-tune the composition of the AS/MP-ZrO2catalyst
with an additional small amount of sulfate or alumina. The
Downloaded by NATIONAL TAIWAN UNIV on August 11, 2009
catalysts are hereafter labeled as xAS/MP-ZrO2,
xAS+A/MP-ZrO2, or xAS+S/MP-ZrO2with x corresponding to the nominal
aluminum sulfate concentration in wt % based on the MP-ZrO2
and A or S corresponding to the extra fixed concentration of nominal aluminum or sulfate based on the total sample weight. The elemental composition of these catalysts and their corre-sponding catalytic activities for n-butane isomerization reaction were summarized in Table 1. For detailed time-dependence behavior, the conversions vs time-on-stream are shown in Figure 2. An important finding in our elemental analysis study is that the amount of Al loaded onto the catalyst has a direct influence on the amount of S in the catalyst. The use of ammonium sulfate is superior over other methods of sulfation of zirconia,24while
the addition of aluminum in the form of aluminum sulfate increases much the loading of sulfur. This observation is apparent by comparing the S loading of catalysts 2AS/MP-ZrO2
and 2AS+A/MP-ZrO2or 1AS/MP-ZrO2and 1AS+A/MP-ZrO2.
The correlation in the loadings of Al and S in these catalysts hinted that there are some kinds of interaction between them. An increase of Al content leads a large increase of S content. The best performing catalysts are 2AS+A/MP-ZrO2and 2AS/
MP-ZrO2with their n-butane conversion reaching about 60%
(TON≈ 1.75 µmol g-1s-1) at 250°C (Figure 2). In addition, the selectivity of main product, isobutane, for these catalysts
was higher than 85% while small amounts of methane, propane, and pentane were also observed.
The time evolution of activity shows general decay to steady activity from initial high conversions with two characteristics: (1) larger deactivation when excess sulfur was present (1AS + S and 2AS +S), and (2) more steady behavior in conversion Figure 1. XRD patterns, N2adsorption-desorption isotherm, and pore size distribution for MP-ZrO2, 2S/MP-ZrO2, and 2AS/MP-ZrO2catalysts calcined at 630°C.
TABLE 1: Elemental Analysis, Texture Properties, and Performance of Selected Sulfated Mesoporous Zirconia-Supported Catalysts in n-Butane Isomerization Reaction at 250°C
loading (wt %) TON (µmol g-1s-1)d
catalysta Alb Sb surface areac(m2/g) surface density (S atom/nm2) 15 min 360 min activitye(loss %)
1S 0.07 114.6 0.1 0.006 2S 0.11 122.4 0.2 0.006 1AS 0.84 1.77 141.6 2.4 1.02 0.77 9 1AS + A 1.45 2.85 188.3 2.8 1.64 1.28 12 1AS + S 0.82 3.40 151.0 4.2 1.30 0.35 32 2AS 1.45 2.89 150.8 3.6 1.74 1.39 11 2AS + A 1.96 4.00 163.8 4.6 1.75 1.37 13 2AS + S 1.22 4.28 157.1 5.1 1.82 0.93 30
aCatalysts used mesoporous zirconia (MP-ZrO2) as support. bDetermined from ICP-AES and EA. cDetermined from N2
adsorption-desorption isotherm at 77 K.dTurnover number, which is the number of micromoles of n-butane converted per gram of catalyst
per second at 250°C.e
Defined by variation of butane conversion at time on stream between 15min and 360min.
Figure 2. Catalytic profiles of mesoporous zirconia-supported catalysts in n-butane isomerization reaction at 250°C. The ratio of n-butane:H2 is 1:10 (V:V) at a total inlet flow rate of 15.5 mL/min and 1 atm.
Downloaded by NATIONAL TAIWAN UNIV on August 11, 2009
when excess aluminum was added (1AS + A and 2AS +A). The initial (at 15 min) and steady (at 360 min) activities are listed in Table 1.
3.3. Acidity of the Catalysts and Promotion Effect of Al. To measure the acidity of SZ catalyst, we used ammonia as the probe molecule. In situ ammonia adsorption-desorption mea-surements describe the acidity of solid acids with oxidative power such as SZ catalyst.25,26Carbon-containing base probes
such as pyridine would have been oxidized by SZ at moderately high temperature. Figure 3 illustrates the in situ DRIFT spectra for 2AS/MP-ZrO2catalyst after treatment of surface-adsorbed
NH3at different temperatures. Two types of acid sites were
found on this catalyst, namely, the Brønsted acid sites (B) and Lewis acid sites (L). The band at 1609 cm-1corresponds to the asymmetric N-H deformation mode (δas) of NH3coordinated
to the Lewis acid sites.27Another intense band at 1428 cm-1
and the weak band at 1675 cm-1correspond to theδsandδas
of NH4+formed on the Brønsted acid sites, respectively.27The
bands in the region below 1373 cm-1 are mainly due to the lattice vibration of MP-ZrO2. All the bands due to adsorbed
NH3decreased in intensity at increasing desorption temperatures,
indicating the loss of NH3adsorbed on these acid sites. The
strength of the Brønsted acid sites in 2AS/MP-ZrO2 catalyst
seems to be stronger than that of Lewis acid sites since there are relatively more NH3molecules remaining adsorbed on the
Brønsted acid sites at 400°C.
27Al MAS NMR technique is used to study the coordination
environment of Al. Here, this technique is employed to study the relationship between the acid sites and Al species on 2AS/ MP-ZrO2catalyst before and after calcination. The27Al MAS
NMR spectra of 2AS/MP-ZrO2catalyst are shown in Figure 4.
Al2(SO4)3 and Al2O3 were used as external standards for
comparison. The spectrum of uncalcined catalyst 2AS/MP-ZrO2(UC) consists of a broader resonance between 30 and -60
ppm centered at∼0 ppm, which is assigned to Al in octahedral sites. The signal for calcined sample 2AS/MP-ZrO2is apparently
sharper than that for 2AS/MP-ZrO2(UC) due to its more
homogeneous chemical environment of octahedral Al species. Besides, the minor resonance center at∼30 ppm in the spectrum of 2AS/MP-ZrO2 has been reported for aluminum in 5-fold
coordination in oxo complexes between 30 to 40 ppm.29
The sample 2A/MP-ZrO2was prepared in a manner similar
to 2AS/MP-ZrO2 but using aluminum nitrate in place of
aluminum sulfate. As shown in Figure 5, they behaved differ-ently in the desorption of adsorbed NH3at 250°C. In the case
of MP-ZrO2, even though the IR bands correspond to the Lewis
and Brønsted acid sites appeared slightly at low temperatures (not shown here), they nearly vanished at a desorption temper-ature of 250°C. On the other hand, the IR bands due to NH3
adsorbed on both acid sites were still observed at this temper-ature for the samples 2S/MP-ZrO2and 2AS/MP-ZrO2. However,
the band intensity corresponding to Brønsted acid sites for 2AS/ MP-ZrO2sample is quantitatively higher than that for
nonpro-moted SZ catalyst 2S/MP-ZrO2.
One notes that the DRIFT spectrum of 2A/MP-ZrO2(uncalcined) is distinctively different from 2A/MP-ZrO2
in that only IR bands due to NH3adsorbed on the Lewis acid
sites are observed (around 1609 cm-1). Therefore, we can Figure 3. DRIFT spectra of the adsorption of NH3at 120°C on 2AS/
MP-ZrO2catalyst and followed by desorption at 250, 350, 400°C. (B ) Brønsted acid sites; L ) Lewis acid sites.)
Figure 4. 27Al MAS NMR spectra of 2AS/MP-ZrO2catalyst before and after calcination. Al2(SO4)3and Al2O3were standard samples for comparison.
Figure 5. DRIFT spectra of the adsorption of NH3at 120°C on (a) 2A/MP-ZrO2, (b) 2A/MP-ZrO2(uncalcined), (c) 2AS/MP-ZrO2, (d) 2S/ MP-ZrO2, and (e) MP-ZrO2catalysts and followed by desorption at 250°C. (B ) Brønsted acid sites; L ) Lewis acid sites.)
Downloaded by NATIONAL TAIWAN UNIV on August 11, 2009
conclude that the formation of Brønsted acid sites requires the participation of Al species with high calcination temperatures such as 630°C. Moreover, the strong sharp peak at 1355 cm-1 in 2A/MP-ZrO2(uncalcined) due to aluminum nitrate, or its
hydrolyzed product formed during the impregnation process, disappeared after calcination at 630 °C. Thus, the extra-framework Al species substitute into the MP-ZrO2framework
after the calcination process.
Although NH3can be used as a probe (e.g., in DRIFT study)
to differentiate between Brønsted and Lewis acid sites on a catalyst, it has a limitation in that no quantitative information could be derived on the distribution of Brønsted acid strength. To overcome this problem, we turned to XPS. Previously, we have measured XPS spectra of the N 1s peak of adsorbed pyridine to investigate the type and strength of acid sites displayed by SZ catalysts by resolving the spectra into three components of different type and strength of acid sites on catalysts.15 Three kinds of acid sites were identified on the
catalyst: a Lewis site, a weak Brønsted site, and a strong Brønsted site. In Figure 6 (left), the XPS N 1s spectra of
catalysts 2AS/MP-ZrO2, 1AS/MP-ZrO2, and 1AS+S/MP-ZrO2
were then fitted by three singlet peaks corresponding to the difference degree of interaction between pyridine and the acid sites. These catalysts were chosen for XPS study because of their special characteristic in catalysis (see Figure 2 and Table 1). The peak of lowest BE (399.8 eV) corresponds to the pyridine atom coordinated to the Lewis acid site. The peaks at medium and highest BEs (401.5 and 402.8 eV) would be due to the pyridinium ions on medium and strong strengths of Brønsted acid sites, respectively. The assignment of these peaks and their respective BEs are in accordance with our previous published work.15Relative concentrations of the three kinds of
acid sites for each catalyst were summarized in the pie charts (inserts of Figure 6, left). They are quite different for different loadings of Al. For probing the Lewis acid site, we used CO as a probe molecule as its FTIR spectra are measured.30In fact,
the DRIFT CO adsorption analysis also showed that Lewis acidity was present in SZ promoted by alumina. This is shown in Figure 6 (right) demonstrating that when probe molecule CO interacts with coordinatively unsaturated cations on surface, a band atνco≈ 2200 cm-1was observed. From literature data,31
this band is ascribed to CO molecules interacting with the surface Zr4+cations that are responsible for Lewis acidity. In
our case here, the fraction of Lewis acid sites is the highest in 1AS+S/MP-ZrO2(with the highest amount of surface density
in S) but moderate in 1AS/MP-ZrO2and 2AS/MP-ZrO2, which
are in close agreement with the pyridine adsorbed XPS results. 3.4. Redox Properties of Catalysts. Besides the structure and acidity described in sections 3.1 and 3.2, attentions also focused on the redox properties for these catalysts. We have checked for paramagnetic sites on our catalysts by using EPR. In pure MP-ZrO2(Figure 7a), Zr3+and O2-paramagnetic sites
are observed, and this finding is in line with the published EPR results of Jentoft et al.32 Zr3+ is a coordinatively unsaturated
site with an oxygen vacancy and O2-could be formed by the
interaction between adsorbed O2with the F-center electron. On
the other hand, in 2AS/MP-ZrO2catalyst (Figure 7d), the Zr3+
sites diminished in number, but much more new O2
-species are now formed. A reasonable explanation for this phenomenon is that Zr3+has been oxidized to the coordinatively saturated
Figure 6. The N1s photoelectron profiles with pyridine (left) and IR spectra with CO (right) adsorbed on (a) 2AS/MP-ZrO2, (b) 1AS/MP-ZrO2, and (c)1AS+S/MP-ZrO2catalysts after activation.
Figure 7. EPR spectra of (a) MP-ZrO2, (b) 2A/MP-ZrO2, (c) 9S/MP-ZrO2, and (d) 2AS/MP-ZrO2catalysts degassed at room temperature under vacuum and recorded at -196°C.
Downloaded by NATIONAL TAIWAN UNIV on August 11, 2009
Zr4+ state and the electron released is responsible for the
formation of new O2- species. Very likely, surface adsorbed
SOxspecies are involved in this oxidation process whereby one
of its oxygens is now filling the oxygen vacancy previously associated with Zr3+. Indeed, we found that the intensity of this
new O2- signal correlates with the amount of sulfur in the
catalyst. Superoxide species may be then correlated with the oxidative dehydrogenation ability of the catalyst. Li et al. have reported that butene species formed by oxidation of n-butane are shown to be the key intermediate during n-butane isomer-ization on SZ catalysts.17 Therefore, we adopted the Baeyer
testsa conventional method to check the oxidation of alkenes with aqueous potassium permanganate (KMnO4)sto explore
whether certain unsaturated olefin species were formed oxida-tively by our catalysts in the initiating step of butane isomer-ization. Since MnO4-is purple and MnO2is light brown, this
color change can be used to assay for the presence of alkenes. Figure 8 illustrates the corresponding time-dependent UV-vis spectra of KMnO4 aqueous solution at room temperature.
Potassium permanganate is a powerful oxidant, and we can attribute the formation of MnO2to the direct redox between
olefin species and KMnO4because there were no other reducing
products. With the elapsed reaction time, it can be easily seen that three peaks at about 315, 525, and 545 nm, which originated from KMnO4gradually disappeared with the concurrence of a
new peak shifted to 370 nm which indicated the formation of MnO2aqueous solution.
One can note that the spectra corresponding to KMnO4
reduction to MnO2by the species generated from 2AS/MP-ZrO2
catalyst (Figure 8a) change much more apparently than those from the other catalysts at the same time interval shown in parts b-d of Figure 8. It implies that the higher radical concentration the catalyst, such as 2AS/MP-ZrO2, the stronger ability it
oxidizes n-butane to the olefin species such as butene. On the other hand, it is noteworthy of mentioning again that the EPR signal to radical correlates directly with the amount of sulfur in the catalyst.
To explore the active sites on 2AS/MP-ZrO2surface
interact-ing with butane molecules durinteract-ing isomerization, in situ
diffuse-reflectance IR was collected to monitor the gas-solid interaction between them. The 2AS/MP-ZrO2sample was first activated at
400 °C for 30 min in an air flow, and then the adsorption of butane was done by flowing butane (g99.9%) through the activated sample at 50°C for 5 min. Finally the system was flushed with dry air for 10 min.
Figure 9 illustrates the IR spectrum of the calcined but not yet activated catalyst is typical of hydrated surface with adsorbed water (1600-1700 cm-1range). Activation in a flowing dry air results in strong reduction of this broadband, and the spectrum of the activated sample exhibits an distinctive band at 1390 cm-1(Figure 9b), which is assigned to the stretching vibration of the SdO bond in adsorbed SO3molecules.28The
band at 2850-3010 cm-1(Figure 9a) and a small shoulder at about 1462 cm-1(Figure 9b) correspond to butane adsorbates on the 2AS/MP-ZrO2surface. In the initial process of desorption,
Figure 8. UV-vis spectra of KMnO4aqueous solution used to check unsaturated olefin species produced by (a) 2AS/MP-ZrO2, (b) 9S/MP-ZrO2, (c) 2A/MP-ZrO2, and (d) MP-ZrO2samples during the n-butane isomerization.
Figure 9. In situ DRIFT spectra of 2AS/MP-ZrO2catalyst recorded before and after activation and followed by desorption at the range from 100 to 250°C after the adsorption of n-butane at 50°C on the activated catalyst.
Downloaded by NATIONAL TAIWAN UNIV on August 11, 2009
even though the IR bands correspond to the butane molecules appeared strongly at low temperatures due to physisorption, they nearly vanished above a desorption temperature of 150 °C. Another interesting feature is that 2AS/MP-ZrO2catalyst with
its stretching vibration of the SdO bond located at 1390 cm-1 shifted downward to 1363 cm-1 when exposed to butane molecules. As the desorption temperature increased the vibration band shifted upward in the range of 1363-1377 cm-1. This phenomenon hints that there are certain kinds of interaction between butane molecules and the active sites on 2AS/MP-ZrO2
catalyst during the isomerization reaction. In other words, it is reasonable for us to visualize that species adsorbed on AS/MP-ZrO2catalyst, a labile SO3surface species, are likely to be the
active sites involving the butane isomerization reaction. The formation of water during desorption process is shown by the increasing intensity of the deformation band at 1600 cm-1 (Figure 9b) with desorption of the butane molecules (Figure 9a). The gradual formation of water on 2AS/MP-ZrO2catalyst
also indicates the butane molecules were activated via oxidative dehydrogenation to form butane and water by the SO3groups.
4. Discussion
Since the discovery of the strong acidic property of SZ, a great deal of work have been reported on their application in n-alkane isomerization reaction. However, there is a diversity of opinions on the mechanism and reactive centers of alkane isomerization reaction over SZ, which is further complicated by that the reactions were carried out under a vast number of conditions and catalyst modifications. In this paper, we set our goal to clarify the role of alumina promotion (active site and reaction pathway) in the two main functions of SZ in the n-butane isomerization reaction, that is, its redox activation of n-butane to butene and the role of acidity in isomerization..
4.1. General Comment on Catalytic Performance. In this study, the main group metal Al was successfully incorporated into S/MP-ZrO2and enhanced substantially the catalytic activity
and stability of n-butane isomerization reaction. By comparison of catalysts with and without Al, Table 1 shows that alumina is highly promoting in catalytic activity. From the sulfur content in Table 1, one can see that Al increases the sulfur loading by at least 10-folds. However, the more than 100-fold increase in activity need consideration other than sulfur loading. Previously, Gao and co-workers reported a 2.2-fold increase in catalytic activity in Al promotion of SZ.33Here, the promotional effect
of Al to S/MP-ZrO2is even more striking. Anyway, the loading
of alumina and then increased sulfates groups has four conse-quences in (1) stabilizing the active tetragonal crystalline phase of zirconia, (2) increasing total surface area, (3) enhancing the Brønsted acidity, and (4) increasing the oxidative dehydrogen-ation. In our previous report, we have discussed the enhanced stability of tetragonal phase (the more active phase) of zirconia upon promotion of Al.8Also, from Table 1 we do see there is
an increase in total surface area of MP-ZrO2, similar to the
observation of Kim et al.34However, the increased surface area
cannot account for the very large increase in catalytic activity. It can only be a minor factor. Thus in this work, we focus on the last two effects of Al on the acidity and redox properties of SZ.
Our discussions will be based on both structural characteriza-tions of the catalyst and the reaction mechanism of butane isomerization. Thus before presenting our discussions, we would like to give summary of the reaction mechanism that will fit nicely with our data. There are two reaction models commonly used to describe the n-alkane isomerization reaction over SZ
solid acids, namely, the monomolecular model and the bi-molecular model. There is still controversy over the operation of the two mechanisms in butane isomerization on SZ.35-39In
monomolecular mechanism,35,36the formation of a protonated
cyclopropane ring from C4 +
carbenium ion on SZ is the crucial step. The monomolecular mechanism can satisfactorily explain the high selectivity for isobutene, especially at initial time-on-stream. On the other hand, the bimolecular mechanism occurs through the formation of C8+carbenium ion, which is resulted
from reacting C4+ with butane.37-39 The C8+ species then
undergoes isomerization andβ-scission. The bimolecular mech-anism is supported for its lower activation energy, isotope scrambling,37,39 and the C
3 and C5 disproportion products.40
Recently, after studying the nonspecific olefin promotional effect, Goodwin and co-workers41 proposed a dual-nature
mechanism that integrates the supporting evidence for the above two mechanisms. In the dual-nature model, the C8+species is
formed but the part from the C4 +
acts “as though” in a monomolecular isomerization. The reaction pathway is sum-marized in Scheme 1, after small modifications. In Scheme 1, one can see in the major route, the isomerization along the circled part is really monomolecular in nature although it is attached to an butane (olefin)-derived C4and become part of a
C8 +
species. Disproportional scission only occurs occasionally. The product distribution is related to the reaction pathway taken; the catalytic performances over the three selected samples of various Al and S loadings, as summarized in Table 2, are quite different in conversion and selectivity. A high selectivity for isobutane implies a dominant pseudomonomolecular mech-anism as indicated by the circles in Scheme 1. In the initial period, the conversions are higher and the selectivities lower. Furthermore, the production of propane is a lot more than pentane. This could be due to the activity of strong Brønsted acid in forming oligomer species of higher carbon number (C12
and beyond) and its cracking leading to the volatile propane and the less desorbing higher alkane species stay on the surface of SZ to form coking species. The cokes eliminate some of the strong Brønsted acid site and decrease to a steady activity. The steady state is probably dominated by the pseudo-mono-molecular mechanism in Scheme 1, and the side products C3
and C5 are found to be in equal amount. After 60 min, the
activity stabilized gradually and selectivity maintained a stable value above 90%. This implies long chain reaction propagation length, probably more than 500 skeleton isomerization of sec-butyl carbenium for each oxidation step.42 We note that
Al-promoted catalysts show only minor decay of activity from initial to steady activity compared to most other reports in literature. Al not only promoted but the catalytic activity but also make it more steady. From Table 1, one can see that increasing the sulfur content without also increasing Al content would increase the initial activity but it would quickly decay. This may be due to the relatively weak acidity, which leads to less coking and the easy transport in mesostructure of alumina-promoted S/MP-ZrO2. This decreases the need of coke
elimina-tion processes, such as repeated calcinaelimina-tions, supercritical fluid processing,43,44or using the expensive platinum.45In our catalytic
process, Pt is not needed to prevent coke formation at all because little coking exists under the action of relatively mild acid sites. 4.2. Strong Brønsted (SB), Weak Brønsted (WB), and Lewis (L) Acid Sites. In our previous study of the acidity of Al-promoted SZ,14we have identified by XPS measurement of
adsorbed pyridine three kinds of acid sites: one Lewis acid (L), one strong Brønsted (SB), and one weak Brønsted (WB) site. We reported the catalytic activity for butane isomerization is
Downloaded by NATIONAL TAIWAN UNIV on August 11, 2009
closely correlated to the concentration of WB sites. But high catalytic performance was also attributed to the simultaneous presence of Brønsted and Lewis acid sites.22Our results from 27Al MAS NMR (Figure 4) and NH
3DRIFT (Figure 5) studies
on 2AS/MP-ZrO2catalyst suggest that Brønsted acid sites are
created by the isomorphous substitution of Zr4+ by Al3+.
However, Al3+substitution may be only minor since it did not
cause a significant change in the coordination environment of Zr4+ in the catalyst judging from the absence of a peak
broadening effect in our EXAFS spectrum (not shown). In other words, Al3+probably occupied only the surface Zr4+sites. Hua
et al. have used XPS to study the composition of alumina-promoted SZ and also found that the atomic ratio of Al/Zr at the surface is much higher than that in the bulk.33Anyway, the
concentration of Brønsted acid sites depends directly on the amount of substituted Al in the surface layer of the catalyst.
According to our pyridine XPS results (Figure 6, left), Brønsted acid sites can be divided into two groups of different acid strength. To be a good catalyst, the concentration of SB and WB (or even the L) sites need to be balanced, and they
complement each other during different stages of reaction as we shall explain below. Generally, the stronger the Brønsted acid sites, the faster the rate of C4H9
+
, C8H17 +
, and isobutane formation. However, the rate of deactivation by coke formation is also faster. Obviously, a larger number of SB sites will promote the initial rate of reaction. However, this will also promote coke formation. In order to sustain the reaction through the later stages of reaction, there must be many WB sites which are less susceptible to deactivation by coke formation. Undoubt-edly, WB sites played an important role in the steady stages of reaction.33
The origin of L sites which have a weaker acid strength is not all clear. On the one hand, our NH3DRIFT study (Figure
5) showed that one of the candidates is the extra-framework Al species. On the other hand, our pyridine XPS study cannot exclude the possible involvement of surface Zr species with oxygen deficiency next to it in the formation of L sites (compare 1AS/MP-ZrO2and 1AS+S/MP-ZrO2in Figure 6). As shown
in Scheme 1, we propose that the olefin species is adsorbed on a L acid site (olefin modified acid site), while a neighboring Brønsted acid site with an adsorbed carbenium ion would promote the formation of the dimer C8
+
species. This explains the necessity of simultaneous presence of L and B sites.
4.3. Dehydrogenation of n-Butane. In all the mechanisms proposed so far, an initial formation of butene species is necessary for the carbenium formation upon reacting with a Brønsted acid site. In the unpromoted or non transition metal-promoted SZ catalysts, the nature of dehydrogenation site(s) for n-alkane is not known for certain. As these catalysts do not possess superacidity, dehydrogenation of n-butane by a pro-tonation barbonium mechanism does not seem to be a feasible proposal. There must be some other site(s) on the SZ catalyst capable of performing the dehydrogenation. Recently, oxidative dehydrogenation has been proposed as a possible route for butene formation since sulfate group does have strong oxidative power.17,18
From the Bayer test (Figure 8), we have substantial production of butene catalyzed by the catalyst with the dehydrogenation process. The in situ DRIFT vibrational spectra in Figure 9 SCHEME 1: Mechanism for n-Butane Isomerization
TABLE 2: Catalytic Conversion, Selectivity, and Yield in
n-Butane Isomerization at 250°C after 15 min, 50 min, and at Steady State Regime over the 3 Selected Samples
yield (%) catalysta conversion (%) selectivity (%) C2 C3 i-C4 i-C5 After 15 min 2AS 58.63 85.1 1.55 5.87 49.92 1.29 1AS 34.22 93.0 0.30 1.15 31.83 0.94 1AS+S 43.98 87.0 0.37 3.99 38.28 1.34 After 50 min 2AS 52.67 92.1 1.05 1.62 48.53 1.47 1AS 34.15 93.5 0.28 0.97 31.94 0.96 1AS+S 35.64 89.5 0.22 2.13 31.88 1.41 After 360 min 2AS 46.91 92.1 0.80 1.47 43.20 1.44 1AS 26.03 95.6 0.19 0.48 24.89 0.47 1AS+S 11.79 96.7 0.07 0.17 11.40 0.15 a
Catalysts used mesoporous zirconia (MP-ZrO2) as support.
Downloaded by NATIONAL TAIWAN UNIV on August 11, 2009
identify that butane interact with the sulfate group on the surface of the 2AS/MP-ZrO2 catalyst. FO¨ ttinger et al.28 assigned
pyrosulfate species to a vibrational wavenumber above 1400 cm-1while (ZrO)3SdO (SO3species) would give peak slightly
below 1400 cm-1. Since our DRIFT spectra gives a peak at 1390 cm-1, we believe we have mainly dispersed SO3species
on the surface. Apparently, this SO3species reacts with butane
to give butene which is the starting species for the rest of acid-catalyzed isomerization reactions42
C4H10+ SO3 fC
4H8+ H2O + SO2
It is quite possible the oxidative dehydrogenation would also involve some radical species to induce hydrogen transfer from butane. We then set out to look for radical species on our 2AS/ MP-ZrO2catalyst. In pure MP-ZrO2, Zr3+and O2
-paramagnetic sites are observed, and this finding is inline with the published EPR results of Jentoft and co-workers.32Zr3+is a coordinatively
unsaturated site with an oxygen vacancy, and O2- may be
formed by the interaction between adsorbed O2with the F-center
electron. Upon the loading of Al and S, in 2AS/MP-ZrO2
catalyst the Zr3+sites diminished in number but much more
new O2-species are now formed. Surely, a reasonable
explana-tion for this phenomenon is that Zr3+has been oxidized to the
coordinatively saturated Zr4+state and the electron released is
responsible for the formation of new O2
-species. Very likely, surface adsorbed SO3species (Figure 9) are involved in this
oxidation process, whereby one of its oxygen atoms is now filling the oxygen vacancy previously associated with Zr3+.
Indeed, we found that the intensity of this new O2
-signal correlates directly to the amount of S in the catalyst. It is tempting for us to assign the O2
-species (or its derivatives formed under reaction conditions) as a possible oxidative agent. In that case, the concentration of dehydrogenation sites should also correlate directly to the S content of a catalyst which indeed we found in comparing parts c and d of Figure 7. Furthermore, one of the roles played by Al is to chemisorb more SO3species
on the surface during the catalyst preparation process as we have mentioned in section 3.2. Indirectly, Al could also increase the concentration of dehydrogenation sites. Therefore, another of the promotional effects of Al (although acted indirectly) is to increase the number of dehydrogenation sites which is very important for the isomerization reaction to initiate. We also note that Lewis acid sites may play some role in the oxidative dehydrogenation. Lewis acid is known to promote oxidation reaction by coordinating with an oxygen species and enhancing the electrophilicity of these oxidizing reagents toward their reaction with weakly nucleophilic substrates. In any case, the concentration of L sites need not be large in the catalytic reaction.
In addition to the formation of butene by the initial oxidative dehydrogenation of butane, there may be more olefinic CHx
formed during the reaction (Scheme 1). This would lead to more active surface “olefin” species during the reaction and may play a secondary (or substitutional) role for n-butane activation. Goodwin and co-workers have shown that these olefinic species can increase the rate of butane isomerization reaction.46Hence,
the pristine dehydrogenation sites and the CHxspecies will both
ensure a continuous supply of olefins for the isomerization reaction.
5. Conclusions
In conclusion, we have investigated the promotional effect of alumina to mesoporous SZ on the butane isomerization. The doping of alumina to SZ increases the extent of sulfur retention
on the surface of zirconia which then promotes the oxidative dehydrogenation to butane and the skeleton isomerization. A good catalyst of Al-SZ thus requires delicate balances of several steps in the reaction chain. Our design for an excellent low-temperature active catalysts for alkane isomerization was benefited from (1) matching rate of isomerization of carbenium species with respect to the oxidative dehydrogenation capability and (2) balanced distribution of Brønsted acid and Lewis acid sites. A good performing catalyst with high initial activity requires high oxidative dehydrogenation rate. However, if it is not matched with enough weak Brønsted (WB) acid sites and Lewis (L) acid sites, the overproduced olefinic species would lead to coking and a decay of catalytic activity. For good steady-state activities, one then should have a balanced distribution of SB, WB, and L sites. We found that the optimum relative concentration of SB:WB:L sites is around 0.33:0.50:0.17 for our best performing 2AS/MP-ZrO2 catalyst. A substantial
deviation from this optimum distribution of acid sites can have a delirious effect on the catalytic performance.
Possessing multifunctional catalytic capabilities, oxidation and acid-base, SZ catalysts may be further applied to other reactions, 47 such as in esterification reaction for biodiesel
production3or as a support for metal catalysts.48We have found
in this work that doping SZ with alumina can fine-tune its catalytic properties. This will make our ability to design zirconia-based catalyst system better.
Acknowledgment. This work was supported by a grant from the National Science Council of Taiwan. We would like to thank technical helps from Dr. J.H. Wang and S.T. Wong.
References and Notes
(1) Hino, M.; Kobayashi, S.; Arata, K. J. Am. Chem. Soc. 1979, 101, 6439.
(2) Hino, M.; Arata, K. J. Chem. Soc. Chem. Commun. 1980, 851. (3) Chen, X. R.; Ju, Y. H.; Mou, C. Y. J Phys. Chem. C 2008, 111, 18731.
(4) Wan, K. T.; Khouw, C. B.; Davis, M. E. J. Catal. 1996, 158, 311. (5) Hino, M; Arata, K. Catal. Lett. 1995, 30, 25.
(6) Gao, Z.; Xia, Y. D.; Hua, W. M.; Miao, C. X. Top. Catal. 1998, 6, 101.
(7) Wang, W; Chen, C. L.; Xu, N. P.; Mou, C. Y. Green Chem. 2002,
4, 257.
(8) Chen, C. L.; Cheng, S.; Lin, H. P.; Wong, S. T.; Mou, C. Y. Appl.
Catal., A 2001, 215, 21.
(9) Wang, J. H.; Mou, C. Y. Abstr. Pap. Am. Chem. Soc. 2002, 224, U45.
(10) Chen, C. L.; Li, T.; Cheng, S. F.; Xu, N.; Mou, C. Y. Catal. Lett. 2002, 78, 223.
(11) Cao, C. J.; Han, S.; Chen, C. L.; Xu, N. P.; Mou, C. Y. Catal.
Commun. 2003, 4, 511.
(12) Cao, C. J.; Yu, X. Z.; Chen, C. L.; Mou, C. Y. React. Kinet. Catal.
Lett. 2004, 83, 85.
(13) Wang, W.; Wang, J. H.; Chen, C. L.; Xu, N. P.; Mou, C. Y. Catal.
Today 2004, 97, 307.
(14) Wang, J. H.; Mou, C. Y. Appl. Catal., A 2005, 286, 128. (15) Wang, J. H.; Mou, C. Y. Microporous Mesoporous Mater. 2008,
110, 260.
(16) Wang, J. H.; Mou, C. Y. Catal. Today 2008, 131, 162. (17) Li, X.; Nagaoka, K.; Simon, L. J.; Olindo, R.; Lercher, J. A.; Hofmann, A.; Sauer, J. J. Am. Chem. Soc. 2005, 127, 16159.
(18) Li, X.; Nagaoka, K.; Simon, L. J.; Lercher, J. A.; Wrabetz, S.; Jentoft, F. C.; Breitkopf, C.; Matysik, S.; Papp, H. J. Catal. 2005, 230, 214.
(19) Ciesla, U.; Fro¨ba, M.; Stucky, G.; Schu¨th, F. Chem. Mater. 1999,
11, 227.
(20) Yang, X; Jentoft, R. E.; Jentoft, F. C.; Girgsdies, F.; Ressler, T.
Catal. Lett. 2002, 81, 25.
(21) Sun, Y.; Yuan, L.; Ma, S.; Hua, Y.; Zhao, L.; Wang, W.; Chen, C. L.; Xiao, F. S. Appl. Catal., A 2004, 268, 17.
(22) Hammache, S.; Goodwin, J.G. J. Catal. 2003, 218, 258. (23) Guisnet, M. R. Acc. Chem. Res. 1990, 23, 392.
(24) Sparks, D. E.; Keogh, R. A.; Davis, B. H. Appl. Catal., A 1996,
144, 205.
Downloaded by NATIONAL TAIWAN UNIV on August 11, 2009
(25) Niwa, M.; Habuta, Y.; Okumura, K.; Katada, N. Catal. Today 2003,
87, 213.
(26) Katada, K.; Tsubaki, T.; Niwa, M. Appl. Catal., A 2008, 340, 76. (27) Baertsch, C. D.; Soled, S. L.; Iglesia, E. J. Phys. Chem. B 2001,
105, 1320.
(28) FO¨ ttinger, K.; Halwax, E.; Vinek, H. Appl. Catal., A 2006, 301, 115.
(29) Bradley, S. M.; Howe, R. F.; Kydd, R. A. Magn. Reson. Chem. 1993, 31, 883.
(30) Breitkof, C.; Papp, H.; Li, X.; Olindo, R.; Lercher, J. A.; Lloyd, R.; Wrabetz, S.; Jentoft, F. C.; Meinel, K.; Foster, S.; Schindler, K.-M.; Neddermeyer, H.; Widdra, W.; Hofman, A.; Sauer, J. Phys. Chem. Chem.
Phys. 2007, 9, 3600.
(31) Stichert, W.; Schu¨th, F.; Kuba, S.; Kno¨zinger, H. J. Catal. 2001,
198, 277.
(32) Jentoft, F. C.; Hahn, A.; Kro¨hnert, J.; Lorenz, G.; Jentoft, R. E.; Ressler, T.; Wild, U.; Schlo¨gl, R.; Ha¨βner, C.; Ko¨hler, C. J. Catal. 2004, 224, 124.
(33) Hua, W.; Xia, Y.; Yue, Y.; Gao, Z. J. Catal. 2000, 196, 104. (34) Kim, S. Y.; Lohitharn, N.; Goodwin, J. G.; Olindo, R.; Pinna, F.; Canton, P. Catal Commun. 2006, 7, 209.
(35) Tomishige, K.; Okabe, A.; Fujimoto, K. Appl. Catal., A 2000, 194, 383.
(36) Chao, K. J.; Wu, H. C.; Leu, L. J. J. Catal. 1995, 157, 289. (37) Liu, H.; Adeeva, V.; Lei, G. D.; Sachtler, W. M. H. J. Mol. Catal.
A: Chem. 1995, 100, 35.
(38) Kim, S. Y.; Goodwin, J. G., Jr.; Hammache, S.; Auroux, A.; Galloway, D. J. Catal. 2001, 201, 1.
(39) Adeeva, V.; Lei, G. D.; Sachtler, W. M. H. Appl. Catal., A 1994,
118, L11.
(40) Lohitharn, N; Goodwin, J. G.; Lotero, E. J. Catal. 2005, 234, 199. (41) Lohitharn, N; Lotero, E; Goodwin, J. G. J. Catal. 2006, 241, 328. (42) Li, X.; Nagaoka, K.; Simon, L. J.; Olindo, R.; Lercher, J. A. J.
Catal. 2005, 232, 456.
(43) Funamoto, T.; Nakagawa, T.; Segawa, K. Appl. Catal., A 2005,
286, 79.
(44) Bogdan, V. I.; Klimenko, T. A.; Kustov, L. M.; Kazansky, V. B.
Appl. Catal., A 2004, 267, 175.
(45) Demirci, U. B.; Garin, F. J. Mol. Catal. 2002, 188, 233. (46) Lohitharn, N.; Goodwin, J. G. J. Catal. 2007, 245, 198. (47) Yadav, G. D.; Nair, J. J. Microporous Mesoporous Mater. 1999,
33, 1.
(48) Aristizabal, B. H.; de Correa, C. M.; Serykh, A. I.; Hetrick, C. E.; Amiridis, M. D. J. Catal. 2008, 258, 95–102.
JP810465N
Downloaded by NATIONAL TAIWAN UNIV on August 11, 2009