RECENT PROGRESS IN ZERO-VALENT IRON NANOPARTICLES FOR
GROUNDWATER REMEDIATION
Hsing-Lung Lien,1 Daniel W. Elliott,2 Yuan-Pang Sun2 and Wei-Xian Zhang2,* 1Department of Civil and Environmental Engineering
National University of Kaohsiung Kaohsiung 811, Taiwan.
2Department of Civil and Environmental Engineering
Lehigh University Bethlehem, Pennsylvania 18015, USA
Key Words: Nanotechnology, nanoparticle, iron, groundwater remediation ABSTRACT
Although still in the emerging phase, iron nanoparticles represent a highly promising new weapon in the arsenal of environmental remediation technologies. Nearly a decade after it was first launched in 1996, the iron nanoparticle technology is at a critical stage of the developmental process. Despite significant innovations in terms of novel synthetic methodologies as well as successes at both the bench and field scales, there remain considerable knowledge gaps on fundamental issues (e.g., fate, transport, environmental impact) and economic hurdles which could determine the acceptance of the technology within the academic community as well as by regulators and the private sector. In this paper, an overview of the iron nanoparticle technology is provided beginning with a description of the process fundamentals. This is followed by a discussion of the synthetic schemes for the nanoparticle types developed at Lehigh University. Next, a summary of the major research findings is provided, highlighting the key characteristics and remediation-related advantages of the iron nanoparticle technology versus the granular/microscale iron technology. A discussion of fundamental issues related to its potential applications and environmental impact is presented.
*Corresponding author Email: [email protected]
INTRODUCTION
With a little more a decade in history, the multi-disciplinary nanotechnology boom has inspired the naissance of powerful new tools in the ongoing chal-lenge of addressing the industrialized world’s legacy of contaminated sites [1]. These might include im-proved analytical and remote sensing methodologies, novel sorbents and pollution control devices, and su-perior soil and groundwater remediation technologies. The iron nanoparticle technology described in this contribution may represent an important, early stage achievement of the burgeoning environmental nanotechnology movement.
Since 1996, our research group at Lehigh Uni-versity has been actively engaged in the developing new nanometal materials, improving the synthetic schemes, performing both bench-scale and field-scale assessments, and extending the technology to increas-ing numbers of amenable contaminant classes. The use of iron nanoparticle technology for groundwater
remediation has therefore received great attention [2-13]. Now, nearly a decade later, the iron nanoparticle technology is at a critical stage of the developmental process. Despite significant innovations in terms of novel synthetic methodologies as well as successes at both the bench and field scales, there remain consider-able knowledge gaps and economic hurdles which could limit the acceptance of the technology within the academic community as well as by regulators and the private sector.
In this paper, an overview of the nanoscale zero-valent iron (nZVI) technology is provided beginning with a description of the process fundamentals. This is followed by a discussion of the synthetic schemes for the nZVI types used during the course of this research. Next, a summary of the major research findings is provided, highlighting the key characteristics and remediation-related advantages of the nZVI technol-ogy versus the granular/microscale ZVI technoltechnol-ogy. A discussion of its potential applications and summary of important data gaps and economic hurdles facing
the nZVI technology are also included.
ZVI GENERAL PROCESS DESCRIPTION
ZVI (Fe0) has long been recognized as an excel-lent electron donor, regardless of its particle size. Fe0 exhibits a strong tendency to donate electrons to suit-able electron acceptors.
− + +
→Fe e
Fe0 2 2 (1)
ZVI and dissolved ferrous iron form a redox couple with a standard reduction potential (E0) of -0.440 V [14]. Metallic ZVI can form redox couples with sev-eral environmentally significant and redox-amenable electron acceptors including hydrogen ions, dissolved oxygen, nitrate, sulfate, and carbonate [14]. Under aerobic conditions typical of vadose zone soils or shallow, oxygenated groundwaters, ZVI can react with dissolved oxygen (DO) as follows:
) ( 2 2 ) ( ) ( 2 ) ( 0 ) ( 4 2 2 2Fes + H+aq +O aq → Feaq+ + H Ol (2) yielding ferrous iron and water. The overall E0 for this reaction (E0rxn) is +1.71 V at 25 °C, indicating a
strongly favorable reaction from a thermodynamics perspective [15]. Implicit in the stoichiometry is the transfer of 4 electrons from the iron surface and asso-ciated increase of solution pH (based on the consump-tion of protons). Assuming that residual DO levels remain, the ferrous iron would be expected to undergo relatively facile oxidation to ferric iron, Fe3+. The in-creasing pH favors the formation of one or more iron hydroxide or carbonate-based precipitates which can have the effect of blinding off or otherwise lessening reactivity at the metal surface [14]. In anaerobic or low DO groundwater environments, which is more typical of deeper aquifers and contaminated plumes, ZVI also forms an effective redox couple with water yielding ferrous iron, hydroxide, and hydrogen gas:
− + + + → + 2 2( ) ( ) ) ( ) ( 2 0 ) (s 2H Ol 2Feaq H g 2OH aq Fe (3)
Unlike the reduction of DO, the iron-mediated reduc-tion of water is not thermodynamically favored as evidenced by the E0rxn of -0.39 V at 25 °C [15].
Ther-modynamic considerations notwithstanding, the kinet-ics of these and other reactions in natural waters tend to be rather sluggish and consequently, chemical equi-librium is generally not attained [16].
In addition to the common environmentally rele-vant electron acceptors, ZVI also readily reacts with a wide variety of redox-amenable contaminants. Using a generalized chlorinated hydrocarbon, RCl, as an ex-ample, the ZVI-mediated transformation of RCl to the corresponding hydrocarbon, RH can be represented as:
− +
+ + → + +
+H Fe RH Fe Cl
RCl 0 2 (4)
From a thermodynamics perspective, the large
positive E0rxn values imply that a spontaneous reaction
with ZVI should occur. In terms of the chlorinated hydrocarbons, the degree of favorability increases with the number of chlorine substituents. For many chlorinated hydrocarbons, the E0rxn is on the order of
+0.5 to +1.5 V at 25 °C [14,17].
Examination of Eq. 2 through 4 reveals that mul- tiple species exist in the systems under investigation which are capable of serving as the electron acceptors. The electron donating potential of ZVI has already been discussed. However, ferrous iron can further serve as a reductant by donating an electron to a suit- able electron acceptor yielding ferric iron, Fe3+.
Hy-drogen gas too is a well known reducing agent, par-ticularly in the realm of microbiology where it is often characterized as the “universal electron donor”.
The roles of these three electron donors in the reduction of chlorinated hydrocarbons has been exten-sively documented [14]. For example, Matheson and Tratnyek proposed three possible mechanisms: (1) di-rect reduction at the metal surface, (2) reduction by ferrous iron and (3) reduction by hydrogen with ca-talysis [14]. They studied the potential roles of these reductants and found that ferrous iron, in concert with certain ligands, could slowly reduce the chlorinated hydrocarbons and that dissolved hydrogen gas, in the absence of a suitable catalytic surface, failed to reduce the chlorinated hydrocarbon.
In the presence of granular ZVI, numerous re-search groups observed the rapid transformation of various contaminants confirming the validity of the di-rect surface reduction model [e.g., 14,18]. Weber [18] elegantly confirmed these findings in a study using 4-aminoazobenzene (4-AAB), an aromatic azo dye which readily undergoes ZVI-mediated reduction. In this work, 4-AAB which was immobilized by electro-philic derivatization to a solid support was not re-duced by ZVI because it could not associate with the iron surface while the control, non-derivatized 4-AAB, was rapidly transformed. Thus, these studies clearly demonstrated the fact that the degradation of contami-nants by ZVI is a surface-mediated process via one or more heterogeneous reactions.
OVERVIEW OF MAJOR NZVI SYNTHETIC SCHEMES
At least three distinct synthetic schemes were utilized at Lehigh University to prepare the nZVI. All involved the reduction and precipitation of ZVI from aqueous iron salts using sodium borohydride as the reductant. These included one approach in which the iron salt was iron(III) chloride, heretofore referred to as “the chloride method” and two schemes where iron(II) sulfate was the principal ZVI precursor salt. The latter was termed the “sulfate method”. Each of these methodologies is discussed in the following paragraphs.
1. Type I nZVI Using the Chloride Method
The chloride method synthesis represents the original means of producing nZVI at Lehigh Univer-sity [19-21]. The chloride method iron, also referred to as Type I nZVI, was the earliest generation of nanoscale iron.
In this synthesis, 0.25 M sodium borohydride was slowly added to 0.045 M ferric chloride hexahy-drate in aqueous solution under vigorously mixed conditions such that the volumes of both the boro-hydride and ferric salt solutions were approximately equal (i.e. a 1:1 v/v). The mixing time utilized was approximately 1 h. This reaction is shown in Eq. 5 as
follows [22]: 100 nm
4Fe3+
(aq) + 3BH4-(aq) + 9H2O(l) Æ 4Fe0(s)
+ 3H2BO3-(aq) + 12H+(aq) + 6H2(g) (5)
The ratio between the borohydride and ferric salt ex-ceeded the stoichiometric requirement by a factor of approximately 7.4. This excess helped to ensure the rapid and uniform growth of the nZVI crystals [19]. The harvested nano-iron particles were then washed successively with a large excess of distilled water, typically > 100 mL g-1. The solid nanoparticle mass was recovered by vacuum filtration and washed with ethanol. The residual water content of the nZVI mass was typically on the order of 40-60%. A Scanning Electron Microscopy (SEM) micrograph of the Type I nZVI is shown in Fig. 1a.
If bimetallic particles were desired, the ethanol-wet nZVI mass was soaked in an ethanol solution con-taining approximately 1% palladium acetate as indi-cated in Eq. 6: + + + 0 → 0 + 2 2 Fe Pd Fe Pd (6)
The chloride method proved to be readily adapted to any standard chemical laboratory assuming that fume hoods and adequate mixing was provided to dissipate and vent the small quantities of H2 produced.
2. Type II and III nZVI Using the Sulfate Method
The development of the sulfate method for producing nZVI arose from two fundamental concerns associated with the chloride method: (1) potential health and safety concerns associated with handling the highly acidic and very hygroscopic ferric chloride salt and (2) the potential deleterious effects of exces-sive chloride levels from the nZVI matrix in batch degradation tests where chlorinated hydrocarbons are the contaminant of concern. In addition, the reduction of the iron feedstock from Fe(II) requires less boro-hydride than the chloride method, in which Fe(III) is the starting material which may favorably enhance overall process economics. Because this synthetic scheme represented the second generation of iron
Fig. 1. EM images of nZVI aggregates. (a) SEM for Type I, (b) TEM for Type II and (c) TEM for Type III.
40 35 30 25 20 45 50 Diffraction Angle 2θ
nanoparticles developed at Lehigh University, the iron is referred to as Type II nZVI.
Sulfate method nZVI was prepared by metering equal volumes of 0.50 M sodium borohydride at 0.15 L min-1 into 0.28 M ferrous sulfate according to the following stoichiometry:
2Fe2+(aq) + BH4-(aq) + 3H2O(l) Æ 2Fe0(s)
+ H2BO3-(aq) + 4H+(aq) + 2H2(g) (7) In te n s it y (a) Fresh (b) After 48 hours 40 35 30 25 20 45 50
The stoichiometric excess of borohydride used in the Type II nZVI synthesis was about 3.6, considerably less than that with the Type I nZVI. In light of this process change, the rate of borohydride addition was extended to approximately 2 h to help control particle size. Thus, the reduction in production costs achieved was partially off-set by the longer synthesis time.
As was the case with the chloride method, the synthesis was carried out in a fume hood in open polyethylene containers (about 18 L) fitted with vari-able speed, explosion-resistant mixers. No attempt was made to exclude air from the reaction mixture. The freshly prepared nZVI particles were allowed to settle for approximately 1 h and were then harvested by vacuum filtration. The finished nanoparticles were washed with copious amounts of distilled water (> 100 mL g-1), then by ethanol, purged with nitrogen, and refrigerated in a sealed polyethylene container under ethanol until use. The residual water content of the Type II nZVI was typically on the order of 45-55%, very similar to that observed for the Type I iron. A representative transmission electron microscopy (TEM) image of the Type II nZVI is shown in Fig. 1b.
The Type III nZVI was also synthesized using the sulfate method. Because the synthetic procedure for the Type III iron was very similar to that for the Type II nZVI, the details are not repeated here. It rep-resented the latest generation of nZVI and exhibited an average particle size of approximately 50-70 nm, very similar to that observed for the Type II iron. However, the moisture content was appreciably lower than observed for the previous nZVI types: 20-30% versus 40-60% for Types I and II. The basis for this difference is not known. A TEM image of the Type III nZVI is shown in Fig. 1c.
CHARACTERIZATION OF NZVI
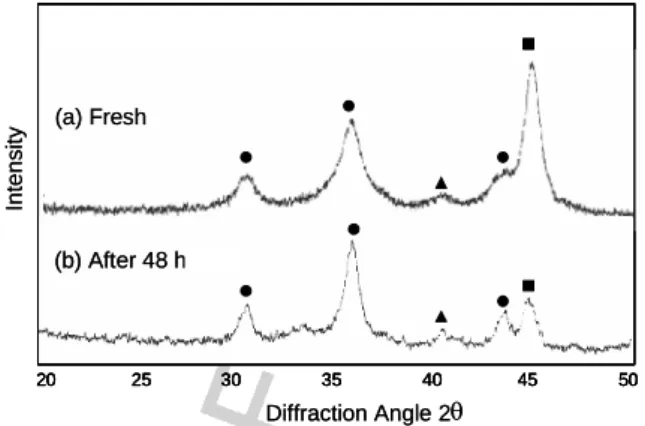
The average particle size of the chloride method nZVI was on the order of 50-200 nm and the specific surface area was measured by mercury porosimetry to be approximately 33.5 m2 g-1 [19]. X-ray diffraction of the Type I nZVI surface composition indicated the major surface species of freshly prepared nanoscale Pd/Fe is Fe0 (44.7o) with lesser quantities of iron(III) oxide, Fe2O3 (35.8o), and Pd0 (40.1o) (Fig. 2). The
presence of iron(III) oxide, which results from air ex-posure of the nZVI, visually appeared as a surficial
(a) Fresh (b) After 48 hours 40 35 30 25 20 45 50 Diffraction Angle 2θ In te n s it y (a) Fresh (b) After 48 hours 40 35 30 25 20 45 50 (a) Fresh (b) After 48 hours
Fig. 2. X-ray diffractogram of Fe/Pd nanoparticles. Solid square, circle and triangle represent Fe0, α-Fe2O3 and Pd0, respectively.
rust patina on the surface of the iron and typically did not extend into the bulk iron mass. Not surprisingly, the XRD diffractogram for nanoscale Pd/Fe aged for 48 h shows relatively larger peaks for the iron oxide but still an appreciable ZVI peak.
The average particle size of the sulfate method nZVI was on the order of 50-70 nm. Analysis of over 150 individual particles and clusters yielded a mean diameter of 67±13 nm with more than 80% being smaller than 100 nm and fully 30% being smaller than 50 nm [23]. Moreover, the BET specific surface area of the iron was 35±3 m2 g-1, approximately equivalent to that of the Type I nZVI [24].
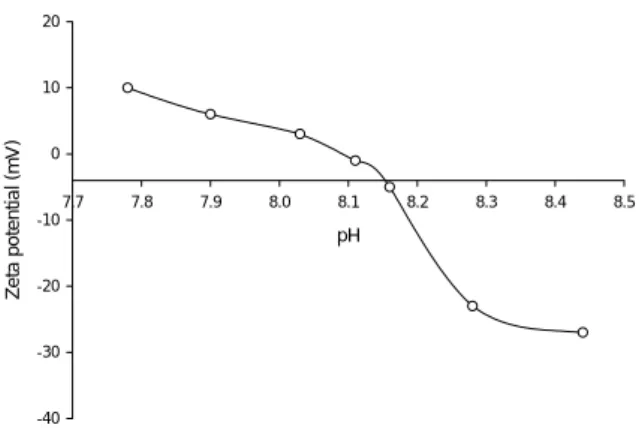
Using an electroacoustic spectrometer (Disper-sion Technologies DT 1200), the zeta potential of a 0.85% (by weight) slurry of Type III nZVI in water was measured to be -27.55 mV at a pH of 8.77. Ac-cording to the Colloidal Science Laboratory, Inc. (Westhampton, NJ), colloidal particles with zeta po-tential values more positive than +30 mV or more negative than -30 mV are considered stable with the maximum instability (i.e. aggregation) occurring at a zeta potential of 0. Thus, using this benchmark, the Type III nZVI would be considered meta-stable.
Theoretically, zeta potential refers to the poten-tial drop across the mobile or diffuse portion of the classical electric double layer surrounding a particle in solution [25]. It provides an indication of the stability of the colloidal suspension and is a function of many variables including the nature of the particle surface, ionic strength, pH, and presence of other substances that can interact with the surface (e.g., surface-active polymers, etc.). It has been well established that solu-tion pH strongly influences the zeta potential of the colloidal particles. Specifically, as the pH increases, the particles tend to acquire additional negative charge which translates into a decreased (i.e. more negative) zeta potential. The pH corresponding to a zeta poten-tial of 0, known as the isoelectric point, represents the area of minimum particle stability. Figure 3 depicts a zeta potential versus pH titration for a chloride method
pH 7.7 7.8 7.9 8.0 8.1 8.2 8.3 8.4 8.5 Zeta potent ial ( m V) -40 -30 -20 -10 0 10 20 00
Fig. 3. Titration of zeta potential versus pH using 2.0 N sulfuric acid for a 10 g L-1 suspension of nZVI in water.
nZVI of 50-70 nm average particle size in water. In the neutral pH range, the relatively low zeta potential of nZVI in solution supports the observation of parti-cle aggregation which could adversely impact subsur-face mobility.
SUMMARY OF THE NZVI APPLICATIONS
As demonstrated in the Zhang group research at Lehigh, the nZVI technology exhibits enhanced reac-tivity and superior field deployment capabilities as compared with microscale and granular iron as well as other in-situ approaches. The enhanced reactivity stems from the appreciably greater specific surface area of the iron. The colloidal size of the iron nanopar-ticles, their amenability to direct subsurface injection via gravity feed conditions (or under pressure, if de-sired), and need for substantially less infrastructure all contribute to the technology’s portability and relative ease of use.
1. Laboratory Studies
Since 1996, the Zhang group has investigated the ability of nZVI to degrade a wide variety of environ-mental contaminants including PCBs, chlorinated ali-phatic and aromatic hydrocarbons, hexavalent chro-mium, chlorinated pesticides, and perchlorate. 1.1 Chlorinated hydrocarbons
Degradation of chlorinated hydrocarbons using nZVI, mZVI (microscale ZVI) and nZVI/Pd (pal-ladized nZVI) has been extensively studied [26-28]. A wide array of chlorinated hydrocarbons including ali-phatic compounds (e.g, carbon tetrachloride, tri-chloroethylene, and hexachloroethane), aliphatic cy-clic compounds (e.g., lindane), and aromatic com-pounds (e.g., PCB and hexachlorobenzene) has been
tested. In general, nZVI/Pd showed the best overall performance followed by nZVI and then mZVI [21,26,27]. Degradation of carbon tetrachloride by dif-ferent types of ZVI represents a typical example. The surface area normalized rate constant (kSA) data
de-rived from the experimental datasets followed the or-der nZVI/Pd > nZVI > mZVI. The surface area nor-malized rate constant of nZVI/Pd was two-order mag-nitudes higher than that of mZVI. Furthermore, same reaction products including chloroform, dichloro-methane, and methane were observed in the use of mZVI, nZVI, and nZVI/Pd; however, product distri- butions were significantly different. Highest yield of methane (55%) and lowest production of dichloromethane (23%) was found in the use of nZVI/Pd whereas the accumulation of dichloro-methane accounting for more than 65% of initial car-bon tetrachloride and less than 25% of methane pro-duction were observed in the case of mZVI study [21].
The superiority of nZVI/Pd can be attributed to two key factors. First, a significant increased surface area of nZVI. Compared to the mZVI, the enhanced reactivity of the nZVI was attributed to the smaller average particle size which translated into a much lar-ger specific surface area, 33.5 m2 g-1 versus < 0.9 m2 g-1, for the irons studied. Second, the presence of ladium resulting in the catalytic power of the pal-ladized nZVI. It should be pointed out that palladium not only enhances the reactivity but alters the product distribution, implying a different reaction mechanisms involved [29,30].
Unlike chlorinated aliphatic compounds studied extensively, chlorinated aliphatic cyclic compounds and aromatic compounds received less attention and lacked a systematic study, partially due to complicated transformation processes as they possess relatively large molecular structures. Limited studies indicated nZVI or nZVI/Ag (iron-silver nanoparticles) still hibited fairly well dechlorinated capacity [31]. For ex-ample, the iron-silver nanoparticles, with a measured specific surface area of 35 m2 g-1, transformed hexa-chlorobenzene (HCB) to a series of lesser chlorinated benzenes including the following principal products: 1,2,4,5-tetrachlorobenzene, 1,2,4-trichlorobenzene, and 1,4-dichlorobenzene [31]. No chlorobenzene or benzene was observed as reaction products. HCB con-centrations were reduced below the detection limit (< 1 μg L-1) after 4 d.
In comparison, conventional microscale iron powder at an iron to solution ratio of 25 g 100 mL-1
produced little reaction with HCB under similar ex-perimental conditions. After 400 h of elapsed time, to-tal conversion of HCB to products was approximately 12% with primarily 1,2,4,5-tetrachlorobenzene and 1,2,4-trichlorobenzene detected as intermediates [31]. Clearly, nZVI is more well-suited than mZVI to de-grade polychlorinated aromatics like HCB.
1.2 Hexavalent chromium 0 50 100 150 200 250
Hexavalent chromium, Cr(VI) is a highly toxic, very mobile, and quite common groundwater con-taminant. The efficacy of the nZVI technology was evaluated in batch aqueous systems containing soils and groundwater impacted by chromium ore process-ing residuals (COPR) at a former manufacturprocess-ing site in New Jersey [32]. The average Cr(VI) concentration in groundwater samples from the site were measured to be 42.8±0.5 mg L-1 while the concentration in air-dried soils was 3,280±90 mg kg-1 [32]. The total chro-mium concentration, that is Cr(III) plus Cr(VI), was determined to be 7,730±120 mg kg-1 [32]. Due to the
presence of lime in the COPR-contaminated media, the pH of groundwater typically exceeded 10-11. The basis for this reaction involves the very favorable re-duction of Cr(VI) to Cr(III), a relatively non-toxic, highly immobile species which precipitates (i.e., Cr(III) oxyhydroxides) from solution at alkaline pH.
3/2Fe0(s) + CrO42-(aq) + 5H+(aq) →
3/2Fe2+(aq) + Cr(OH)3(s) + H2O(l) (8)
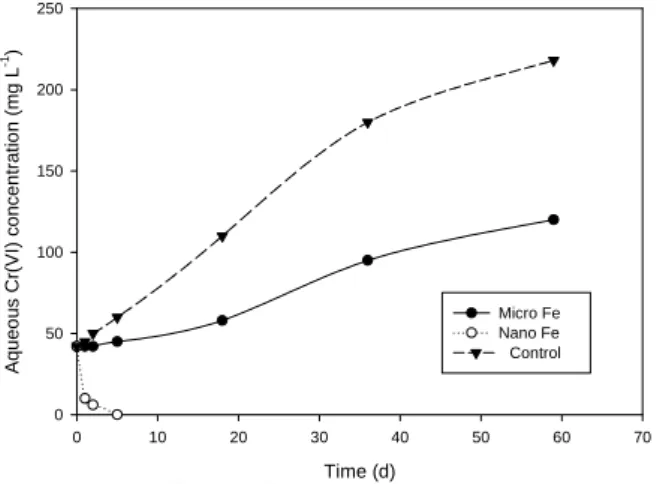
Batch solutions containing 10 g of COPR-impacted soils and 40 mL groundwater were exposed to Type I nZVI concentrations of 5-50 g L-1 (89.5-895 mM) under well-mixed conditions (Fig. 4). Not sur-prisingly, once the COPR-contaminated soils were placed in the reactors, the Cr(VI) concentration in-creased from 42 mg L-1 to approximately 220 mg L-1, demonstrating that substantial desorption and dissolu-tion of hexavalent chromium occurs during the course of the experiment. Within a timeframe of up to 6 d, Cr(VI) concentrations in solution were generally less than the detection limit, < 10 μg L-1 (Fig. 4). By
com-parison, the Cr(VI) concentration was observed to in-crease substantially in the reactors containing micro-scale iron (Fisher, 10 μm) during the course of the re-action.
The reductive capacity of the Type I nZVI was found to be on the order of 84-109 mg Cr(VI) per g of iron, approximately two orders of magnitude greater than the reaction with mZVI. Given the highly hetero-geneous nature of the COPR materials and the highly alkaline pH, it is likely that Cr(VI) desorbing from the matrix is reduced and rapidly precipitated as Cr(OH)3.
This precipitate enmeshes the COPR soils, forming a shell that tends to encapsulate any Cr(VI) remaining in the potentially still reactive core region.
1.3 Perchlorate
Perchlorate, ClO4-, emerged as a high profile
en-vironmental contaminant in the late 1990s when im-proved analytical techniques revealed widespread and previously undetected contamination in water supplies, particularly in the western U.S. [33]. Perchlorate con-centrations in excess of 100 μg L-1 have been detected
Time (d) Aqueo us C r(V I) concen tr at io n (mg L -1) Micro Fe Nano Fe Control 0 10 20 30 40 50 60 70
Fig. 4. Results of batch Cr(VI) reduction by mZVI and nZVI. The doses of mZVI and nZVI were 150 g L-1 and 5 g L-1, respectively. Time (d) 0 5 10 15 20 25 30 Ln(C /C 0 ) -1.2 -1.0 -0.8 -0.6 -0.4 -0.2 0.0 0.2 Nano Fe Micro Fe Control
Fig. 5. Degradation of 200 mg L-1 aqueous perchlorate by nZVI (20 g L-1) at 25 °C.
in Nevada’s Lake Mead, well beyond USEPA guid-ance levels of 1 μg L-1 [33].
While the iron-mediated reduction of perchlorate, shown below in Eq. 9, is a strongly thermodynamically favored process, based on the large, negative value for the standard Gibbs free energy of the reaction, ΔGrxn, of -1,3878 kJ mol-1, one
recent study reported that it is generally not reactive with ZVI [34]. In general,
O H Fe Cl H Fe ClO4−+4 0+8 + → − +4 2++4 2 (9) ZVI transformations require either direct contact with the reactive iron surface or through suitable bridging groups. The structure of perchlorate makes this diffi-cult by virtue of the fact that its reactive chlorine cen-tral atom, Cl(VII), is shielded by a tetrahedral array of bulky oxygen substituents which also fully delocalize the oxyanion’s negative charge.
Representative results of the reaction at 25 °C are shown in Fig. 5. Batch reactors contained nitro-gen-purged deionized water spiked with 1-200 mg L-1
Time (d) 0 5 10 15 20 25 30 Ln ( C /C 0 ) -2.5 -2.0 -1.5 -1.0 -0.5 0.0 25 oC 40 oC 75 oC
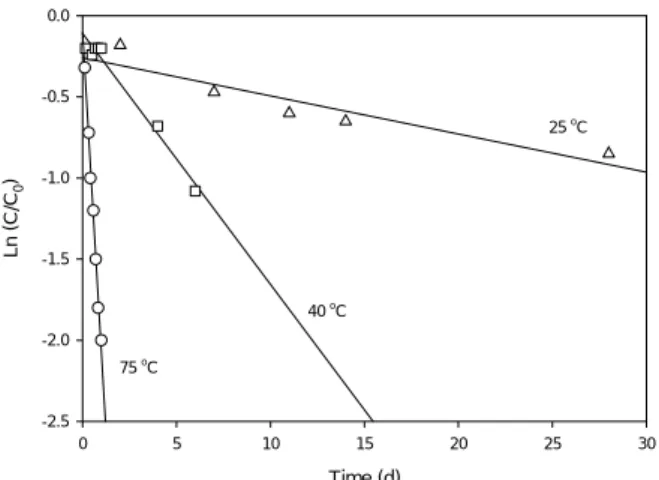
Fig. 6. A plot of Ln (C/C0) vs time for the reduction of perchlorate by nZVI (20 g L-1) at various temperature conditions.
method nZVI. After 28 d of elapsed time, negligible reaction with 20 g L-1 microscale iron (Aldrich, 4.95
μm) was observed while significant removal of per-chlorate was measured in the reactor containing 20 g L-1 Type II nZVI. As depicted in Fig. 6, progressively better removal of perchlorate was observed as the am-bient temperature was increased to 40 °C and finally to 75 °C. The most interesting feature of these plots is the dramatic reduction in the timescale achieved with increasing temperature from 25 to 75 °C. The half-life for perchlorate declined from 18-20 d at 25 °C to about 80-100 h at 40 °C and finally to approximately 8 h at 75 °C. Temperature was clearly helping to cata-lyze the removal of perchlorate from aqueous solution.
Chloride, produced as the principal product of the reaction, demonstrated the occurrence of actual transformation rather that mere sorption or other sur-face-associated sequestration. Besides chloride, the only intermediate identified was chlorate, ClO3-, and
only at trace levels [34]. Values of 0.013, 0.10, 0.64, and 1.52 mg g-1 h-1 were calculated at temperatures of 25, 40, 60, and 75 °C, respectively [34]. The experi-ments conducted at different temperatures enabled calculation of the activation energy for the nZVI-mediated degradation process. The activation energy was determined to be 79±8 kJ mol-1, a relatively for-midable value for aqueous reactions.
2. Field Testing Demonstration
Several in-situ field demonstrations of the nZVI technology have been conducted in a contaminated groundwater site since 2000 including the first field pilot test in Trenton New Jersey from May 8 to July 18, 2000 [35]. Among them, the field demonstration of the technology at an industrial and research facility in Research Triangle Park, NC during the Fall of 2002 is by far the largest testing scale [24]. In the most sig-nificant synthetic effort up to that time, approximately 12 kg of chloride method nZVI/Pd was synthesized at
Lehigh University during June and July. The test area was situated approximately 38 m down gradient of a former waste disposal area which has impacted groundwater with chlorinated solvents [24]. Site hydrogeology is quite complex with much of the im-pacted groundwater traversing through fractures in the sedimentary sandstones and siltstones [24]. In addition to the injection well, the test area infrastructure in-cluded three monitoring wells at distances of 6.6, 13, and 19 m down gradient of the injection well [24].
Approximately 6,100 L of a 1.9 g L-1 nZVI slurry in tap water was injected at a rate of 2.3 L min-1
over a nearly 2-d period from September 13 through September 15, 2002 [24]. The slurry was prepared on-site in a 1,500 L heavy-duty polyethylene tank and mixed during the course of the injection process. Within approximately 7 d from the injection, a reduc-tion of more than 90% of the pre-injecreduc-tion TCE con-centration of 14 mg L-1 was observed at the injection well and nearest monitoring well. These results are comparable to those from the first nZVI field demon-stration conducted in New Jersey two years earlier [35]. Groundwater quality standards for TCE, PCE, and cis-DCE were generally met within 6 weeks of the injection at these two monitoring wells without in-creases in VC being observed. Although pre-injection groundwater ORP levels were in the range of +50 to -100 mV, indicative of iron-reducing conditions, post-injection ORP readings plunged to -700 mV in the in-jection well and -500 in nearby monitoring wells. The radius of influence measured at the injection well was approximately 6-10 m.
This demonstration successfully showed that nZVI can travel distances of more than 20 m in groundwater, that reactivity seems to persist for peri-ods of greater than 4-8 weeks in the field, and that very high degrees of contaminant removal can be achieved.
CHALLENGE AHEAD
As this technology is moving rapidly from labo-ratory research to real world implementation, there are still a number of fundamental issues limiting the ap-plications of iron nanoparticles for environmental remediation including (1) mobility, (2) environmental impact and (3) cost-effectiveness. An important at-tribute of nanoparticles is the potential high mobility in environmental media, especially in the subsurface environment. However, evidence suggests that iron nanoparticles have strong tendency to form much lar-ger aggregates. Recent work at Lehigh has produced a fully stable dispersion of iron nanoparticles by modi-fying the iron surface [36]. The surface-modified (i.e., charged) iron nanoparticles could remain in suspen-sion for extended periods [36-39].
So far there is no report on the eco-toxicity of low-level iron in soil and water. It is the authors’
opin-ion that systematic research on the environmental transport, fate and ecotoxicity is needed to overcome increasing concerns in the environmental use of nanomaterials, and minimize any unintended impact. Iron nanoparticles may actually provide a valuable opportunity to demonstrate the positive effect on envi-ronmental quality. Iron is the fifth most used element; only hydrogen, carbon, oxygen and calcium were con-sumed in greater quantities. It has been found at the active center of many biological molecules and likely plays an important role in the chemistry of living or-ganisms being. It is well documented that iron is an essential constituent of the blood and tissues of the animal body. Iron in the body is mostly present as iron porphyrin or heme proteins, which include hemoglo-bin in the blood, myoglohemoglo-bin and the heme enzymes [40]. The heme enzymes permit the reversible combi-nation with molecular oxygen. By this mechanism the red blood cells carry oxygen from one part of the body to another. Nature has evolved highly organized sys-tem of iron uptake (e.g., the siderophore-mediated iron uptake), transport (via transferrins) and storage of iron (e.g., in the form of ferritin). The intracellular concentration of free iron, i.e., iron not bound to or-ganic ligands, is tightly controlled in animals, plants, and microbes. This is partly a result of the poor solu-bility of iron under aqueous and aerobic conditions: the solubility production of Fe(OH)3 is 4×10-38.
Within the cell, free iron generally precipitates as polymeric hydroxides. The challenge for us is to de-termine the transport, aquatic and biochemistry, ulti-mate fate of manufactured iron nanoparticles in the environment.
CONCLUSIONS
In this paper, we present a comprehensive re-view for current progress in ZVI nanoparticles for groundwater remediation. Three types of nZVI have been developed. The chloride method was used to prepare the Type I iron and exhibited an average par-ticle diameter on the order of 100-200 nm. Types II and III were more recent types produced using the sul-fate method. Types II and III nZVI were appreciably smaller than Type I with average particle size ranges of 60-70 and 50-70 nm, respectively. The nZVI has been tested with more than 75 different environmental contaminants from a wide variety of chemical classes. Among them, we review chlorinated aliphatic hydro-carbons, chlorinated pesticides (Lindane), hexavalent chromium, and perchlorate in this paper. Generally speaking, the reactivity of the nZVI is between 1-3 orders of magnitude greater as regards calculated kSA.
Field tests demonstrated that (1) nZVI can travel dis-tances of more than 20 m in groundwater, (2) reactiv-ity seems to persist for periods of greater than 4-8 weeks in the field and (3) very high degrees of con-taminant removal can be achieved. As this technology is moving rapidly from laboratory research to real
world implementation, understanding fundamental is-sues limiting the applications of iron nanoparticles for environmental remediation is still in great need. They include (1) mobility, (2) environmental impact and (3) cost-effectiveness.
REFERENCES
1. Masciangioli, T. and W.X. Zhang, Environmental technologies at the nanoscale. Environ. Sci. Technol., 37(5), 102A-108A (2003).
2. Carpenter, E.E., S. Calvin, R.M. Stroud and V.G. Harris, Passivated iron as core-shell nanoparticles. Chem. Mater., 15(17), 3245-3246 (2003).
3. Miehr, R., P.G. Tratnyek, J.Z. Bandstra, M.M. Scherer, M.J. Alowitz and E. Bylaska, Diversity of contaminant reduction reactions by zero valent iron: Role of the reductate. Environ. Sci. Technol., 38(1), 139-147 (2004).
4. Mondal, K., G. Jegadeesan and S.B. Lelvani, Removal of selenate by Fe and NiFe nanosized particles. Ind. Eng. Chem. Res., 43(16), 4922-4934 (2004).
5. Ponder, S.M., J.G. Darab, J. Bucher, D. Caulder, I. Craig, L. Davis, N. Edelstein, W. Lukens, H. Nitsche, L.F. Rao, D.K. Shuh and T.E. Mallouk, Surface chemistry and electrochemistry of supported zerovalent iron nanoparticles in the remediation of aqueous metal contaminants. Chem. Mater., 13(2), 479-486 (2001).
6. Ponder, S.M., J.G. Darab and T.E. Mallouk, Remediation of Cr(IV) and Pb(II) aqueous solutions using supported, nanoscale zero-valent iron. Environ. Sci. Technol., 34(12), 2564-2569 (2000).
7. Nurmi, J.T., P.G. Tratnyek, V. Sarathy, D.R. Baer and J.E. Amonette, Characterization and properties of metallic iron nanoparticles: Spectroscopy, electrochemistry, and kinetics. Environ. Sci. Technol., 39(5), 1221-1230 (2005).
8. Schrick, B., J.L. Blough, A.D. Jones, and T.E. Mallouk, Hydrodechlorination of trichloroethylene to hydrocarbons using bimetallic nickel-iron nanoparticles. Chem. Mater., 14(12), 5140-5147 (2002).
9. Liu, Y.Q., S.A. Majetich, R.D. Tilton, D.S. Sholl and G.V. Lowry, TCE dechlorination rates, pathways, and efficiency of nanoscale iron particles with different properties. Environ. Sci. Technol., 39(5), 1338-1345 (2005).
10. Lowry, G.V. and K.M. Johnson, Congener-specific dechlorination of dissolved PCBs by microscale and nanoscale zero valent iron in a water/methanol
solution. Environ. Sci. Technol., 38(19), 5208-5216 (2004).
11. Joo, S.H., A.J. Feitz, D.L. Sedlak, and T.D. Waite, Quantification of the oxidizing capability of nanoparticulate zero-valent iron. Environ. Sci. Technol., 39(5), 1263-1268 (2005).
12. Kanel, S.R., B. Manning, L. Charlet, and H. Choi, Removal of arsenic(III) from groundwater by nanoscale zero-valent iron. Environ. Sci. Technol., 39(5), 1290-1298 (2005).
13. Feng, J. and T. Lim, Pathways and kinetics of carbon tetrachloride and chloroform reductions by nano-scale Fe and Fe/Ni particles: Comparison with commercial micro-scale Fe and Zn. Chemosphere, 59(9), 1267-1277 (2005).
14. Matheson, L.J. and P.G. Tratnyek, Reductive dehalogenation of chlorinated methanes by iron metal. Environ. Sci. Technol., 28(12), 2045-2053 (1994).
15. Snoeyink, V.L. and D. Jenkins, Water Chemistry. John Wiley, New York, NY (1980).
16. Langmuir, D. Aqueous Environmental
Geochemistry. Prentice Hall, Upper Saddle River, NJ (1997).
17. Vogel, T.M., C.S. Criddle and P.L. McCarty, Transformations of halogenated aliphatic compounds. Environ. Sci. Technol., 21(8), 722-736 (1987).
18. Weber, E.J., Iron-mediated reductive
transformations: Investigation of reaction mechanism. Environ. Sci. Technol., 30(2), 716-719 (1996).
19. Wang, C.B. and W.X. Zhang, Synthesizing nanoscale iron particles for rapid and complete dechlorination of TCE and PCBs. Environ. Sci. Technol., 31(7), 2154-2156 (1997).
20. Zhang, W.X., C.B. Wang and H.L. Lien,
Treatment of chlorinated organic contaminants with nanoscale bimetallic particles. Catal. Today, 40(4), 387-395 (1998).
21. Lien, H.L. and W.X. Zhang, Transformation of chlorinated methanes by nanoscale iron particles. J. Environ. Eng.-ASCE, 125(11), 1042-1047 (1999). 22. Glavee, G.N., K.J. Klabunde, C.M. Sorensen and
G.C. Hadjipanayis, Chemistry of borohydride reduction of iron(II) and iron(III) ions in aqueous and nonaqueous media- Formation of nanoscale Fe, FeB, and Fe2B powders. Inorg. Chem., 34(1),
28-35 (1995).
23. Glazier, R., R. Venkatakrishnan, F. Gheorghiu, L. Walata, R. Nash and W.X. Zhang, Nanotechnology takes root. Civil Eng., 73(5), 64-69 (2003).
24. Zhang, W.X., Nanoscale iron particles for environmental remediation: An Overview. J. Nanoparticle Res., 5(3-4), 323-332 (2003).
25. Stumm, W. and J.J. Morgan, Aquatic Chemistry. 3rd Ed. John Wiley, New York, NY (1996).
26. Lien, H.L. and W.X. Zhang, Nanoscale iron particles for complete reduction of chlorinated ethenes. Colloid. Surface. A, 191(1-2), 97-105 (2001).
27. Lien, H.L. and W.X. Zhang, Hydrodechlorination of chlorinated ethanes by nanoscale Pd/Fe bimetallic particles. J. Environ. Eng.-ASCE, 131(1), 4-10 (2005).
28. Johnson, T.L., M.M. Scherer and P.G. Tratnyek, Kinetics of halogenated organic compounds degradation by iron metal. Environ. Sci. Technol., 30(8), 2634-2640 (1996).
29. Li, T. and J. Farrell, Reductive dechlorination of trichloroethene and carbon tetrachloride using iron and palladized-iron cathodes. Environ. Sci. Technol., 34(1), 173-179 (2000).
30. Brewster, J.H., Mechanisms of reductions at metal surfaces. I: A general working hypothesis. J. Am. Chem. Soc., 76(24), 6361-6363 (1954).
31. Xu, Y. and W.X. Zhang, Subcolloidal Fe/Ag particles for reductive dehalogenation of chlorinated benzenes. Ind. Eng. Chem. Res., 39(7), 2238-2244 (2000).
32. Cao, J. and W.X. Zhang, Stabilization of chromium ore processing residue (COPR) with nanoscale iron particles. J. Hazard. Mater., 132(2-3), 213-219 (2006).
33. Urbansky, E.T., Perchlorate in the Environment, Kluwer Academic/Plenum Publishers, New York, NY (2000).
34. Cao, J., D.W. Elliott and W.X. Zhang, Perchlorate reduction by nanoscale iron particles. J. Nanoparticle Res., 7(4-5), 499-506 (2005).
35. Elliot, D.W. and W.X. Zhang, Field assessment of nanoscale bimetallic particles for groundwater treatment. Environ. Sci. Technol., 35(24), 4922-4926 (2001).
36. Sun, Y.P., Dispersion of Nanoscale Iron Particles. Ph.D. Dissertation, Dept. Civil Environ. Eng., Lehigh University, Bethlehem, PA (2006).
37. Schrick, B., B.W. Hydutsky, J.L. Blough and T.E. Mallouk, Delivery vehicles for zerovalent metal nanoparticles in soil and groundwater. Chem. Mater., 16(11), 2187-2193 (2004).
38. Li, F., C. Vipulanandan and K.K. Mohanty, Microemulsion and solution approaches to nanoparticle iron production of degradation of
trichloroethylene. Colloid. Surface. A, 223(1-3), 103-112 (2003).
39. He, P. and D.Y. Zhao, Preparation and
characterization of a new class of starch-stabilized bimetallic nanoparticles for degradation of chlorinated hydrocarbons in water. Environ. Sci. Technol., 39(9), 3314-3320 (2005).
40. Ballou, D.P. (Ed.), Essays in Biochemistry. Vol. 34: Metalloproteins. Princeton University Press, Princeton, NJ (1999).
Discussions of this paper may appear in the discus-sion section of a future issue. All discusdiscus-sions should be submitted to the Editor-in-Chief within six months of publication.
Manuscript Received: July 5, 2006 Revision Received: September 28, 2006 and Accepted: October 5, 2006