行政院國家科學委員會專題研究計畫 成果報告
化學與光能轉換的基礎研究
計畫類別: 個別型計畫 計畫編號: NSC93-2119-M-009-001- 執行期間: 93 年 01 月 01 日至 93 年 12 月 31 日 執行單位: 國立交通大學應用化學系(所) 計畫主持人: 林明璋 報告類型: 完整報告 報告附件: 出席國際會議研究心得報告及發表論文 處理方式: 本計畫可公開查詢中 華 民 國 94 年 3 月 18 日
中文摘要 國科會於 2004 年以「國科會講座」延聘美國 Emory 大學講座教授林明璋院士自美返台 主持交通大學分子科學研究中心以致力發展最先進的研究計劃。除了在互益的基礎上,與台 灣物理、化學方面的學者就相關聯的研究主題進行數個合作研究計劃之外,為將分子科學研 究中心的研究發展維持在一定的水準上,近期內已提交以再生能源以及奈米粒子輔以超快電 子動力學與電子在系統中轉換方面之生物化學研究為主題的三個大型研究計劃。這些以理論 計算為輔的合作研究實驗包括: (1)由王念夏教授與兩位研究生研製以 pump-probe 雷射來發射螢光的 NCN 與 NO 反應動力學 實驗。 (2)由李遠鵬教授與一個博士生在清大進行並輔以 Emory 大學理論計算結果的氧原子與 CH3OH 在巨烈衝擊波反應的動力學實驗。 (3)由李遠哲與原分所同仁進行 C6H5NO 的光分裂動力學。 (4)建立一套低壓有機金屬化學蒸鍍設備與以研究 InN/TiO2奈米粒子薄膜系統中反覆轉換特 性為主的飛秒與奈秒雷射探測系統。 截至目前為止,本計劃已有四篇論文在科學期刊上發表或即將出刊,除此之外,並有十篇 在交通大學準備或撰寫的論文將會陸續以國科會贊助的名義在國際期刊上發表。 關鑑詞:NCN 與 NO 反應、O+CH3OH 反應、C6H5NO 光分解、InN/TiO2 太陽能研究。
英文摘要
The 2004 visit of M. C. Lin from Emory University, supported by National Science Council for the full year, had been devoted to the development of a forefront research program for the Center for Interdisciplinary Molecular Science (CIMS) at National Chiao Tung University, and to studies of several collaborative research projects with chemical physicists in Taiwan on selected topics of mutual interest. For the development of a sustainable research program for CIMS in the near future, 3 major proposals have been submitted for the Center with focus on renewable energy research and on biochemical systems on nanoparticles aided by studies on the dynamics of fast electron and energy transfer in those systems. In the collaborative studies, experiments aided by theoretical simulations have been carried out on (1) the kinetics of NCN reaction with NO by pump-probe laser-induced fluorescence with N. S. Wang and two graduate students; (2) the kinetics of O-atom reaction with CH3OH in shock waves with Y.-P. Lee and a PhD student at NTHU, aided by theoretical
calculations carried out at Emory University; (3) the photo-fragmentation dynamics of C6H5NO with
Y. T. Lee and coworkers at IAMS; and (4) the establishment of a low pressure organometallic chemical vapor deposition apparatus and femtosecond and nanosecond laser probing systems for forward and back electron transfer characterization in the InN/TiO2 nanoparticle film systems. Four
papers have been published or accepted for publication in SCI journals. In addition, 10 papers which were prepared or edited at NCTU by the PI have been credited to NSC for the support of the effort.
關鑑詞:CIMS’s program、NCN+NO kinetics、O+CH3OH kinetics、C6H5NO fragmentation
一、前言:
The objectives of this project for the period Jan. 1 – Dec. 31, 2004 centered on: (1) Establishment of a forefront research program at the Center for Interdisciplinary Molecular Science (CIMS) with emphasis on studies of electron and energy transfer dynamics in nano-particles and biological systems and (2) Collaboration with scientists in Taiwan on kinetics and dynamics of chemical reactions of interest to atmospheric chemistry, combustion and photo fragmentation dynamics, as well as the establishment of a research laboratory for preparation of InN/TiO2 nanoparticle films for solar energy conversion applications.
CIMS, established officially at NCTU in July 2003, has now over 10 faculty members with a broad spectrum of research fields. The Center’s major focus is placed on the applications of ultra-fast spectroscopy to study energy and electron transfer dynamics over a wide range of the spectrum (covering from the infrared to the UV). On account of the national need in renewable energy research to alleviate the acute shortage in Taiwan’s energy resources, the Center’s near-term research objectives will be placed on solar energy conversion and the catalytic studies of ethanol to H2 conversion for fuel cell applications.
In the proposed collaborative research projects, 4 full papers have been completed and submitted for publications (2 published and 2 accepted for publication), with additional 10 papers credited to NSC for works carried out at Emory University but written or edited by the PI during the past year visiting NCTU.
二、報告內容:
(一) Establishement of research programs at the Center for Interdisciplinary Molecular Science (CIMS) in National Chiao Tung University
CIMS was officially established at NCTU in July 2003. It has now over 10 faculty members from the campus and neighboring research institutions such as NSRRC with a broad spectrum of research fields. The most recent addition was Prof. Yuan-Pern Lee who moved to NCTU to be University Distinguished Professor on August 1, 2004. The Center’s major focus is placed on the applications of ultra-fast spectroscopy to study energy and electron transfer dynamics over a wide range of the spectrum (covering from the infrared to the UV). In order to train physical chemists with a broad range of expertise, fundamental gas phase spectroscopy and chemical kinetics with a solid training in quantum chemistry and statistical mechanics are essential. This area of research will be continually emphasized also.
On account of the national need in renewable energy research to alleviate the acute shortage in Taiwan’s energy resources (we import as much as 98% of our energy supplies) the Center’s research focus in the near future will be placed on solar energy conversion and the catalytic studies of H2 conversion from ethanol, a truly “green fuel” which may be
derived from sugar and other biomasses including celluloses with advanced fermentation processes, for H2 fuel cell applications.
In the past year, three major proposals have been submitted from CIMS which, if funded, should strengthen our current interdisciplinary collaborative projects funded by NCTU in the areas of bio- and nano-materials research.
(二) Progress in collaborative studies
The proposed research milestones for Jan. – Dec. 2004 as put forth in the original proposal include:
a. Kinetics of NCN reaction with NO studied by pump-probe LIF measurements with N. S. Wang and two PhD students at CIMS/NCTU to carry out experiment and theoretical calculations.
b. Kinetics of O-atom reaction with CH3OH in shock waves with Y.-P. Lee and a PhD student
at NTHU, aided by theoretical calculations to be carried out at CIMS/NCTU.
c. Photo-fragmentation study of C6H5NO with C.-K. Ni and Y. T. Lee at IAMS, aided by
theoretical calculations to be carried out at CIMS/NCTU.
d. Photoluminescence measurement for the bandgap and electron mobility of InN/TiO2
nano-structures deposited at Emory using a femtosecond laser at CIMS /NCTU with E. W. G. Diau and a PhD student.
The progress in these collaborative research projects are briefly summarized below: 1. Atmospheric chemistry
a. NCN + NO kinetics
In collaboration with N.S. Wang’s group at NCTU, the kinetics for the reaction of NO with the NCN radical, a key prompt NO precursor reaction intermediate in the new CH + N2 mechanism put forth by Lin and coworkers,[1,2] has been investigated. The rate
constants for the reaction have been measured by laser photolysis/laser induced fluorescence technique in the temperature range 254 – 353 K in the presence of He (40 – 600 Torr) and N2 (30 – 528 Torr) buffer gases. The NCN radical was produced from the
photodissociation of NCN3 at 193 nm and monitored with a dye laser at 329.01 nm. The
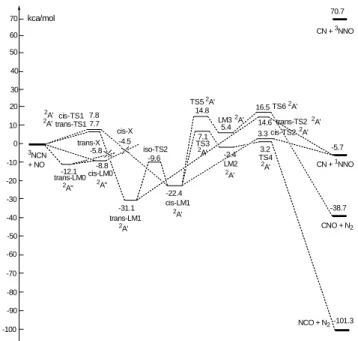
reaction was found to be strongly positive-pressure dependent with negative-temperature dependence, as was reported previously. The experimental data could be reasonably accounted for by dual channel RRKM calculations based on the predicted potential energy surface using the G2M method (see Figs. 1 and 2). The reaction is predicted to occur via weak intermediates, cis- and trans-NCNNO, in the 2A” state which crosses with the 2A’ state containing more stable cis- and trans-NCNNO isomers. The high barriers for the fragmentation of these isomers and their trapping in the 2A’ state by collisional stabilization give rise to the observed positive pressure dependence and negative temperature effect. The predicted energy barrier for the fragmentation of the cis-NCNNO (2A’) to CN + N2O
also allows us to quantitatively account for the rate constant previously measured for the reverse process CN + N2O → NCN + NO.[3]
b. Kinetics of the S + O2 reaction
In collaboration with Y-P Lee’s group, we have investigated the kinetics of the S + O2 reaction, which has not been well characterized to date. The rate coefficients of the
reaction at 50 Torr Ar pressure in the temperature range 298−878 K were determined with the laser photolysis technique. S atoms were generated by photolysis of OCS with a KrF excimer laser at 248 nm; their concentration was monitored via resonance fluorescence
excited by atomic emission of S produced from microwave-discharged SO2. Our
measurements show that k(298 K) = (1.92±0.29)×10−12
cm3 molecule−1 s−1, in satisfactory agreement with previous reports. New data determined for 505−878 K show non-Arrhenius behavior; combining our results with data reported at high temperatures, we derive an expression k(T) = (9.02±0.27)×10−19
T 2.11±0.15 exp[(730±120)/T] cm3 molecule−1 s−1 for 298 ≤ T/K ≤ 3460. Theoretical calculations at the G2M(RCC2) level, using geometries
products relative to those of the reactants (see Fig. 3). Rate coefficients predicted with multichannel RRKM calculations agree satisfactorily with experimental observations (see Fig. 4); the reaction channel via SOO(1A') dominates at T < 500 K, whereas channels involving formation of SOO(3A") followed by isomerization to SO2 before dissociation,
and formation of SOO(1A") followed by direct dissociation, become important at high temperatures, accounting for the observed rapid increase in rate coefficient.
Fig. 1. The potential energy surface of the NCN + NO system
CN + 1NNO CNO + N2 NCO + N2 0 10 20 -10 -20 -30 -40 -50 -60 -70 -80 -90 3NCN + NO cis-TS1 cis-LM1 LM2 LM3 cis-TS2 TS3 TS4 TS5 TS6 7.8 -22.4 7.1 -2.4 3.2 -101.3 -100 14.8 5.4 16.5 -38.7 3.3 -5.7 kca/mol 7.7 -31.1 trans-TS1 trans-LM1 -8.8 -12.1 cis-X trans-X cis-LM0 trans-LM0 -4.5 -5.8 30 40 50 60 70 70.7 CN + 3NNO -9.6 iso-TS2 2A" 2A" 2A' 2A' 2A' 2A' 2A' 2A' 2A' 2A' 2A' 2A' 2A' 14.6 trans-TS22A'
Fig. 2. Comparison of the predicted and measured rate constant for NCN + NO
1.0 1.5 2.0 2.5 3.0 3.5 4.0 -14.0 -13.5 -13.0 -12.5 -12.0 -11.5 -11.0 -10.5 -10.0 Experimental values: 3 Torr, ref. 8 80 Torr, this paper 300 Torr, ref. 8 400 Torr, this paper 600 Torr, ref. 8 Log (k/[cm 3molecule -1s -1]) 1000/T [K-1 ]
Theoretical values of this paper: 3 Torr 80 Torr 300 Torr 400 Torr 600 Torr 1.0 1.5 2.0 2.5 3.0 3.5 4.0 -13.0 -12.5 -12.0 -11.5 -11.0 -10.5 -10.0 Log (k/[cm 3mo lecule -1s -1]) 1000/T [K-1 ]
Theoretical values of this paper: 100 Torr
400 Torr
Experimental values of this paper: 100 Torr
400 Torr
(a)
Fig. 3. Potential energy diagrams of the S-O2 system
2. Combustion reactions—High temperature kinetics of the O + CH3OH reaction.
CH3OH is an important alternate fuel to gasoline. In collaboration with Y-P Lee’s
group, we have studied the kinetics of the O + CH3OH reaction, which is not available
under the combustion conditions. The rate coefficients of the reaction in the temperature range 835−1777 K were determined using a diaphragmless shock tube. O atoms were generated by photolysis of SO2 with a KrF excimer laser at 248 nm or an ArF excimer laser
at 193 nm; their concentrations were monitored via atomic resonance absorption excited by emission from a microwave-discharged mixture of O2 and He. Rate coefficients determined
for the temperature range can be represented by the Arrhenius equation: k(T) = (2.29±0.18)×10−10 exp [−(4210±100)/T] cm3
molecule−1 s−1; unless otherwise noted, all listed errors represent one standard deviation in fitting. Combination of these and previous data at lower temperature shows a non-Arrhenius behavior described as the three-parameter equation k(T) = (2.74±0.07)×10−18
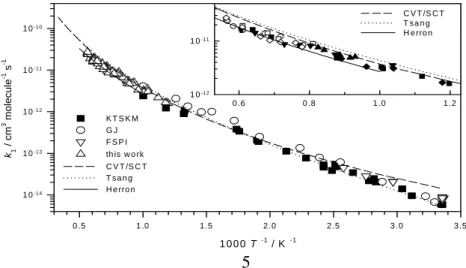
T 2.25±0.13 exp [−(1500±90)/T] cm3 molecule−1 s−1. Theoretical calculations at the B3LYP/6-311+G(3df, 2p) level locate three transition states (see Fig. 5). Based on the energies computed with CCSD(T)/6-311+G(3df, 2p)//B3LYP/6-311+G(3df, 2p), rate coefficients predicted with canonical variational transition state theory with small curvature tunneling corrections agree satisfactorily with experimental observations (see Fig. 6). The branching ratios of two accessible reaction channels forming CH2OH + OH (1a) and CH3O + OH (1b) are predicted to vary strongly with temperature. At
300 K, reaction (1a) dominates, whereas reaction (1b) becomes more important than reaction (1a) above 1700 K.
Fig. 4. Comparison of the predicted and measured rate constant for the S + O2 reaction k / cm 3 mo lec u le -1 s -1 1 0- 1 5 1 0- 1 4 1 0- 1 3 1 0- 1 2 1 0- 1 1 1 03 T- 1 / K- 1 0 1 2 3 1 0- 1 2 1 0- 1 1 f r o m t h e r e v e r s e r x . ( O + S O ) A B 1 2 3 4
Fig. 5. Energy diagram of the O-CH3OH system
0 . 0 1 0 . 6 6 . 3 5 2 . 7 T S 3 T S 2 T S 1 O (3 P ) + C H3O H H O O + C H 3 C H3O + O H C H2O H + O H 2 8 . 1 2 . 4 - 5 .5
Fig. 6. Comparison of the predicted with measured rate constant for O + CH3OH reaction
0 .5 1 .0 1 .5 2 .0 2 .5 3 .0 3 .5 k1 / cm 3 mo le c u le -1 s -1 1 0-1 4 1 0-1 3 1 0-1 2 1 0-1 1 1 0-1 0 K T S K M G J F S P I th is w o rk C V T /S C T T s a n g H e rro n 0 .6 0 .8 1 .0 1 .2 1 0-1 2 1 0-1 1 C V T /S C T T s a n g H e rro n
3. Phtofragmentation of C6H5NO
Nitrosobenzene is a key photolytical source of the phenyl radical. In collaboration with Y. T. Lee, C. K. Ni et al. at IAMS, the dynamics of the photofragmentation of C6H5NO
has been studied using multimass ion imaging techniques. Photodissociation at 248 nm shows that there is only one dissociation channel, i.e., C6H5NO → C6H5 + NO, regardless of
the fact that the other channel C6H5NO → C6H4 + HNO is energetically accessible in
agreement with theoretically predicted result (see Fig. 7). Photodissociation at 193 nm also shows the same dissociation channel. However, about 10% of the C6H5 radicals produced at
this wavelength further decomposed into benzyne and H atom, and the dissociation rates of phenyl radical as a function of internal energies were measured. The averaged photofragment translational energies released from the dissociation of nitrosobenzene at 193 nm and 248 nm are 10.2 and 6.9 kcal/mol, respectively, and fragment distributions are almost isotropic at both wavelengths (see Fig. 8) In addition, the thermal rate constant for dissociation of C6H5NO has been computed and compared with experimental data; the
agreement between theory and experiment is excellent, confirming the most recently reported unusually high A-factor (see Fig. 9).[4]
Fig. 7. Energy diagram of the C6H5NO system
C6H5 + NO TS C6H4 + HNO C6H5NO, X1A' C6H4N(H)O Ere l (k ca l/ mol ) 0.0 86.2 56.6 66.6 60.3 24 8 n m 1 9 3 nm C6H4 + H + NO 134.0 C6H5NO, 3A" C6H5NO, 1A" 35.3 23.8 S0 S1 S2 h ν 90.4 (89.3) 35.9 (32.9) 0.0
Fig. 8. (a) Photofragment ion image intensity profiles of m/e = 77 from two different photolysis laser polarizations at 248 nm. Thick and thin lines represent the polarization of UV laser perpendicular and parallel to the VUV laser beam, respectively. (b) Anisotropy parameter β as a function of fragment translational energy.
0 100 200 300 400 500 0 20 40 60 80 100 In te n s it y (arb .) CCD pixel 0 5 10 15 20 -1.0 -0.5 0.0 0.5 1.0 1.5 2.0 β
Translational energy (kcal/mol)
(a)
(b)
Fig.9. Experimental and predicted thermal rate constants for the dissociation of C6H5NO at
the high-pressure limit. Points are experimental data; solid, dash-dotted, and dotted lines correspond to the prediction for the states of X 1A′, 1A″, and 3A″, respectively. Their sum gives k∞ = 1.52 × 1017 exp[-55200/RT] s-1
1.0 1.2 1.4 1.6 1.8 2.0 -25 -20 -15 -10 -5 0 5 10 15 ln (k ; s -1 ) 1000 / T (K-1)
4. Deposition and characterization of InN/TiO2 nanoparticle films
The PI’s group at Emory University has recently demonstrated that the chemically
robust InN film can be directly deposited on TiO2 nanoparticles by OMCVD
(organomettalic chemical vapor deposition) with very good physical adhesion.[5] The films exhibit a very broad UV/vis absorption covering 400 – 800 nm region of the spectrum. In
order to explore the possibility of developing the InN/TiO2 system for solar energy
conversion devices, efforts have been made through a series of experiments at CIMS, NCTU. In the following summary, we report the progress made in these efforts.
a. Construction of a small OMCVD system and a photoluminescence apparatus
A new OMCVD system has been constructed at CIMS for a new series of InN thin-film deposition on TiO2 nanoparticles with emphasis on the improvement of chemical
connectivity between InN and TiO2 for enhancement of electron transfer rates. The
interface materials or functional groups may include electron withdrawing or electron providing elements such as B or P, respectively. Other potential materials include semi-conductive metal oxide quantum dots and nanoparticles. The schematic diagram of the OMCVD system is shown in Fig. 10. In addition, a photoluminescence measurement apparatus has been assembled for thin-film characterization as shown in Fig. 11. These systems will be used in our continuing exploration of the new generation of cheap solar energy conversion devices.
Fig. 10. Sample holder and CVD chamber of CIMS’s OMCVD apparatus
Fig. 11. Photoluminescence apparatus for solid surface characterization at 4 K or 77 K.
b. Establishment of a femtosecond pump-probe system for electron and energy transfer dynamics studies
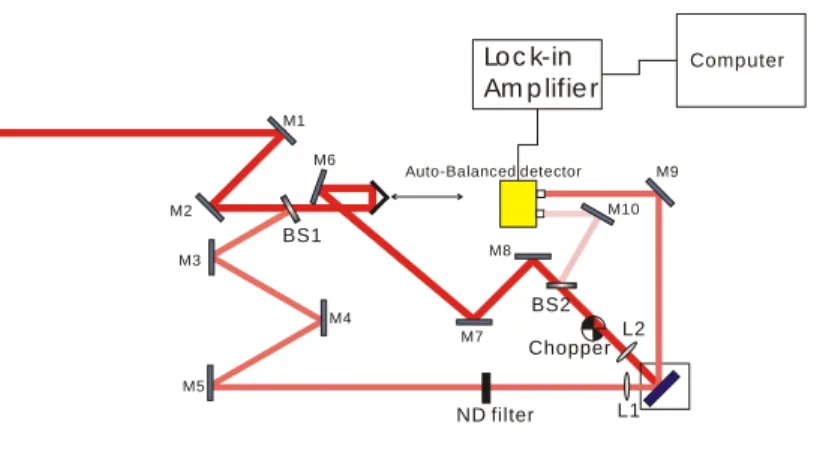
In this year, we have successfully built a new fs pump probe system, and the experimental set up is shown in Fig 12. Briefly, a fs mode-locked Ti-Sapphire oscillator
UV lamp To vacuum Sapphire window In(CH3)3 View port HN3 1-torr head Deposition plate Ti foil feed-thermocouple Thermal resist
purpose of studying solar energy systems, the laser wavelength can be further extended to the visible region (525-665 nm) via coupling the oscillator with an OPO (Optical Parametric Oscillator) device. The laser beam was divided into two parts via a beam splitter (BS1) and used as pump (~80%) and probe (~ 20%). A computer-controlled optical delay line was set in pump path to vary the delay time between pump and probe pulses. A small portion of the pump beam was reflected by BS2 and used as the reference of the auto-balanced detector. The major part of the pump beam was modulated by a mechanical chopper (~1 kHz) and then focused onto the thin-film sample; the average power of pump beam was ~400 mW at 800 nm. For the probe beam, the power was attenuated to ~1/10 (40 mW) of the pump beam by a neutral-density filter, and focused onto the sample overlapping with pump beam at exactly the same position. The reflected probe beam was collected as the signal in auto-balanced detector and then the signal was sent to lock-in amplifier controlled by a computer.
In the present stage, we have successfully measured the ultrafast carrier dynamics of the reference compound (GaAs) by the constructed fs pump-probe system. Fig 13 shows our initial test data for the transient signal representing the photo-reflectance change of GaAs as a function of time at 800 nm.
Fig. 12. Experimental set-up of fs pump-probe system
M1 M2 M3 M4 M5 M6 M7 M8 M9 M10 BS2 BS1 Chopper L1 L2 ND filter Loc k-in Am p lifie r Computer Auto-Balanced detector
Fig 1 : Experimental set-up of fs pump-probe system
Fig. 13. Initial test data for the transient signal representing the photo-reflectance change of GaAs as a function of time at 800 nm
c. Establishment of nanosecond transient absorption system
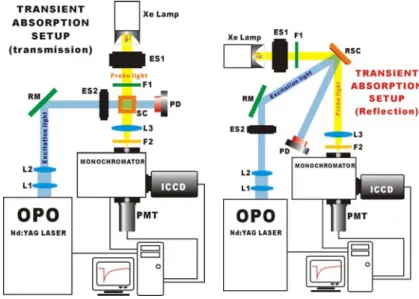
A nanosecond transient absorption spectrometer has been reconstructed for the kinetics measurements in the condensed phase. The arrangement of the setup is summarized as follows (see fig. 14). The excitation source generates excited-state species under investigation; a ns Nd:YAG/OPO system (funded by the NSC project) was used as the light source to cover a broad excitation wavelength range (210-2300 nm). A pulsed Xe lamp (1-5 µs duration), synchronously triggered by the excitation laser, was used for measurements of the time-dependent absorption spectra of the sample; the transient absorption spectra at a certain delay time can be recorded by the gated intensified charged-coupled devices (ICCD). Alternatively, the time-evolution profile of the sample at a certain absorption wavelength can be obtained by a sensitive photomultiplier tube (PMT). The current experimental setup is suitable for samples dissolved in solution (a transmission design shown in the left-hand side); in the future the system can be modified to a reflection design (shown in the right-hand side of the figure) for the purpose of electron back transfer kinetics measurements on samples such as InN functionalized on nanocrystaline TiO2 thin films.
Fig. 14. Nanosecond transient absorption setup for electron back transfer kinetic studies.
RM: reflective mirror; L: lens; ES: electronic shutter; F: filter; SC: sample cell; RSC: reflective sample cell; PD: photodiode sensor.
References:
[1] L. V. Moskaleva, W. S. Xia and M. C. Lin, "The CH + N2 Reaction over the Ground
Electronic Doublet Potential Energy Surface: A Detailed Transition State Search", Chem. Phys. Lett., 331, 269-77 (2000).
[2] L. V. Moskaleva and M. C. Lin, "The Spin-Conserved Reaction CH + N2→H + NCN: A
Major Pathway to Prompt NO Studied by Quantum/Statistical Theory Calculations and Kinetic Modeling of Rate Constant", Proc. Combust. Intst., 28 (Part II), 2393-401 (2000). [3] N. S. Wang, D. L. Yang, M. C. Lin and C. F. Melius, "Kinetics of CN Reactions with N2O
and CO2", Int. J. Chem. Kinet., 23, 151 (1991).
[4] J. Park, I. V. Dyakov, A. M. Mebel, and M. C. Lin, "Experimental and Theoretical Studies of the Unimolecular Decomposition of Nitrosobenzene: High-Pressure Rate Constants and the C-N Bond Strength", J. Phys. Chem. A, 101, 6043 (1997).
三、計畫成果自評:
All of the proposed collaborative projects described above had been successfully carried out with 2 papers already published in the world’s top chemical physics journals and 2 more accepted for publications. In addition, 10 papers written or edited by the PI during the visit at NCTU have been credited to NSC. They are listed below:
Papers published or accepted for publications:
a. Cheng-Ming Tzeng,Y. M. Choi, Cheng-Liang Huang,Chi-Kung Ni, Yuan T. Lee, and M. C. Lin, “Photodissociation of nitrosobenzene and decomposition of phenyl radical”, J. Phys. Chem., A, 108, 7928-35, (2004).
b.Chih-Wei Lu and Yu-Jong Wu, Yuan-Pern Lee, R. S. Zhu and M. C. Lin, “Experimental and Theoretical Investigations of Rate Coefficients of the Reaction S(3P) + O2 in the Temperature
range 298−878 K”, J. Chem. Phys., 121, 8271-78 (2004).
c.Chih-Liang Huang, Shiang Yang Tseng, Tzu Yi Wang, N. S. Wang, Z. F. Xu and M. C. Lin, “Reaction Mechanism and Kinetics of the NCN + NO Reaction: Comparison of Theory and Experiment”, J. Chem. Phys., in press.
d.Chih-Wei Lu and Shen-Long Chou,Yuan-Pern Lee, Shucheng Xu, Z. F. Xu and M. C. Lin, “Experimental and Theoretical Studies of Rate Coefficients for the Reaction O (3P) + CH3OH at High Temperatures”, J. Chem. Phys., in press.
Other publications prepared at NCTU during 2004 with acknowledgments to NSC:
1.R. S. Zhu and M. C. Lin, “RateConstants of ClO + NO for the Forward and Reverse Processes, ChemPhysChem, 5, 1864-70 (2004).
2.R. S. Zhu and M. C. Lin, “Ab Initio Study of the Oxidation of NCN by O2”, Int. J. Chem.
Kinet., in press.
3.J. H. Wang and M. C. Lin, “Adsorption and Reaction of N2H4 on Si(100)-2 × 1: A
Computational Study with Single- and Double-Dimer Cluster Models”, Surf. Sci., in press. 4. Shucheng Xu and M. C. Lin, “A Computational Study on the Kinetics and Mechanism for
the Unimolecular Decomposition of C6H5NO2 and the Related C6H5 + NO2 and C6H5O + NO
Reactions”, J. Phys. Chem., A, in press.
5.R. S. Zhu and M. C. Lin, “Ab initio Studies of ClOx Reactions: XI. Prediction of the Rate
Constants of ClO + NO2 for the Forward and Reverse Processes”, ChemPhysChem,
submitted.
6.Jenghan Wang, M. C. Lin and Ying-Chieh Sun, “Reactions of Hydrazoic Acid on TiO2
Nanoparticles: An Experimental and Computational Study”, J. Phys. Chem, B, in press. 7.Y. M. Choi and M. C. Lin, “Kinetics and Mechanisms for Reactions of HNO with CH3 and
C6H5 Studied by Quantum-chemical and Statistical-theory Calculations”, Int. J. Chem. Kinet.,
in press.
8.Jenghan Wang and M. C. Lin, “Reactions of Trimethyl Indium on TiO2 Nanoparticles: An
Experimental and Computational Study”, J. Phys. Chem, B, submitted..
9.R. S. Zhu, J. Park and M. C. Lin, “Ab initio Kinetic Study of the Low Energy Paths of the HO + C2H4 Reaction”, Chem. Phys. Lett., submitted.
10. Z. F. Xu, H.-C. Hsu and M. C. Lin, “Ab Initio Kinetics of the HCO Reaction with NO: Abstraction vs. Association/Elimination Mechanism”, J. Chem. Phys., Submitted
Photodissociation of nitrosobenzene and decomposition of phenyl radical
Cheng-Ming Tzeng,1 Y. M. Choi,2 Cheng-Liang Huang,1,3 Chi-Kung Ni1*,Yuan T. Lee,1,4 and M. C. Lin,2,5*
1. Institute of Atomic and Molecular Sciences, Academia Sinica, P. O. Box: 23-166, Taipei, Taiwan
2. Department of Chemistry, Emory University, Atlanta, Georgia 30322 USA
3. Present address: Department of Applied Chemistry, National Chiayi University, Chiayi, Taiwan
4. Department of Chemistry, National Taiwan University, Taipei, 106, Taiwan
5. Center for Interdisciplinary Molecular Science, National Chiao Tung University, Hsinchu 300, Taiwan.
---
Abstract
Photodissociation of nitrosobenzene in a molecular beam has been studied using multimass ion imaging techniques. Photodissociation at 248 nm shows that there is only one dissociation channel, i.e., C6H5NO → C6H5 + NO, regardless of the fact that the other channel C6H5NO →
C6H4 + HNO is energetically accessible in agreement with theoretically predicted result.
Photodissociation at 193 nm also shows the same dissociation channel. However, about 10% of the C6H5 radicals produced at this wavelength further decomposed into benzyne and H atom, and
the dissociation rates of phenyl radical as a function of internal energies were measured. The averaged photofragment translational energies released from the dissociation of nitrosobenzene at 193 nm and 248 nm are 10.2 and 6.9 kcal/mol, respectively, and fragment distributions are almost isotropic at both wavelengths. In addition, the thermal rate constant for dissociation of C6H5NO has been computed and compared with experimental data; the agreement between
theory and experiment is excellent, confirming the most recently reported unusually high A-factor (>1017 s-1).
I. Introduction
Nitrosobenzene has been popularly employed photolytically as the precursor of phenyl radicals in many kinetic and spectroscopic studies because of its large extinction coefficient in the UV region.1-8 The molecule has a large energy gap between the S
1 and S2 states.9,10 Hence,
the possibility of fluorescence from the S2 state has been discussed.11 However, no fluorescence
has been observed. 12-14 The existence of a fast decay channel in the S
2 state is therefore
expected. Indeed, it was found that the dissociation of nitrosobenzene in argon matrices at 12 K is very efficient upon irradiation of UV photons.15
The dynamics of photodissociation of nitrosobenzene in the gas phase have been studied recently.16-22 However, conclusions are very different from these studies. Dick and coworkers
measured the absorption spectrum of the S0 - S2 transition of the ultracold nitrosobenzene in a
supersonic jet.17 A lifetime of the S
2 state determined directly by the homogeneous linewidth of
the absorption spectrum was found to be 60 ± 3 fs. They also measured the alignment, velocity distribution, and populations of the rotational and vibrational states of the NO fragments via laser induced fluorescence and ion imaging technique at various UV wavelengths.18-20 Fragments
have an isotropic velocity distribution and no alignment was observed. In addition, the NO rotational population has a statistical distribution and only 10 % of the NO fragments are
2) decays rapidly through internal conversion to the S1 or S0 state, and the dissociation occurs
through a statistical mechanism on the potential surface of the lower state on a timescale much slower than rotation of the parent molecule. On the other hand, Han and coworkers also have studied the photodissociation of nitrosobenzene at 266 nm recently.21,22 The anisotropy
parameter β = -0.64 was found from the time-of-flight spectra of NO and C6H5 photofragments.
Their laser induced fluorescence study also demonstrated that more than 60 % of the NO fragments are populated in the vibrational excited state and the NO fragment rotational temperature is much higher than that measured by Dick et al.
In all existing studies, including those kinetic and spectroscopic papers cited above, only one dissociation channel, C6H5NO → C6H5 + NO, has been assumed and/or detected in the
photodissociation of nitrosobenzene in the UV region, despite the fact that the C6H4 + HNO
product channel is accessible at 248 nm while the C6H4 + H + NO product channel becomes
energetically accessible at 193 nm.
In this report, photodissociation of nitrosobenzene at 193 nm and 248 nm were studied using multimass ion imaging techniques. Particular attention has been focused on the search of the other possible dissociation channel and the fragment anisotropy measurement. In addition, the decomposition of the phenyl radical produced from the photodissociation reaction and the
thermal decomposition of nitrosobenzene were also studied; the data will be discussed in reference to the ab initio MO and statistical theory results.
II. Experimental and Computational Methods
Experimental setup. The experimental techniques have been described in detail in our previous reports on other aromatic molecules,23-26 and only a brief description is given here.
Nitrosobenzene vapor was formed by flowing ultrapure He at pressures of 500 Torr through a reservoir filled with liquid sample at 293 K. The nitrobenzene/He mixture was then expanded through a 500 µm pulsed nozzle to form the molecular beam. Molecules in the molecular beam
were photodissociated by an UV laser pulse. Due to the recoil velocity and center-of-mass velocity, the fragments were expanded to a larger sphere on their flight to the ionization region, and then ionized by a VUV laser pulse. The distance and time delay between the VUV laser pulse and the UV photolysis laser pulse were set such that the VUV laser beam passed through the center-of-mass of the dissociation products, and generated a line segment of photofragment ions by photoionization. The length of the segment was proportional to the fragment recoil velocity in the center-of-mass frame multiplied by the delay time between the photolysis and the ionization laser pulses. To separate the different masses within the ion segment, a pulsed electric
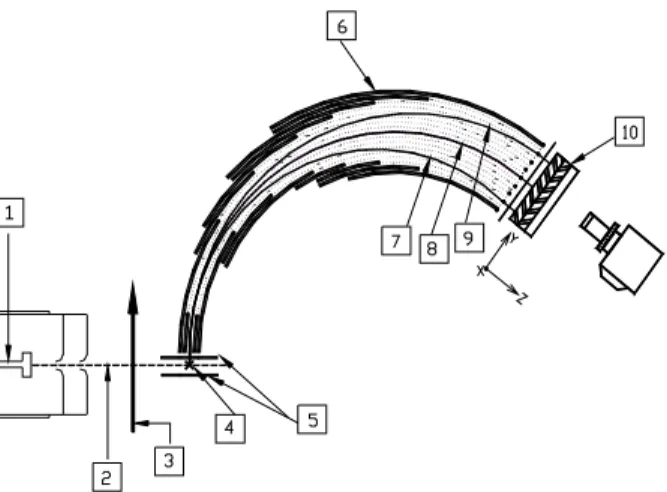
field was used to extract the ions into a mass spectrometer after ionization. While the mass analysis was being executed in the mass spectrometer, the length of each fragment ion segment continued to expand in the original direction according to its recoil velocity. At the exit port of the mass spectrometer, a two-dimensional ion detector was used to detect the ion positions and intensity distribution. In this two-dimensional detector, one direction was the recoil velocity axis and the other was the mass axis. The schematic diagram of the experimental set up is shown in Fig.1.
Fig. 1. Schematic diagram of the multimass ion imaging detection system. (1) nozzle; (2) molecular beam; (3) photolysis laser beam; (4) VUV laser beam, which is perpendicular to the plane of the paper; (5) ion extraction plates; (6) energy analyzer; (7), (8) and (9) simulation ion trajectories of m/e = 16, 14, 12; (10) Two-dimensional detector, where Y-axis is mass axis, and X-axis (perpendicular to the plane of the paper) is the velocity axis.
Computational method. The hybrid density functional B3LYP27-29 with three split
valence basis sets, 6-31G(d), 6-31+G(d), and 6-31G(2df,p), has been applied to optimize the geometries of the reactant, intermediate, transition state, and products with tight convergence criterion. All the stationary points were identified by the number of imaginary frequencies (NIMG) with NIMG = 0 for stable species and NIMG = 1 for transition states, as well as by the normal mode analysis. To continue to study the unimolecular reaction of C6H5NO based on our
earlier work30 at the G2M(rcc, MP2) and G2M(RCC, MP2) levels,31 in this report we also
performed quantum-chemical calculations with the G2M(RCC, MP2) scheme. A series of single-point energy calculations for the G2M (RCC, MP2) composite scheme using the geometries and zero-point energy (ZPE) corrections from the B3LYP/6-31G(d) and
B3LYP/6-31+G(d) levels of theory have been carried out to obtain more reliable information on energetics for the potential energy surface (PES) and the rate constant prediction by the
following scheme:
E0[G2M] = RCCSD(T)/6-311G(d,p) + MP2/6-311+G(3df,2p) – MP2/6-311G(d,p)
+ ∆HLC + ZPE
The empirical ∆HLC is given by -5.3nβ - 0.19nα in mhartree, where nα and nβ are the numbers of
α and β valence electrons, respectively. Some of the energies have also been calculated by the G3SX composite method32 with the geometries optimized at the B3LYP/6-31G(2df,p) level.
Since the method does not include any empirical correction, this method has been proposed to apply for the system whose number of spins is changed like the present system. Therefore, we can compare the two composite schemes for the issue on the spin change. In addition,
time-dependent density functional theory (TD-DFT)33-35 at the B3LYP/6-31+G(d) level has been
used to calculate the vertical excitation energies and oscillator strengths for the S1 and S2 states
of C6H5NO. The electronic structure calculations were carried out with the Gaussian 9836 and
MOLPRO 9837 programs.
III. Results and Discussions
(a) Dissociation of nitrosobenzene at 248 nm
Ions of m/e = 30, 77 and 78 were the only two fragments we observed from the photodissociation of nitrosobenzene at this wavelength. m/e = 78 is the corresponding 13C
isotope of fragment m/e = 77. Photofragment ion images are shown in Figs. 2(a) and 2(b). Photolysis laser intensity in the region between 2.5 ~ 30 mJ/cm2 were used. They showed the
same shapes of image. It suggests that one-photon absorption is the dominant process in this laser intensity region. Since there was no HNO and C6H4 detected, we can conclude that there is
only one dissociation channel at 248 nm, i.e., C6H5NO → C6H5 + NO, in spite of the fact that the
0 10 20 30 40 50 60 70 0.0 0.2 0.4 0.6 0.8 1.0 1.2 1.4 Probability (a rb .)
Translational energy (kcal/mol) Velocity (a) (b) m30 m77 (c)
Fig. 2. Photofragment ion image of (a) m/e = 30, (b) m/e = 77 and 78, and (c) photofragment translational energy distribution at 248 nm. Arrow indicates the maximum available energy.
nitrosobenzene is a “clean” precursor to generate phenyl radical at this wavelength.
The photofragment translational energy distribution obtained from the images is illustrated in Fig. 2(c). It shows that the probability monotonically decreases with the increasing
translational energy. The average released translational energy is about 6.9 kcal/mol, and it is about 11 % of the total available energy. Compared to the average translational energy measured
in previous studies, our value is much smaller than 29 % of the fragment translational
spectroscopy measurement at photolysis wavelength 266 nm,22 and it is very close to 7.3 % and 6
~ 11 % of velocity map ion imaging measurement at 290.5 and 225.96 nm, respectively.20
The C6H5 photofragment ion image profiles at two different photolysis laser polarizations are
presented at Fig. 3(a). The shapes and the intensities of the profiles at the polarization directions parallel and perpendicular to the VUV probe laser beam are very close to each other. The anisotropy parameter β for the fragments with different translational energy is illustrated in Fig. 3(b). At low translational energy region where most of the fragments are produced, the values of β are very close to zero, indicating the isotropic distribution of the fragments. At high
translational energy region, the values of β fluctuate between 0 and 1. The poor S/N ratios are due to the small amount of high translational energy fragments produced at high translational energy region. We can conclude that most of the fragments are isotropically distributed. If there is any anisotropic distribution, it must be from the fragments with large translational energy. However, the amount of those fragments is very small, and the value of β is positive.
The anisotropy parameter β we measured at 248 nm is very close to the value of 0.05 from the single line measurement at 225.96 nm.20 It is also very close to the value of 0.03 ~ –0.05
0 100 200 300 400 500 0 20 40 60 80 100 In ten s it y (a rb .) CCD pixel 0 5 10 15 20 -1.0 -0.5 0.0 0.5 1.0 1.5 2.0
β
Translational energy (kcal/mol) (a)
(b)
Fig. 3. (a) Photofragment ion image intensity profiles of m/e = 77 from two different photolysis laser polarizations at 248 nm. Thick and thin lines represent the polarization of UV laser perpendicular and parallel to the VUV laser beam, respectively. (b) Anisotropy parameter β as a function of fragment translational energy.
from the measurement of several rotational states of NO v = 0 at photolysis wavelength 290.5 nm.20 However, it is very different from the value of –0.64 obtained from the photofragment
translational energy measurement at 266 nm.22 Both the translational energy distribution and
anisotropy parameter β measurement suggest that our results are similar to that of Dick’s, but they are different from that of Han’s.
(b) Dissociation of nitrosobenzene at 193 nm
Fragment ions of m/e = 30, 76, 77 and 78 were observed from the photodissociation of C6H5NO at 193 nm. Photolysis laser intensity in the region between 0.2 ~ 1.7 mJ/cm2 were used,
and all of these fragment intensities showed linear dependence on the laser intensity. The images are shown in Fig. 4. As the delay time between pump and probe laser pulses increased, the relative intensity between m/e = 76 and 77 also changed, as shown in Fig. 5. Since no HNO fragment was detected, fragments of m/e = 76 must result from the slow dissociation of the energetic fragments m/e = 77. Consequently, we can conclude that there is still only one
dissociation channel of nitrosobenzene at 193 nm, even though the photon energy is much higher than the dissociation barrier of C6H5NO → C6H4 + HNO. This observation is consistent with the
significantly smaller decay rate predicted for this product channel as will be presented later. The photofragment translational energy distribution obtained at this wavelength is shown in Fig. 4(c). The average energy released is 10.2 kcal/mol, which is slightly larger than that at 248 nm.
0 10 20 30 40 50 0.0 0.2 0.4 0.6 0.8 Pr obab ility (arb.)
Translational energy (kcal/mol)
Velocity
(a)
(b)
m30
m77
(c)
m76
Fig. 4. Photofragment ion image of (a) m/e = 30, (b) m/e = 76, 77 and 78, and (c) photofragment translational energy distribution at 193 nm.
C6H5 photofragment ion image intensity profiles at two different photolysis laser polarizations
was found to be very close to each other, as presented in Fig. 6(a). The values of β as a function of translational energy are shown in Fig. 6(b). It suggests that most of the fragments are iso-
tropically distributed, and only a small amount of fragments with large translational energy have a positive value of β. The dissociation mechanism at 193 nm must be similar to that at 248 nm.
100 200 300 400 0 50 100 In te ns it y (a rb .) CCD pixel 100 200 300 400 0 1k 2k Intens it y (arb.) CCD pixel 0 100 200 300 400 500 0 1k Int e n s it y (a rb .) CCD pixel 100 200 300 400 0 50 100 150 In te n s it y ( a rb .) CCD pixel (a) (b) (c) (d)
Fig. 5. Fragment ion image intensity profiles at various delay times. The thin solid line and thick solid line represent m/e=77 and 76, respectively. (a) t = 5 µs, (b) t = 9 µs,
(c) t = 15 µs, (d) t = 18 µs.
(c) Decomposition of phenyl radical
The phenyl radical plays a very important role in the combustion of small aromatic
hydrocarbons.38-40 There has been considerable interest in its reactions with combustion species
as well as its decomposition kinetics and mechanism. The ab initio calculation of a previous study41 showed that the decomposition of phenyl radical producing benzyne + H was
0 100 200 300 400 500 0 10 20 30 40 50 60 70 In te nsit y ( a rb.) CCD pixel 0 5 10 15 20 25 -1.0 -0.5 0.0 0.5 1.0 1.5 2.0
β
Translational energy (kcal/mol) (a)
(b)
Fig. 6. (a) Photofragment ion image intensity profiles of m/e = 77 from two different photolysis laser polarizations at 193 nm. Thick and thin lines represent the polarization of UV laser perpendicular and parallel to the VUV laser beam, respectively. (b) Anisotropy parameter β as a function of fragment translational energy.
endothermic by 76.0 kcal/mol, and the reaction was reported to occurs without a distinct transition state based on geometry optimization with the B3LYP method. This loose structure, also confirmed by MP2 and CCSD optimizations with the 6-31+G(d,p) basis set, however, over-estimated the high-temperature thermal decomposition rate constant determined by Braun-Unkoff et al,42 as was also shown by Wang and co-workers.43 The new optimization
carried out in this work with G96LYP/6-31+G(d,p) method located a low energy transition state. A G2M calculation based on this structure gives the barrier for the decomposition reaction at 79.4 kcal/mole above C6H5. For phenyl radicals produced from the photodissociation of
nitrosobenzene at 193 nm, the maximum internal energy for phenyl radical is 93.8 kcal/mol. The decomposition into benzyne + H therefore is energetically allowed, as shown in Fig. 7.
The decomposition of phenyl radical into benzyne and H atom can be clearly seen from the image intensity changes of m/e = 77 and 76 at various delay times, as shown in Fig. 5. The central part of the fragment images in the velocity axis corresponds to small translational energy released, i.e., most of the available energy is distributed in the fragments’ internal degrees of freedom. For the phenyl radicals located at the central part of the image, they contain a lot of internal energy and quickly decompose into benzyne and H atom. Since the mass ratio between benzyne and H atom is large and the available energy for the decomposition process is small, benzyne produced from this part of phenyl radicals is still located at the same position in the
velocity axis. The fast decomposition of this part of phenyl radicals can be seen from the fast rise of the central part of the image m/e = 76 at short delay time. On the other hand, phenyl radicals located on both sides contain less internal energy, and they have slow decomposition rates. The corresponding benzyne only appears at long delay time images. For the phenyl radicals
positioned at both ends of the image in velocity axis, the translational energy released is large
C6H5 + NO TS C6H4 + HNO C6H5NO, X1A' C6H4N(H)O Ere l (k ca l/ mol ) 0.0 86.2 56.6 66.6 60.3 2 4 8 n m 19 3 n m C6H4 + H + NO 134.0 C6H5NO, 3A" C6H5NO, 1A" 35.3 23.8 S0 S1 S2 h ν 90.4 (89.3) 35.9 (32.9) 0.0
Fig. 7. Energy level scheme from the TD-DFT calculations at the B3LYP/6-31+G(d) level and schematic energy diagram for the isomerization and dissociation reactions of C6H5NO calculated
at the G2M(RCC, MP2)//B3LYP/6-31+G(d) method in kcal/mol. Note that the energy of the 1A″ state, 35.3 kcal/mol, includes ZPE-correction.. The energy levels in parentheses are from ref 18.
and the energy left in the internal degrees of freedom is too small for the radical to be further dissociated into benzyne and H atom. They remain as phenyl radicals and no analogous benzyne is produced. Therefore, the length of the image m/e = 76 is shorter than that of m/e = 77 even at very long delay time.
The decomposition rate of phenyl radical as a function of internal energies can be obtained from the image intensity changes of each part of the image along the fragment velocity axis at various delay times. The internal energy of phenyl radical can be calculated from the following equation.
E(int)phenyl = hν - D0 - E(tran) - E(int)NO
The photon energy hν is 148 kcal/mol, the dissociation energy D0 is 54 kcal/mol,30 and the
amount of translational energy released, E(tran), can be calculated directly using the fragment velocity obtained from the image. The only uncertainty in this equation is the internal energy of fragment NO. However, a proper estimation of the NO fragment internal energy could be made according to the previous studies and our measurement. Previous studies showed that the rotational and vibrational state distributions of NO fragments are statistical, and most of the energy is located in phenyl radical vibrational degrees of freedom. For example, the average vibrational energy of NO fragment is only 1.3 % of the total available energy at photolysis laser
wavelength 255 nm, and the rotational energies of both phenyl radical and NO are only 0.2 % and 3.7 %, respectively due to the transition state geometry. For nitrosobenzene
photodissociation at 193 nm, both the value of anisotropy parameter β and the translational energy distribution suggest that dissociation mechanism is similar to that at 255 nm, i.e., the dissociation occurs after internal conversion to the lower electronic state. Energy must be redistributed among various vibrational degrees of freedom before dissociation occurs and the energy distribution is statistical. Therefore, we estimated that about 95 % of the available energy (total available energy after the subtraction of translational energy) is distributed in phenyl radical vibrational degrees of freedom.
For a given internal energy, the decomposition of phenyl radical into benzyne and H atom can be described by the equation m77(T1) = exp(- kT1) and the growth of the corresponding
benzyne can be described by m76(T1) = 1 - exp(- kT1). m77(T) is the phenyl radical
concentration for a given internal energy at delay time T. m76(T) is the benzyne concentration
produced from the corresponding phenyl radical. k is the dissociation rate of phenyl radical. The ion intensity of m/e = 77 and 76 thus can be described by the following equations
I77 (T1) = A (1 - γ) exp(- kT1) (1)
A and B are the ionization cross sections of fragment mass 77 and 76, respectively. γ is the branching ratio of benzyne produced from the dissociative ionization of phenyl radical, described by the following reaction.
C6H5 + hνvuv → C6H4+ + H (3)
The dissociation rate can be calculated from the ion intensity ratios of fragment m/e = 77 and 76 at two different delay times using the following equation.
γ γ γ γ γ γ γ γ γ γ − + − + = + + = 1 ) 1 -( ) -A(1 B 1 ) 1 -( ) -A(1 B ) -A(1 A ) -B(1 ) -A(1 A ) -B(1 ) ( I ) ( I ) ( I ) ( I 2 1 2 2 2 1 1 1 2 m77 2 m76 1 m77 1 m76 kT kT -kT -kT -kT -kT -kT -kT e e e e e e e e T T T T (4)
At short delay time when no benzyne is produced from the decomposition of phenyl radical, ion intensity ratio between I76 and I77 is close to the value of γ/(1-γ). The upper limit of γ/(1-γ) was
obtained from the image at the available shortest delay time (5 µs), and final result of the decomposition rate k was calculated from the numberical solution of equation (4). The phenyl radical dissociation rates as a function of internal energy obtained from this equation are shown in Fig. 8. It shows that if the translational energy released in the photodissociation of
nitrosobenzene is less than 1 kcal/mol, the decomposition rate of corresponding phenyl radical is in the region between 105 - 104 s-1. However, if the translational energy release is more than
0 10 20 30 40 102 103 104 105 106 107 108 109 Rate (s -1 )
Phenyl radical internal energy (kcal/mol)
Fig. 8. Phenyl radical dissociation rate as a function of internal energy. Open circles, solid squares, and open diamonds represent the experimental values assuming 18 %, 5 % and 0 % of the available energy (93 kcal/mol) distributed in NO internal degrees of freedom. Solid line is the prediction at the G2M(RCC, MP2)//G96LYP/6-31+G(d) level in this work.
comparison of absolute ion image intensities of m/e = 77 between short delay time and long delay time indicate that about 10 % of phenyl radicals decompose into benzyne and H atom within the time scale (<25 µs) of our experiment. Compared to the decomposition rate obtained from G2M(RCC, MP2)//G96LYP/6-31+G(d) ab initio MO and statistical-theory calculations, the agreement between theory and experiment is satisfactory.
C6H5NO system has been calculated with the G2M method31 based on the geometry optimized at
the B3LYP/6-31+G(d,p) level of theory. The optimized structures of the reactant, transition state and intermediate are presented in Fig. 9. All the geometries of the C6H5NO (S0, X 1A′), C6H5NO
(T1, 3A″), and C6H5NO (S1, 1A″) are planar with CS symmetry. The equilibrium C-N bond
lengths of T1 and S1 states are shorter than that of the S0 state as shown in Fig. 9. The predicted
energetics and potential energy diagram for the formation of products accessible to 193 nm photon energy are presented in Table 1 and Fig. 7, respectively. At the G2M level, the
4-centered transition state leading to C6H4 + HNO was predicted to locate at 67 kcal/mol above
O N C C C C C C C C O C C N C C C C O C C C N C
C
6H
5NO, C
sTS
C
6H
4N(H)O
1.223 (1.232) {1.233} 1.439 (1.381) {1.371} 115.4 (128.1) {128.1} 121.2 (120.5) {120.3} 1.218 1.389 1.251 1.623 1.234 1.042 1.365 129.3 127.9 127.8 S0,X1A' (T1,3A") {S1,1A"}Fig. 9. Optimized structures of the reactant, transition state, and intermediate at the B3LYP/6-31+G(d) level of theory.
Table 1. Relative energiesa for the isomerization and decomposition reactions of C6H5NO.
species or reaction B3LYP/
6-31G(d) G2M b B3LYP/ 6-31+G(d) G2M c B3LYP/ 6-31G(2df,p) G3SX d C6H5NO 0.0 0.0 0.0 0.0 0.0 0.0 C6H5 + NO 53.3 56.5 52.2 56.6 53.0 55.6 TS 66.1 66.7 65.6 66.6 63.3 66.0 C6H4N(H)O 60.5 60.2 57.8 60.3 58.4 59.6 C6H4 + HNO 96.2 86.2 93.2 86.2 94.8 86.3 C6H4 + H + NO 140.4 134.0 138.4 134.0 140.2 134.1
a. Relative energies are ZPE-corrected in kcal/mol.
b, c, and d. Based on the optimized geometries calculated at B3LYP/6-31G(d), B3LYP/6-31+G(d), and B3LYP/6-31G(2df,p), respectively.
C6H5NO, about 19 kcal/mol below the dissociated products. This suggests that a stable
molecular complex must exist between C6H5NO and C6H4 + HNO. A detailed IRC calculation
indeed confirms the presence of a benzyne-HNO complex, C6H4N(H)O, lying 60 kcal/mol above
C6H5NO (see Fig. 9 for its structure). Further decomposition of the C6H4N(H)O complex was
found to take place without a well-defined intrinsic barrier, similar to the fragmentation of C6H5NO to C6H5 and NO.
As shown in Table 1, the energetics predicted by both G2M and G3SX agree very closely. The latter, which does not include “high-level corrections”, is believed to be more reliable for applications to reactions with spin changes. In Table 2, the result of our TD-DFT calculations at the B3LYP/6-31+G(d) level for the S1 and S2 states confirms the energy gap between them, 54.5
Table 2. Vertical energies in kcal/mol and oscillator strengths (f) in parenthesis for C6H5NO with
estimated by TD-DFT at the B3LYP/6-31+(d) level.
state sym B3LYP/6-31+G(d) experimentala
S1 1A″ 35.9 (0.0003) 32.9
S2 1A′ 90.4 (0.0183) 89.3
a. from ref 18.
Decomposition of photo-excited C6H5NO. Assuming that the internal conversion is
fast comparing with fragmentation lifetimes, we can predict the fragmentation rates of C6H5NO
following the excitation at 248 and 193 nm on the basis of the RRKM theory. As both product channels giving NO and HNO occur barrierlessly, the energy-dependent specific constants (kE)
were evaluated with the flexible transition state approach44-47 using the Variflex code.48 The
minimum energy paths (MEP) representing the dissociation processes were approximated with the Varshni potential,49 V(r) = De{- a exp[- b(R2 - R02)]} - De, where R is the reaction coordinate,
R0 is the equilibrium value of R, a = R0/R, and De is the bond energy excluding the zero-point
energy correction. Along the reaction coordinate, we stretched the bond length of C-N with the step size of 0.2 Å along the path from the equilibrium value to 5 Å. Each geometry was fully optimized at the B3LYP/6-31+G(d) level of theory and its energy was then scaled by the G2M(RCC, MP2) value for the Varshni potential. The approximated bond energy and b for the ground state with 1A′ are 60.4 kcal/mol and 0.522 Å-2, respectively, using R
calculations for the dissociation of C6H4N(H)O, C6H5NO (T1, 3A″) and C6H5NO (S1, 1A″) gave
rise to De = 30.3, 36.0, and 20.0 kcal/mol; b = 0.863, 0.834, and 0.991 Å-2; R0 = 1.365, 1.381 and
1.371Å, respectively.
Table 3. Decay rate of C6H5NO via different pathways.
Pathway 248 nm 193 nm
C6H5NO → C6H5 + NO 2.5 × 1010 3.2 × 1010
C6H5NO → C6H4 + HNOa 1.0 × 104 1.3 × 107
C6H5NO → C6H4N(H)O 2.9 × 105 1.8 × 107
a. The rate constant for C6H5NO → VTS → C6H4 + HNO with VTS as the limiting step.
Using the molecular parameters given above, the specific rate constants for the
dissociation of C6H5NO at 248 and 193 nm (approximately 115 and 148 kcal/mol, respectively)
provide the lifetimes of the excited molecule along various pathways as summarized in Table 3. The lifetimes of C6H5NO are predicted to about 40 ps following the excitation at 248 nm and 33
ps at 193 nm producing exclusively C6H5 + NO as was observed experimentally. The formation
of HNO, slower by 105 at 248 nm and 103 at 193 nm, is experimentally undetectable. As the
formation of HNO occurs via both the tight 4-centered TS1 and the variatioanl TS near the dissociation threshold, with the latter being slightly slower due to its higher energy (see Table 3), we have examined the effect of multiple reflections between the two TS’s; the effect was found to be negligible because of the high photon energies. The predicted lifetimes are much longer
than that derived from the linewidth measurement suggesting that internal conversion is considerably faster than the fragmentation process.
Thermal decomposition rate constant. The high-pressure, first-order rate constant for the decomposition of C6H5NO producing C6H5 + NO, k∞ = (1.42 ± 0.13) × 1017 exp[-(55060 ±
1080)/RT] s-1,30 is seen to have an unusually large A-factor. The extrapolated rate constants in the
temperature range of 800 - 1000K using the equation lies between the result of Horn and coworkers48 (after high-pressure-limit correction by the RRKM theory) and that of Choo et al.49
determined by the very-low-pressure pyrolysis method. Theoretically, the rate constant can be computed with the Varshni potential evaluated for the 3 MEP’s producing NO. The thermal unimolecular dissociation rate constant defined by the rate equation,
d[C6H5]/dt = k∞ [C6H5NO]0 = Σki∞ Xi = kX∞ [X] + kA∞ [A] + kB∞ [B] (5)
where [C6H5NO]0, [X], [A] and [B] are, respectively, the initial concentration of nitrosobenzene
entirely at its ground electronic state and those at the ground electronic, 3A″ and 1A″ states under
thermal equilibrium, and the ki∞ are the first-order rate constants for the dissociation from these
states. Assuming KA and KB to be the equilibrium constants for X ↔ A and X ↔ B, respectively,
equation (5) leads to the total unimolecular rate constant,
1.0 1.2 1.4 1.6 1.8 2.0 -25 -20 -15 -10 -5 0 5 10 15 ln (k ; s -1 ) 1000 / T (K-1)
Fig. 10. Experimental and predicted thermal rate constants for the dissociation of C6H5NO
at the high-pressure limit.. (□) is from ref 50, (∆) with NO added and (○) without NO added are from ref 30, and dashed line is from ref 51. Solid, dash-dotted, and dotted lines correspond to the prediction for the states of X 1A′, 1A″, and 3A″, respectively. Their sum gives k∞ = 1.52 × 1017
exp[-55200/RT] s-1
The predicted individual rate constants presented in Fig. 10 gave k∞ = 1.52 × 1017 exp[-55200/RT]
s-1, which is in excellent agreement with the result obtained by Park et al.30, k∞ = (1.42 ± 0.13) ×
1017 exp[-(55060 ± 1080)/RT] s-1, confirming the unusually high A-factor. Notably, the result
presented in Fig 10 indicates that the contributions from both T1 and S1 states are insignificant in
have recently shown that contributions from 3 excited states to its second-order rate constants are significant.52
IV. Conculsions
We demonstrate that there is only one dissociation channel of nitrosobenzene at both 248 and 193 nm. No products corresponding to the dissociation channel of C6H5NO → C6H4 + HNO
were observed, in full agreement with theoretical prediction. The dissociation rate of
nitrosobenzene is on a timescale much longer than the rotation of molecule, and the fragment distribution is almost isotropic. The results at 248 nm are close to that of Dick and the
coworkers’ measurement. In addition, about 10% of phenyl radical produced at 193 nm further decompose into benzyne and H atom, and the dissociation rates of phenyl radical as a function of internal energies were measured. The results for the dissociation of the excited C6H5NO and the
C6H5 radicalagree well with theoretically predicted values. In addition, the rate constant for the
thermal decomposition of nitrosobenzene has been calculated with the variational RRKM theory by including the small contributions from the two low-lying excited states (T1 and S1). The
agreement between theory and the experiment of Park et al.30 was also found to be excellent,
Acknowledgements
CKN acknowledges the support from the National Science Council, Taiwan under contract no. NSC 91-2113-M-001-023. MCL and YMC are grateful to Dr. J. Park for his preliminary optimization of the TS in Figure 7 and for the support of this work from the Basic Energy Sciences, Department of Energy, under contract no. DE-FG02-97-ER14784 and the Cherry L. Emerson Center of Emory University for the use of its resources, which are in part supported by a National Science Foundation grant (CHE-0079627) and an IBM shared University Research Award. MCL also acknowledges the support from the National Science Council for a Distinguished Visiting Professorship at National Chiao Tung University in Hsichu, Taiwan.
References
(1) Yu, T.; Lin, M. C. J. Am. Chem. Soc. 1993, 115, 4371. (2) Lin, M. C.; Yu, T. Int. J. Chem. Kinet. 1993, 25, 875. (3) Yu, T.; Lin, M. C. J. Phys. Chem. 1994, 98, 2105. (4) Yu, T.; Lin, M. C. Int. J. Chem. Kinet. 1994, 26, 771.
(5) Yu, T.; Lin, M. C.; Melius, C. F. Int. J. Chem. Kinet. 1994, 26, 1095. (6) Yu, T.; Lin, M. C. J. Am. Chem. Soc. 1994, 116, 9571.
(7) Yu, T.; Lin, M. C. J. Phys. Chem. 1995, 99, 8599.
(8) Tonokura, K.; Norikane, Y.; Koshi, M.; Nakano, Y.; Nakamichi, S.; Goto, M.; Hashimoto, S.; Kawasaki, M.; Andersen Sulbaek, M. P.; Hurley, M. D.; Wallington, T. J. J.Phys. Chem. A 2002, 106, 5908.
(9) McCoustra, M. R. S.; Pfab, J. Chem. Phys. Lett. 1985, 122, 395.
(10) Engert, J. M.; Slenczka, A.; Kensy, U.; Dick, B. J. Phys. Chem. 1996, 100, 11883. (11) Bhujle, V.; Wild, U. P.; Baumann, H.; Wagniere, G. Tetrahedron 1976, 32, 467. (12) Bhujle, V. V. Spectrosc. Lett. 1997, 10, 587.
(13) Chernoff, D. A.; Hochstrasser, R. M. Chem. Phys. Lett. 1980, 70, 213. (14) Condirston, D. A.; Knight, A. R.; Steer, R. P. J. Photochem. 1980, 14, 257.
(15) Hatton, W. G.; Hacker, N. P.; Kasai, P. H. J. Chem. Soc., Chem. Commun. 1990, 227. (16) Niles, S.; Wight, C. A. Chem. Phys. Lett. 1989, 154, 458.
(17) Keβler, A.; Kensy, U.; Dick, B. Chem. Phys. Lett. 1998, 289, 516.
(18) Keβler, A.; Slenczka, A.; Seiler, R.; Dick, B. Phys. Chem. Chem. Phys. 2001, 3, 2819. (19) Seiler, R. D., B. Chem. Phys. 2003, 288, 43.
(20) Obernhuber, T. J.; Kensy, U.; Dick, B. Phys. Chem. Chem. Phys. 2003, 5, 2799. (21) Li, Y. M.; Sun, J. L.; Han, K. L.; He, G. Z. Chem. Phys. Lett. 2001, 338, 297.
(22) Huang, J. H.; Wang, G. J.; Gu, X. B.; Han , K. L.; He, G. H. J. Phys. Chem. A 2000, 104, 10079.
(23) Tsai, S. T.; Lin, C. K.; Lee, Y. T.; Ni, C. K. Rev. Sci. Instrum. 2001, 72, 1963. (24) Tsai, S. T.; Lee, Y. T.; Ni, C. K. J. Phys. Chem. A. 2000, 104, 10125.
(25) Tsai, S. T.; Lin, C. K.; Lee, Y. T.; Ni, C. K. J. Chem. Phys. 2000, 113, 67. (26) Tsai, S. T.; Huang, C. L.; Lee, Y. T.; Ni, C. K. J. Chem. Phys. 2001, 115, 2449. (27) Becke, A. D. J. Chem. Phys. 1992, 96, 2155.
(28) Becke, A. D. J. Chem. Phys. 1992, 97, 9173. (29) Becke, A. D. J. Chem. Phys. 1993, 98, 5648.
(30) Park, J.; Dyakov, I. V.; Mebel, A. M.; Lin, M. C. J. Phys. Chem. 1997, 101, 6043. (31) Mebel, A. M.; Morokuma, K.; Lin, M. C. J. Chem. Phys. 1995, 103, 7414.
(32) Curtiss, L. A.; Redfern, P. C.; Raghavachari, K.; Pople, J. A. J. Chem. Phys. 2001, 114, 108. (33) Bauernschmitt, R.; Ahlrichs, R. Chem. Phys. Lett. 1996, 256, 454.
(34) Stratmann, R. E.; Scuseria, G. E.; Frisch, M. J. J. Chem. Phys. 1998, 109, 8218.
(35) Casida, M. E.; Jamorski, C.; Casida, K. C.; Salahub, D. R. J. Chem. Phys. 1998, 108, 4439. (36) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgomery, J. A., Jr.; Stratmann, R. E.; Burant, J. C.; Dapprich, S.; Millam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Malick, D. K.; Rabuck, A. D.;
Raghavachari, K.; Foresman, J. B.; Cioslowski, J.; Ortiz, J. V.; Baboul, A. G.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Andres, J. L.; Gonzalez, C.; Head-Gordon, M.; Replogle, E. S.; Pople, J. A. Gaussian 98; Revision A.7; Gaussian, Inc., Pittsburgh PA, 1998. (37) Amos, R. D.; Bernhardsson, A.; Berning, A.; Celani, P.; Cooper, D. L.; Deegan, M. J. O.; Dobbyn, A. J.; Eckert, F.; Hampel, C.; Hetzer, G.; Knowles, P. J.; Korona, T.; Lindh, R.; Lloyd, A. W.; McNicholas, S. J.; Manby, F. R.; Meyer, W.; Mura, M. E.; Nicklass, A.; Palmieri, P.; Pitzer, R.; Rauhut, G.; Schutz, M.; Schumann, U.; Stoll, H.; Stone, A. J.; Tarroni, R.; Thorsteinsson, T.; Werner, H.-J. MOLPRO; version 98.1; University of Birmingham, Birmingham, U.K., 1998. (38) Glassman, I. Combustion, 2nd ed.; Academic Press: New York, 1986.
(39) Haynes, D. S. Fossil Fuel Combustion; Wiley-Interscience: New York, 1991. (40) Bockhorn, H. Soot Formation in Combustion; Springer-Verlag: New York, 1993. (41) Madden, L. K.; Moskaleva, L. V.; Kristyan, S.; Lin, M. C. J. Phys. Chem. A. 1997, 101, 6790.
(42) Braun-Unkoff, M.; Frank, P.; Just, T. 22nd Sym. (Int.) Combust. 1988, 1053. (43) Wang, H.; Laskin, A.; Moriaty, N.W., Frenklach, M., Proc. Combust. Inst. 2000, 28, 1545.
(44) D. M. Wardlaw and R. A. Marcus, Chem. Phys. Lett. 110, 230 (1984); J. Chem. Phys. 83, 3462 (1985)
(45) Klippenstein, S. J. Chem. Phys. Lett. 1990, 170, 71; Klippenstein, S. J. J. Chem. Phys. 1991, 94, 6469.
(46) Klippenstein, S. J. J. Chem. Phys. 1992, 96, 367.
(47) Robertson, S. H.; Wagner, A. F.; Wardlaw, D. M. J. Chem. Phys. 1995, 103, 2917. (48) Klippenstein, S. J.; Wagner, A. F.; Dunbar, R. C.; Wardlaw, D. M.; Robertson, S. H. Variflex; version 1.00, July 16, 1999.
(50) Horn, C.; Frank, P.; Tranter, R. S.; Schaugg, J.; Grotheer, H.-H.; Just, T. 26th Sym. (Int.) Combust., 1996.
(51) Choo, K.-L.; Golden, D. M.; Benson, S. W. Int. J. Chem. Kinet. 1975, 7, 713.