A Web Application for Biomedical Entities and Relations Annotation
Using the Unstructured Information Management Architecture
Pei-Hsuan Chou
1,3, Hong-Jie Dai
1,2, Chi-Hsin Huang
1,
Richard Tzong-Han Tsai
4*and Wen-Lian Hsu
1,21
Institute of Information Science, Academia Sinica, Taipei, Taiwan, R.O.C.
2
Dept. of Computer Science, National Tsing-Hua Univ., Hsinchu, Taiwan, R.O.C.
3
Dept. of Information Management, National Central Univ., Taoyuan, Taiwan, R.O.C.
4
Dept. of Computer Science & Engineering, Yuan Ze Univ., Taoyuan, Taiwan, R.O.C.
[email protected],{hongjie,sinyuhgs}@iis.sinica.edu.tw,
[email protected], [email protected]
Abstract
-BIOSMILE Web Search (BWS), a web-based NCBI-PubMed search application, which can analyze articles for selected biomedical verbs and give users relational information, such as subject, object, location, time, etc. After receiving keyword query input, BWS retrieves matching PubMed abstracts, annotates named entities in abstracts and lists them along with snippets by order of relevancy to protein-protein interaction. BWS was assembled using the unstructured information management architecture. BWS is accessible free of charge at http://bioservices.cse.yzu.edu.tw/BWS.Keywords: named entity recognition, semantic role labeling, protein-protein interaction, unstructured information management architecture
1. Introduction
Taking advantage of the large, well-curated biomedical resources, today’s biologists are able to search through a massive volume of online articles in their research. For example, a user can retrieve from the PubMed database of over eighteen million articles. Unfortunately, users of basic search engines may need to further scan or read retrieved articles in more detail to pick out specific information of interest. Consider the sentence "KaiC enhanced KaiA-KaiB interaction in vitro and in yeast cells," which describes an enhancement relation. Needless to say, search services that can identify elements in this relation, such as the action "enhanced", the enhancer "KaiC", the enhanced "KaiA-KaiB interaction" and the location "in vitro and in yeast cells", as well as biomedical named entities (NEs), KaiA/B/C, can save biologists much time.
Several advanced services have already been developed in biomedical community. iHOP [1] website searches sentences containing specified genes and identifies other genes in them with a graphic user interface. [2, 3] provide enhancements to PubMed's retrieval by organizing the results or highlighting specific information in text. MEDIE1 identifies subject-verb-object (SVO) relations and biomedical NEs in sentences. Our proposed system, BIOSMILE web search (BWS), has similar features to the above systems. It can label biomedical NEs in sentences, including DNA, RNA, cell, protein and disease names, and summarize recognized relations.
This task of recognizing NEs is referred to named entity recognition (NER). NER in biomedical articles is a challenging task due to there is no community-wide agreement on how a particular biomedical NEs should be named [4]. To tackle this problem, our previous NER system, NERBio [3, 5], which was developed for the BioCreAtIvE II Gene Mention (GM) tagging task [6] is integrated. Furthermore, for researchers interested in protein-protein interaction (PPI), BWS classifies articles as PPI-relevant or -irrelevant using the system [3, 7], we developed for the BioCreAtIvE II PPI Article Sub-task [8].
After identifying NEs, a state-of-art semantic relation analysis technique, semantic role labeling (SRL) [9], is applied to extract complex semantic relations between biomedical verbs and sentence components, such as agent2, patient3, time and
location. These relations can be important for
* Corresponding author
1 http://www-tsujii.is.s.u-tokyo.ac.jp/medie/
2 Deliberately performs the action (e.g., Bill drank his soup quietly).
3 Experiences the action (e.g. The falling rocks crushed the
precise definition and clarification of complex
Figure 1. BWS search interface biomedical relations.
To integrate above features into manageable processes, the Unstructured Information Management Architecture (UIMA) [10], originally developed by IBM, is adopted. UIMA enables BWS to be decomposed into components and transformed the natural language processing (NLP) processes into a manageable pipeline, for example, "document retrieval"→"sentence boundary detection"→"NE detection." The UIMA framework can facilitate developers to manage components and the data flow between them.
2. Usage
Figure 1 shows the BWS search interface. It accepts either PubMed identifier (PMID) or keyword input (Figure 1, No. 1), so BWS search queries are compatible with PubMed search. Upon entering a query, users will receive output sorted by PMID, including the title, authors and abstract. Recognized NEs, including DNA, RNA, cell, protein and disease, appear in different colored text in the search results (Figure 1, No. 2.) A graduated bar meter on the right-hand side of the abstract (Figure 1, No. 3) in the "Protein-Protein Interaction" column indicates PPI relevance.
Once search results appear, users can perform relation analysis for a single abstract by clicking the "Analyze" button (Figure 1, No. 4), which appears below the abstract’s PubMed ID in the PMID column. For multiple abstracts, they can check off abstracts of interest and then click the "Analyze" button (Figure 1, No. 5) at the top of the search results pane.
Figure 2 shows the results of relation analysis. Action verbs representing biomedical relations are marked red (Figure 2, No. 1). Clicking on the one of the verbs in the right-hand pane will open a list
all the elements of the relation, including agent, patient, location, manner, time, etc. (Figure 2, No. 2).
Figure 2. Relation analysis
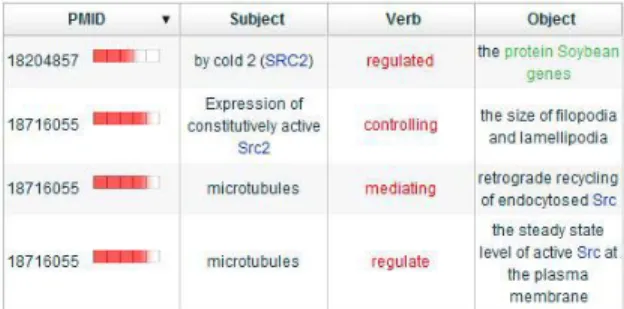
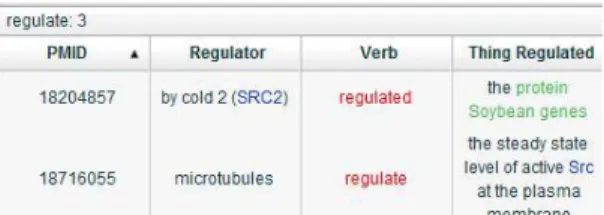
In addition to displaying relations article by article, we provide an analysis summary table that contains all relations in abstracts. Figure 3 shows the simple version which lists six major elements in a relation, including subject, verb, object, location, and extent. This table provides users a brief summary of relations.
Figure 3. Analysis summary table (simple) The summary table also provides an advanced display (Figure 4), which lists all elements in a relation. The description of each element corresponding to the verb is also displayed. Relations are classified by their main verbs, making it easy to browse through all the relations in an article verb by verb.
Figure 4. Analysis summary table (advanced)
3. Methods
In the following section, we describe the core NLP components of BWS. Then we describe the UIMA framework which provides the platform for creating and integrating information on our BWS.
3.1. The core components of BWS
The BWS is composed of three NLP components: a NE recognizer, a PPI abstract classifier, and a relation analyzer.
3.1.1. Component 1: NE Recognizer. The biomedical NE recognizer based on NERBio [3, 5] is employed to label NEs in all retrieved abstracts. NER is formulated as a word-by-word sequence labeling task, where the assigned tags delimit the boundaries of any NE names. The underlying machine learning (ML) model used by our NE recognizer is conditional random fields (CRF) [11] with a set of features selected by a sequential forward search algorithm.
3.1.2. Component 2: PPI Abstract Classifier. The PPI abstract classifier assigns each retrieved abstract a score that indicates its relevance to PPI. This score ranges from -1 (least relevant) to +1 (most relevant.) In PPI abstract classification, some words have different levels of information in different contexts. For example, "bind" is more informative when it appears in a sentence that has at least two protein names. Accordingly, we divide the general word bag into several contextual bags. The words in each sentence are bagged according to the number of NEs in the sentence. If there are 0 NEs, the words are put into contextual bag 0; if 1 NE, then bag 1; and if 2 or more NEs, then bag 2. We employ support vector machines (SVM) [12] as the machine learning model to build our PPI abstract classifier [13, 14].
3.1.3. Component 3: Relation Analyzer. The biomedical semantic relation analyzer extracts relations among selected biomedical verbs and phrases from sentences.
The component was developed based on semantic role labeling (SRL) technology. In SRL, sentences are represented by one or more
predicate-argument structures (PASs). Each PAS is composed of a predicate (e.g., a verb) and several arguments (e.g., noun phrases) that have different semantic roles, including main arguments such as agent or patient, as well as adjunct arguments, such as time, or location. Here, the term argument refers to a syntactic constituent of the sentence related to the predicate; and the term semantic role refers to the semantic relationship between a predicate and an argument of a sentence. For example, the sentence "KaiC enhanced KaiA-KaiB interaction in vitro and in yeast cells," describes an enhancement relation. It can be represented by a PAS as follows:
[KaiC agent] [enhanced predicate] [KaiA-KaiB
interaction patient] [in vitro and in yeast cells location]
in which "enhanced" is the predicate, "KaiC" the agent in which its semantic role is "causer of greatness, agent", "KaiA-KaiB interaction" the patient in which its semantic role is "thing enhanced", and "in vitro and in yeast cells" the location. Thus, the agent, patient, and location are the arguments of the predicate.
A collection of PASs forms a proposition bank, which is essential in building a ML based SRL system. In 2006, we constructed the first ever biomedical proposition bank, BioProp [15], by annotating semantic role information on GENIA’s full parse trees. The full lists of the BioProp’s
predicates are available at http://bioservices.cse.yzu.edu.tw/BioProp/.
Using BioProp as the training corpus, we constructed our biomedical semantic relation analyzer [16] which uses the maximum entropy model [17] as the underlying ML model.
3.2. Using UIMA to Integrate and Manage Components
Above three core components require processing and transferring data internally. For example, the NER process in named entity recognizer can simplify as follows: the abstract is firstly tokenized by a tokenizer and detected sentence boundary by the LingPipe [18] sentence model. For each detected sentence, the GENIA tagger [19] is applied to generate part-of-speech (POS) information. The information is then feed to CRF model to generate NE annotating result. Finally, the result will be transformed into human-readable format for displaying in BWS interface. In order to make the process easier, we adopt UIMA as a middleware layer to facilitate the smooth interaction of many NLP sub-components that may not be originally designed to interoperate
with each other. In the following section, we describe three main UIMA components adopted in this paper.
3.2.1. Common Analysis Structure. In UIMA, the original document, such as an abstract from PubMed, and its analysis are represented in a structure called the common analysis structure (CAS). The CAS is conceptually analogous to the annotations [20]. In general, annotations associate some metadata with a region in the original document. For example, the annotation associates a label with a span of text in the document by giving the span’s start and end positions. The label could be a NE tag in NER task, or a semantic role tag in SRL task. Such annotations are maintained separately from the document itself; this is a flexible strategy since the raw text in a document can keep unchanged during the analysis process.
The analysis results represented by CAS may be thought of as a collection of metadata that is enriched as it passes through successive stages of analysis. At a specific stage of analysis, for example, the NER stage. The CAS may include the POS information (metadata) generated by the GENIA tagger. The NE recognizer receiving the CAS may consider the information to identify NEs. The enriched information then may be inputted to other analysis engines that produce the relation summaries or classifications of the document. For example, input the information to PPI abstract classifier to generate PPI ranking score.
3.2.2. Analysis Engine. Once initialized, the CAS is sent down processing pipelines. Components that act on the contents of the CAS, and in particular, those that add content to the CAS, are known as analysis engines (AEs.)
AE come in two forms: primitive and aggregate. An example of a primitive AE would be a tokenizer, which takes the raw text as its input and produces as output a set of annotations that describe the boundaries of tokens. Aggregate AEs consist of combinations of primitive AEs where downstream AEs may rely on annotations created during upstream processing. An example of an aggregate AE would be the GENIA tagger that uses token annotations created by a tokenizer as its input and adds POS tags to the tokens.
3.2.3 CAS consumer. The final major component in UIMA is the CAS Consumer. A CAS Consumer is any program that takes in a CAS as part of its input,however, they are not assumed to update the CAS. CAS consumers represent the end of the process. An example CAS consumer, particularly
relevant to this paper, would be the program which transforms the annotations into human-readable format.
4. Results and Discussion
4.1. Performance Evaluation
In this section, the prediction performance, including NERBio, PPI abstract classifier and SRL, for our BWS system was reported. The performance is evaluated in terms of precision, recall, and F-measure, which are defined as follows: items recognized of number the items recognized correctly of number the Precision= items true of number the items recognized correctly of number the Recall=
(
Precision Recall)
Recall Precision measure F + × × = − 2The datasets provided by the BioCreAtIvE II GM tagging task [6] was used to evaluate the NER and PPI article classifier, respectively. The precision, recall and F-measure of NER are 82.59%, 89.12% and 85.76%, respectively. Our PPI abstract classifier achieved an F-measure of 80.85% (with a precision of 91.2% and a recall of 78.4%).
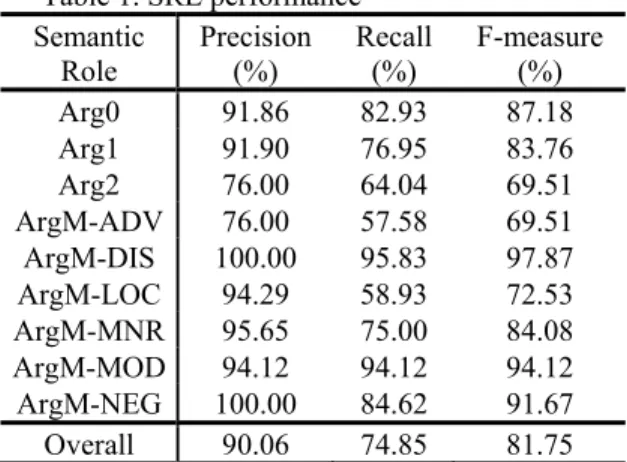
To evaluate the performance of our biomedical SRL on online retrieved sentences, our in-lab biologists annotated a gold-standard dataset which is composed of 100 randomly selected PubMed abstracts with 315 PASs. Table 1 shows the evaluation results.
Table 1. SRL performance Semantic
Role Precision (%) Recall (%) F-measure (%) Arg0 91.86 82.93 87.18 Arg1 91.90 76.95 83.76 Arg2 76.00 64.04 69.51 ArgM-ADV 76.00 57.58 69.51 ArgM-DIS 100.00 95.83 97.87 ArgM-LOC 94.29 58.93 72.53 ArgM-MNR 95.65 75.00 84.08 ArgM-MOD 94.12 94.12 94.12 ArgM-NEG 100.00 84.62 91.67 Overall 90.06 74.85 81.75 Table 2. Argument types and their descriptions
Type Description Arg0 Agent Arg1 Direct object/theme/patient
Arg2 Not fixed
ArgM-DIS Discourse connectives ArgM-LOC Location ArgM-MNR Manner
ArgM-MOD Modal verb
ArgM-NEG Negation marker As you can see in Table 1, our system achieved satisfactory F-measures (87.18% and 83.76%) for Arg0 and Arg1; in most cases, Arg0 is the subject and Arg1 is the object of a sentence. It shows that we can identify SVO relations with high accuracy. The description of each semantic role is described in Table 2. As to the overall performance, our biomedical SRL system achieved an F-measure of 81.75%, with a precision of 90.06% and a recall of 74.85%, which are slightly lower than the performance achieved by Tsai et al. [16] under the conditions in which gold-standard parses are given. This performance is close to state-of-the-art ML-based SRL systems in other specific domains [21].
4.2. The Benefits of adopting UIMA
Adopting UIMA framework into system development is not a trivial work; UIMA is not a lightweight architecture, and it requires software developers with mature software engineering skill. Despite these costs, the use of UIMA in our work does provide gains in efficiency over time. Following UIMA to define standard application programming interfaces (API) between different NLP components promotes the sharing of NLP components and eases the workload typically involved with integrating third-party software. For example, the GENIA tagger [19] which can provide the POS information, and LingPipe [18] which provides the sentence detection function are integrated into our system with little effort even though they were not originally designed be interoperable. In the following paragraph, we point out some advantages of applying UIMA framework for developing large-scale NLP systems based on our experience.
With the UIMA development paradigm, the common interface for passing data among components removes the need to write customized code for stitching together various processing modules. For software development, the processing modules can be isolated from the communications and data transfer mechanisms. This promotes more modular code and facilitates applying unit testing for individual components. For system integration, the standard API among components not only enables tools that were not originally designed to be integrated as a
subcomponent integrable and interoperable, but also promotes the sharing of those components among developers, and perhaps more importantly, among the NLP community. In fact, there are already some research groups starting to use UIMA, such as Tsujii’s UIMA repository4, the
JULIE Lab5 and the BioNLP UIMA component
repository6 and share their UIMA components
with NLP community. We believe that by using UIMA framework, developers can focus on tuning the performance of their individual components and make use of disparate resources easier to build complex interconnecting workflows.
5. Conclusion
In this paper, we have described the features of BWS, which include (1) NER including DNA, RNA, cell, protein and disease names; (2) PPI relevance ranking for abstracts; (3) semantic relation analysis of abstracts for selected biomedical verbs and extraction of a wide variety of relational information between sentence components such as agent, patient, negation, location, and time; and our experiences in developing BWS with the UIMA framework.
In the near future, BWS will allow users to specify the semantic role of each query term (agent, predicate, patient, etc.) to facilitate searching for specific biomedical relations. The system will also retrieve related sentences instead of entire abstracts to improve readability. In addition, the system will allow users to construct biomedical relation networks from single or multiple retrieved abstracts. Such networks will be presented in a navigable interface to allow visual browsing of complex relations such as biomedical pathways.
References
[1] J. M. Fernández, R. Hoffmann, and A. Valencia, "iHOP web services," Nucleic Acids Res., vol. 35, pp. W21-W26, 2007. [2] A. Eaton, "HubMed: a web-based
biomedical literature search interface," Nucleic Acids Research, vol. 34, p. W745, 2006.
[3] H.-J. Dai, H.-C. Hung, R. T.-H. Tsai, and W.-L. Hsu, "IASL Systems in the Gene Mention Tagging Task and Protein Interaction Article Sub-task," in Proceedings of Second BioCreAtIvE Challenge Evaluation Workshop, Madrid,
4 http://www-tsujii.is.s.u-tokyo.ac.jp/uima/ 5 http://www.julielab.de/
Spain, 2007, pp. 69-76.
[4] U. Leser and J. Hakenberg, "What makes a gene name? Named entity recognition in the biomedical literature," Briefings in Bioinformatics, vol. 6, p. 357, 2005. [5] R. T.-H. Tsai, C.-L. Sung, H.-J. Dai, H.-C.
Hung, T.-Y. Sung, and W.-L. Hsu, "NERBio: using selected word conjunctions, term normalization, and global patterns to improve biomedical named entity recognition," BMC Bioinformatics, vol. 7 Suppl 5, p. S11, 2006.
[6] L. Smith, L. K. Tanabe, R. J. n. Ando, C.-J. Juo, I.-F. Chung, C.-N. Hsu, Y.-S. Lin, R. Klinger, C. M. Friedrich, K. Ganchev, M. Torii, H. Liu, B. Haddow, C. A. Struble, R. J. Povinelli, A. Vlachos, W. A. B. Jr., L. Hunter, B. Carpenter, R. T.-H. Tsai, H.-J. Dai, F. Liu, Y. Chen, C. Sun, S. Katrenko, P. Adriaans, C. Blaschke, R. T. Perez, M. Neves, P. Nakov, A. Divoli, M. Mana, J. Mata-Vazquez, and W. J. Wilbur., "Overview of BioCreative II Gene Mention Recognition," Genome Biology, 2008.
[7] R. T.-H. Tsai, H.-C. Hung, H.-J. Dai, and Y.-W. Lin, "Exploiting Likely-Positive and Unlabeled Data to Improve the Identification of Protein-Protein Interaction Articles," 6th InCoB - Sixth International Conference on Bioinformatics, 2007.
[8] M. Krallinger and A. Valencia, "Evaluating the Detection and Ranking of Protein Interaction Relevant Articles: the BioCreative Challenge Interaction Article Sub-task (IAS)," Proceedings of Second BioCreAtIvE Challenge Evaluation Workshop, pp. 29-39, 2007.
[9] N. Xue and M. Palmer, "Calibrating features for semantic role labeling," Proceedings of EMNLP, vol. 4, 2004. [10] D. Ferrucci and A. Lally, "Building an
example application with the Unstructured Information Management Architecture," IBM Systems Journal, vol. 43, pp. 455-475, 2004.
[11] J. Lafferty, A. McCallum, and F. Pereira, "Conditional random fields: Probabilistic models for segmenting and labeling sequence data," in International Conference on Machine Learning (ICML), 2001, pp. 282–289.
[12] T. Joachims, "Text Categorization with
Suport Vector Machines: Learning with Many Relevant Features," in Proceedings of ECML-98, 10th European Conference on Machine Learning, 1998.
[13] H.-J. Dai, H.-C. Hung, R. T.-H. Tsai, and W.-L. Hsu, "IASL Systems in the Gene Mention Tagging Task and Protein Interaction Article Sub-task," in Proceedings of Second BioCreAtIvE Challenge Workshop, 2007.
[14] R. T.-H. Tsai, H.-C. Hung, H.-J. Dai, and W.-L. Hsu, "Exploiting Likely-Positive and Unlabeled Data to Improve the Identification of Protein-Protein Interaction Articles," in 6th InCoB - Sixth International Conference on Bioinformatics, 2007.
[15] W.-C. Chou, R. T.-H. Tsai, Y.-S. Su, W. Ku, T.-Y. Sung, and W.-L. Hsu, "A Semi-Automatic Method for Annotating a Biomedical Proposition Bank," Proceedings of ACL Workshop on Frontiers in Linguistically Annotated Corpora, pp. 5-12, July 22 2006.
[16] R. T.-H. Tsai, W.-C. Chou, Y.-S. Su, Y.-C. Lin, C.-L. Sung, H.-J. Dai, I. T. Yeh, W. Ku, T.-Y. Sung, and W.-L. Hsu, "BIOSMILE: A semantic role labeling system for biomedical verbs using a maximum-entropy model with automatically generated template features," BMC Bioinformatics, vol. 8, p. 325, 2007.
[17] A. L. Berger, V. J. Della Pietra, and S. A. Della Pietra, "A maximum entropy approach to natural language processing," Computational Linguistics, vol. 22, pp. 39-71, 1996.
[18] B. Baldwin and B. Carpenter, "LingPipe," in http://www.alias-i.com/lingpipe/. [19] Y. Tsuruoka, Y. Tateishi, J. D. Kim, T.
Ohta, J. McNaught, S. Ananiadou, and J. Tsujii, "Developing a robust part-of-speech tagger for biomedical text," Lecture notes in computer science, pp. 382-392, 2005.
[20] R. Grishman, "TIPSTER Architecture Design Document Version 2.2," DARPA, available at http://www.tipster.org, 1996. [21] X. Carreras and L. ı. M`arquez,
"Introduction to the CoNLL-2005 Shared Task: Semantic Role Labeling," Proceedings of CoNLL-2005, 2005.