Synthesis and Characterization of Light-Emitting H-Bonded
Complexes and Polymers Containing Bis(pyridyl) Emitting Acceptors
Hong-Cheu Lin,*,‡Chien-Min Tsai,‡ Guan-Hao Huang,‡ and Yu-Tai Tao†Department of Materials Science and Engineering, National Chiao Tung UniVersity, Hsinchu, Taiwan (ROC), and Institute of Chemistry, Academia Sinica, Taipei, Taiwan (ROC)
ReceiVed September 9, 2005; ReVised Manuscript ReceiVed NoVember 10, 2005
ABSTRACT: A series of bis(pyridyl) acceptor emitters are synthesized and the LC and photophysical (PL and EL) properties of their supramolecular structures, including H-bonded polymer networks and trimers, are explored. Unique mesomorphic properties may be introduced into these H-bonded complexes and polymers containing nonmesogenic acceptor emitters. In addition, the emission properties of bis(pyridyl) acceptor emitters can be adjusted by their surrounding nonphotoluminescent proton donors. Compared with pure bis(pyridyl) acceptor emitters, red-shifts of PL emission wavelengths were observed in most of the H-bonded complexes and polymers. Because of the higher acidity value of m-PC10BA (16) and its stronger H-bonded effect in H-bonded polymer
complexes, the largest red-shift of 93 nm inλmaxvalue of PL has occurred in the fully H-bonded polymer network
p-PBBBP-OC8(8)/m-PC10BA (16) in contrast to pure emitter p-PBBBP-OC8(8). A very narrow full width at
half-maximum (fwhm) value (38 nm) of EL spectra can be obtained in the device of H-bonded complexes blended with PVK, and a higher brightness of EL is produced at an appropriate annealing temperature.
Introduction
Recently, hydrogen bonds (H-bonds) have been shown to be useful in the development of applications of electronic and photonic devices.1-4The energies of H-bonds (25-40 kJ/mol) are stronger than van der Waals interactions (5-10 kJ/mol) but weaker than ionic bonds (200-400 kJ/mol).5 Many kinds of H-bonds and building elements have been developed in the H-bonded structures to stabilize liquid crystalline phases. It has been demonstrated that liquid crystalline (LC) polymers,6-9 copolymers,10,11 and monomers12-16 containing H-bonds are appropriate candidates for obtaining functionalized LC systems. Since the report of Burroughes et al.,17 rapid progress has been made in improving the efficiencies and lifetimes of light-emitting diodes (LED) based on conjugated polymers. A number of reports demonstrate polarized electroluminescence (EL) with the aim of incorporating such polymer LEDs as backlights into liquid crystal displays.18 For instance, cyanobiphenyl liquid crystals can be aligned by rubbing polyimide and thus to have dichroic ratios in absorption spectra.19 The frequently used materials are polyacrylate,20,21 polyfluorene,18,22-28 and their oligomer derivatives.29Additionally, lyotropic liquid crystals, which possess a hexagonal mesophase to solubilize the monomer and direct its electropolymerization, are utilized to prepare poly-(3,4-ethylenedioxythiophene) (PEDOT) films that replicate the texture and birefringence of the LC template.30 In particular, liquid crystalline conducting polymers containing mesogenic parts in their side chains31-33or dyes doped in liquid crystalline structure34 have attracted much interest in their molecular alignment, which is anticipated for use in polarized emissions. Moreover, polarized OLEDs were constructed using heptafluo-rene lightly doped with monodisperse conjugated oligomers for an efficient emission of blue light, and a high polarization ratio was obtained.35Besides, some other higher polarization ratios and dichroic ratios of emissions are reported as well.23,36
Polymer light emitting diodes containing pyridine37,38and thiophene39repeat units are of considerable interest in the areas of electron transporting. The photoluminescence (PL) of the polymers8,40and copolymers41,42due to hydrogen bonds within and between the polymer chains are reported. For instance, hierarchical self-assemblies are formed in comb-shaped su-pramolecules43consisting of poly(2,5-pyridinediyl), acid dopants, and H-bonded alkyl side chains. The observation of the thermoreversible gelation of fluorescent p-phenylenevinylene derivatives44 shows that they control their gelation as a consequence of cooperative hydrogen bonds and self-assembly induced byπ-π stacking. Among blue, green, and red emis-sions, one of the main problems of good polymer emitters is the demand for good color purity. Reducing the values of the full width at half-maximum (fwhm) of polymer emissions, i.e., producing good color purity, in blend films was reported in the literature.45-48
To our knowledge, H-bonded effects on PL and electrolu-minescent (EL) properties of liquid crystalline supramolecules containing acceptor emitters have been scarcely reported.14Our preliminary results show that a red shift occurs in the bis(pyridyl) emitters (as proton acceptors) when proton donor acids are H-bonded to the supramolecules (H-bonded trimers). Distinct liquid crystalline properties can be introduced by complexation of different acceptor emitters and complementary donors. However, the H-bonded trimers could not be utilized for LED applications due to the poor film-forming quality of the small molecules in our previous work.14To solve this problem, we have surveyed new H-bonded complexes containing photolu-minescent bis(pyridyl) acceptors, including H-bonded complexes blended with poly(9-vinylcarbozole) (PVK) matrixes, and this leads to high quality films of supramolecular LED polymer blends. Therefore, we also would like to illustrate the H-bonding effects on the LC, PL, and EL properties of these light-emitting supramolecules.
Experimental Section
General Information. NMR spectra were carried out on a
Bruker AC 300 M Hz spectrometer. The thermal transition
* Author for correspondence. Telephone: 8863-5712121, ext. 55305. Fax: 8863-5724727. E-mail: [email protected].
‡National Chiao Tung University. †Academia Sinica.
10.1021/ma051967w CCC: $33.50 © 2006 American Chemical Society Published on Web 12/21/2005
temperatures and textures of all products were obtained from a Perkin-Elmer DSC-7 and Leitz Laborlux S polarizing optical microscope (POM) equipped with a THMS-600 heating stage. The heating and cooling rates were 10°C min-1for all measurements in N2unless mentioned. UV-vis absorption spectra were recorded
in dilute solutions (10-6 M) on a HP 8453 spectrophotometer. Fluorescence spectra were obtained on a Hitachi F-4500 spectro-photometer. The textures of mesophases were studied using a polarizing optical microscope (Leica DMLP) equipped with a hot stage. The highest occupied molecular orbital (HOMO) energy levels were estimated from the optical response on a Riken Photoelectron spectrometer AC-2. The work function and ionization potential can be measured by the photoelectron spectrometer for the OLED materials in air. To align the liquid crystals into a monodomain, these materials were sealed in antiparallel rubbing LC cells with a cell gap of 9µm, and the polarized PL emission
spectra were measured at corresponding mesomophic temperatures. Number-average and weight-average molecular weights, Mn and Mw, respectively, were determined by polystyrene-calibrated gel
permeation chromatography (GPC; Jasco PU-1580 and Jasco RI-930) using THF as the eluent.
Device Fabrication. Prepatterned ITO substrates with an
effec-tive individual device area of 3.14 mm2were cleaned by ultrasonic
machine in various detergent solutions and D. I. water. After that
the ITO substrates were treated with oxygen plasma for 2 min before spin-coating. The emission layer consists of synthesized emitters doped into poly(9-vinylcarbozole) (PVK) matrix with a concentra-tion of 5 wt %, and polymer thin films were made by spin-coating from THF solution with a concentration of 1.65 wt %. The spin-coating rate of 3000 rpm was proceeded for 40 s, and the thickness of the emitting layer was about 60 nm (with a total thickness of 100 nm, including 40 nm of electron transporting layer). The electron transporting layer (40 nm) of 2,2′,2′′ -(1,3,5-phenylene)-tris[1-phenyl-1H-benzimidazole] (TPBI) was deposited thermally at a rate around 0.1-0.3 Å/s under a pressure of∼2 × 10-5Torr in an Ulvac Cryogenic deposition system. One layer of magnesium and silver alloy (ca. 10:1, 50 nm) was deposited as a cathode, which was capped with 100 nm of silver. The current-voltage-luminescence characteristics were measured on ambient conditions by a Keithley 2400 source meter and a Newport 1835C optical meter equipped with 818ST silicon photodiode.
Materials. Chemicals and solvents were reagent grades and
purchased from Aldrich, ACROS, TCI, and Lancaster Chemical Co. THF was distilled to keep anhydrous before use. The other chemicals were used without further purification. The synthetic routes of oligo(p-phenylene-vinylene)s are shown in Schemes 1 and 2 (for compounds 1-9), and all proton donors (compounds
10-16) are illustrated in Chart 1, and all products were identified Scheme 1
as the required materials by1H and13C NMR spectroscopy and
elemental analyses.
Synthesis of 1,4-Bis(triphenylphosphoniomethyl)benzene Di-bromide (a). A 10.0 g (94.2 mmol) sample of xylene of was slowly
added into a solution containing 33.5 g (188.4 mmol) of N-bromosuccinimide (NBS) and a catalytic amount of 1.55 g (9.42 mmol) of 2,2′-azobis(isobutyronitrile) (AIBN) in 100 mL of anhydrous CCl4. The reaction mixture was slowly warmed to 70 °C and refluxed for 4 h. After cooling to room temperature the mixture was filtered (isolation of succinimide). The obtained filtrate was removed under reduced pressure. The resulting residue was used without further purification (because unstable) as a brown oil and then directly added into 67.6 g (207.2 mmol) of triphenylphos-phine in 100 mL of anhydrous xylene at 0°C under nitrogen. The reaction mixture was slowly warmed to 110°C and refluxed for
18 h. Upon cooling of the reaction to room temperature, the precipitate formed was filtered off and the crude product purified was washed from hexane to give a white solid. Yield: 34.2 g (46%).
1H NMR (300 MHz, CDCl
3, ppm): δ 5.35 (d, J ) 20.0 Hz, 4H),
6.92 (s, 4H), 7.63-7.78 (m, 30H).
Synthesis of p-PBP (1). A 4.26 g (38.1 mmol) sample of t-BuOK
was slowly added into a solution containing 10.0 g (12.7 mmol) of compound a and 2.98 g (27.9 mmol) of pyridine-4-carboxaldehyde (b) in 50 mL of anhydrous THF at 0°C under nitrogen. The reaction mixture was slowly warmed to 100°C and refluxed for 20 h. Upon cooling to room temperature, the reaction was quenched with water and extracted with dichloromethane. After evaporation of the solvent, the residue was purified by silica gel (eluent: ethyl acetate/ hexane ) 5:1) to get yellow solids. Yield: 1.26 g (16%).1H NMR
(300 MHz, CDCl3, ppm): δ 7.02 (d, J ) 16.4 Hz, 2H), 7.26 (d, J Scheme 2
) 16.4 Hz, 2H), 7.36 (d, J ) 6.0 Hz, 4H), 7.55 (s, 4H), 8.58 (d, J ) 6.0 Hz, 4H).13C NMR (CDCl
3, ppm): 120.82, 126.46, 127.45,
132.38, 136.50, 144.38, 150.21. Anal. Calcd for C20H16N2: C,
84.48; H, 5.67; N, 9.85. Found: C, 84.11; H, 5.89; N, 9.88.
Synthesis of m-PBP (2). This compound was obtained from
compound a, 10.0 g (12.7 mmol), and pyridine-3-carboxaldehyde (c), 2.98 g (27.9 mmol), following a similar procedure as described for compound p-PBP (1). This crude product was isolated as a brown solid. Yield: 1.18 g (15%).1H NMR (300 MHz, CDCl
3, ppm): δ 6.81 (d, J ) 16.5 Hz, 2H), 6.89 (d, J ) 16.5 Hz, 2H), 6.99-7.03 (m, 2H), 7.26 (s, 4H), 7.56 (d, J ) 7.9 Hz, 2H), 8.21 (d, J ) 3.8 Hz, 2H), 8.46 (d, J ) 1.9 Hz, 2H).13C NMR (CDCl 3, ppm): 123.54, 125.10, 127.07, 130.20, 132.64, 132.90, 136.54, 148.54, 148.60. Anal. Calcd for C20H16N2: C, 84.48; H, 5.67; N,
9.85. Found: C, 84.08; H, 5.80; N, 9.88.
Synthesis of o-PBP (3). This compound was obtained from
compound a, 10.0 g (12.7 mmol), and pyridine-2-carboxaldehyde (d), 2.98 g (27.9 mmol), following a procedure similar to that described for compound p-PBP (1). This crude product was isolated as a brown solid. Yield: 1.34 g (17%).1H NMR (300 MHz, CDCl
3,
ppm): δ 7.13-7.17 (m, 2H), 7.2 (d, J ) 10.8 Hz, 2H), 7.39 (d, J ) 7.7 Hz, 2H), 7.59 (s, 4H), 7.62-7.69 (m, 4H), 8.60 (d, J ) 4.7
Hz, 2H).13C NMR (CDCl
3, ppm): 122.07, 122.17, 127.48, 128.10,
132.22, 136.53, 136.70, 149.68, 155.54. Anal. Calcd for C20H16N2:
C, 84.48; H, 5.67; N, 9.85. Found: C, 84.08; H, 5.89; N, 9.83.
Synthesis of 1,4-Bis(chloromethyl-2,5-dimethoxy)benzene (e).
A 60 mL aliquot of HCl was slowly added into 50.0 g (36.2 mmol) of 1,4-dimethoxybenzene and 55.0 g (181.8 mmol) of paraform-aldehyde in 300 mL of CH3COOH at 0°C under nitrogen. The
reaction mixture was slowly warmed to room temperature and refluxed for 48 h. The aqueous layer was collected and added into water, and recrystallized with acetone to get white solids. Yield: 38.2 g (45%).1H NMR (300 MHz, CDCl
3, ppm): δ 3.85 (s, 6H),
4.64 (s, 4H), 6.93 (s, 2H).
Synthesis of 1,4-Bis(triphenylphosphoniomethyl)-2,5-dimeth-oxybenzene Dichloride (f). A 10 g (42.5 mmol) sample of
compound e was slowly added into 24.6 g (94.0 mmol) of triphenylphosphine in 100 mL of xylene. The reaction mixture was slowly warmed to 110°C and refluxed for 18 h. Upon cooling to room temperature, the mixture was washed with hexane to get brown solids. Yield: 29.7 g (92%).1H NMR (300 MHz, CDCl
3,
ppm): δ 2.95 (s, 6H), 5.24 (d, J ) 19.5 Hz, 4H), 6.91 (s, 2H),
7.63-7.79 (m, 30H).
Synthesis of p-PBP-OMe (4). A 14.30 g (128 mmol) sample
of t-BuOK was slowly added into a solution containing 32.7 g (43.0 mmol) of compound f and 11.3 g (106.0 mmol) of
pyridine-4-carboxaldehyde (b) in 50 mL of anhydrous THF at 0 °C under nitrogen. The reaction mixture was slowly warmed to 100°C and refluxed for 20 h. Upon cooling to room temperature, the reaction was quenched with water and extracted with dichloromethane. After evaporation of the solvent, the residue was purified by silica gel (eluent: ethyl acetate/hexane ) 5:1) to get brown solids. Yield: 1.03 g (7%).1H NMR (300 MHz, CDCl 3, ppm): δ 3.95 (s, 6H), 7.07 (d, J ) 22.5 Hz, 2H), 7.15 (s, 2H), 7.26-7.33 (m, 2H), 7.50 (d, J ) 24.7 Hz, 2H), 7.87-7.92 (m, 2H), 8.47-8.50 (m, 2H), 8.74 (d, J ) 2.8 Hz, 2H).13C NMR (CDCl 3, ppm): 56.22, 109.47,
120.92, 126.73, 127.63, 150.08, 151.83. Anal. Calcd for C22H20N2O2:
C, 76.72; H, 5.85, N, 8.13. Found: C, 76.45; H, 5.98, N, 8.21.
Synthesis of m-PBP-OMe (5). This compound was obtained
from compound f 32.7 g (43.0 mmol) and pyridine-3-carboxalde-hyde (c) 11.3 g (106.0 mmol), following a similar procedure as described for compound p-PBP-OMe (4). This crude product was isolated as a yellow solid. Yield: 1.32 g (9%).1H NMR (300 MHz,
CDCl3, ppm): δ 3.95 (s, 6H), 7.10 (d, J ) 15.0 Hz, 2H), 7.14 (s,
2H), 7.26-7.33 (m, 2H), 7.54 (d, J ) 16.5 Hz, 2H), 7.87 (d, J ) 7.9 Hz, 2H), 8.48 (d, J ) 3.8 Hz, 2H), 8.74 (d, 2H).13C NMR
(CDCl3, ppm): 56.17, 109.13, 123.48, 125.24, 125.30, 126.33,
132.58, 133.39, 148.28, 148.54, 151.49. Anal. Calcd for C22H20N2O2:
C, 76.72; H, 5.85, N, 8.13. Found: C, 76.50; H, 6.02, N, 8.32.
Synthesis of o-PBP-OMe (6). This compound was obtained
from compound f 32.7 g (43.0 mmol) and pyridine-2-carboxalde-hyde (d) 11.3 g (106.0 mmol), following a procedure similar to that described for compound p-PBP-OMe (4). This crude product was isolated as a yellow solid. Yield: 3.5 g (24%).1H NMR (300
MHz, CDCl3, ppm): δ 3.91 (s, 6H), 7.11-7.14 (m, 2H), 7.18 (s, 2H), 7.23 (d, J ) 16.4 Hz, 2H), 7.47 (d, J ) 8.1 Hz, 2H), 7.61-7.65 (m, 2H), 7.87 7.04 (d, J ) 16.4 Hz, 2H), 8.7 (d, J ) 4.6 Hz, 2H). 13C NMR (CDCl 3, ppm): 56.08, 109.77, 121.52, 121.82, 126.40, 127.33, 129.00, 136.35, 149.60, 151.85, 156.18. Anal. Calcd for C22H20N2O2: C, 76.72; H, 5.85, N, 8.13. Found: C, 76.24; H, 6.00, N, 8.03. Synthesis of 1,4-Dioctyloxybenzene (g) (n ) 8). A 675 mL
aliquot of ethyl alcohol and 75 mL of water were slowly added into a mixture containing 22.8 g (207.0 mmol) of hydroquinone and 99.9 g (518.0 mmol) of 1-bromooctane. The reaction mixture was slowly warmed to 80°C and refluxed for 48 h. Upon cooling to room temperature, the mixture was extracted with toluene. After evaporation of the solvent, the residue was washed by hexane to get white solid. Yield: 24.2 g (35%).1H NMR (300 MHz, CDCl
3,
ppm): δ 0.88 (t, J ) 6.6 Hz, 6H), 1.28-1.43 (m, 20H),
1.70-1.79 (m, 4H), 3.89 (t, J ) 6.6 Hz, 4H), 6.81 (s, 4H).
Synthesis of 1,4-Didodecyloxybenzene (h) (n ) 12). This
compound was obtained from hydroquinone, 22.8 g (207.0 mmol), and 1-bromododecane, 129.0 g, (518.0 mmol), following a proce-dure similar to that described for compound g. This crude product was isolated as a white solid. Yield: 49.9 g (54%).1H NMR (300
MHz, CDCl3, ppm): δ 0.87 (t, J ) 6.6 Hz, 6H), 1.26-1.43 (m,
36H), 1.70-1.77 (m, 4H), 3.89 (t, J ) 6.6 Hz, 4H), 6.81 (s, 4H).
Synthesis of 1,4-Bis(chloromethyl)-2,5-dioctyloxybenzene (i) (n ) 8). A 35 mL aliquot of HCl was slowly added into 24.4 g
(73.0 mmol) of compound g and 10.9 g (360.0 mmol) of paraformaldehyde in 200 mL of CH3COOH at 0°C under nitrogen.
The reaction mixture was slowly warmed to room temperature and refluxed for 100 h. The aqueous layer was collected and added into water, and it was recrystallized with toluene to get white solids. Yield: 11.6 g (37%).1H NMR (300 MHz, CDCl
3, ppm): δ 0.89
(t, J ) 6.6 Hz, 6H), 1.29-1.60 (m, 20H), 1.75-1.84 (m, 4H), 3.97 (t, J ) 6.4 Hz, 4H), 4.63 (s, 4H), 6.90 (s, 2H).
Synthesis of 1,4-Bis(chloromethyl)-2,5-didodecyloxybenzene (j) (n ) 12). This compound was obtained from compound h 32.6
g (73.0 mmol) and paraformaldehyde 10.9 g (360.0 mmol), following a procedure similar to that described for compound i. This crude product was isolated as a white solid. Yield: 17.1 g (43%).1H NMR (300 MHz, CDCl
3, ppm): δ 0.88 (t, J ) 6.6 Hz,
6H), 1.26-1.31 (m, 36H), 1.70-1.79 (m, 4H), 3.98 (t, J ) 6.4 Hz, 4H), 4.63 (s, 4H), 6.90 (s, 2H).
Synthesis of 2,5-Dioctyloxy-1,4-xylenebis(diethyl phospho-nate) (k) (n ) 8). A 15.1 g (35.0 mmol) sample of compound i
and 57.76 g (350.0 mmol) of P(OEt)3, were slowly warmed to 120 °C and stirred for 100 h. After evaporation of the solvent, the residue was recrystallized with petroleum ether (boiling range 40-65°C) and dried to get white solids. Yield: 18.9 g (85%).1H NMR (300
MHz, CDCl3, ppm): δ 0.89 (t, J ) 6.6 Hz, 6H), 1.20-1.43 (m,
20H), 1.73-1.79 (m, 16H), 3.21 (d, J ) 20.2 Hz, 4H), 3.88-4.09 (m, 12H), 6.91 (s, 2H).
Synthesis of 2,5-Didodecyloxy-1,4-xylenebis(diethyl phospho-nate) (l) (n ) 12). This compound was obtained from compound j 19.0 g (35.0 mmol) and P(OEt)310.9 g (350.0 mmol), following a similar procedure as described for compound k. This crude product was isolated as a white solid. Yield: 22.7 g (87%).1H
NMR (300 MHz, CDCl3, ppm): δ 0.88 (t, J ) 6.6 Hz, 6H),
1.20-1.38 (m, 36H), 1.66-1.79 (m, 16H), 3.24 (d, J ) 20.2 Hz, 4H), 3.88-4.19 (m, 12H), 6.91 (s, 2H).
Synthesis of p-PBP-OC8(7). A 1.68 g (15.0 mmol) sample of
t-BuOK was slowly added into a solution containing 3.8 g (6.0 mmol) of compound k and 1.41 g (13.2 mmol) of pyridine-4-carboxaldehyde (b) in 100 mL of anhydrous THF at 0°C under nitrogen. The reaction mixture was slowly warmed to 100°C and refluxed for 12 h. Upon cooling to room temperature, the mixture was extracted with water and CH2Cl2. After evaporation of the
solvent, the residue was recrystallized by THF and hexane to get yellow solids. Yield: 0.45 g (14%).1H NMR (300 MHz, CDCl
3, ppm): δ 0.88 (t, J ) 6.6 Hz, 6H), 1.26-1.62 (m, 20H), 1.80-1.92 (m, 4H), 4.07 (t, J ) 6.6 Hz, 4H), 7.10 (d, J ) 16.0 Hz, 2H), 7.13 (s, 2H), 7.43 (d, J ) 5.7 Hz, 4H), 7.79 (d, J ) 16.5 Hz, 2H), 8.58 (d, J ) 5.1 Hz, 4H).13C NMR (CDCl 3, ppm): 14.08, 22.65, 26.27, 29.29, 29.38, 29.41, 31.80, 69.52, 110.99, 120.82, 126.65, 126.72, 127.86, 145.08, 150.17. Anal. Calcd for C36H48N2O2: C,
79.96; H, 8.95, N, 5.18. Found: C, 80.02; H, 9.08, N, 4.86.
Synthesis of 4-(trans-2-[4]-pyridylvinyl)benzaldehyde (m). A
4.66 g (50.0 mmol) sample of 4-picoline was slowly added into a solution containing 10.1 g (75.0 mmol) of terephthaldicarboxalde-hyde in 7 mL of acetic acid and 16 mL of acetic anhydride. The reaction mixture was slowly warmed refluxed for 6 h. Upon cooling to 50°C, the mixture was extracted with HCl (6 N) solution. The aqueous layer was collected and added into NaOH (3 N) solution, and extracted with ethyl acetate. After evaporation of the solvent, the residue was recrystallized with isopropyl alcohol to get orange solids. Yield: 4.7 g (45%).1H NMR (300 MHz, CDCl
3, ppm): δ
7.17 (d, J ) 16.3 Hz, 1H), 7.29 (d, J ) 16.9, 2H), 7.41 (d, J ) 6.1 Hz, 2H), 7.69 (d, J ) 8.2 Hz, 2H), 7.91 (d, J ) 8.3 Hz, 2H), 8.62 (d, J ) 6.1 Hz, 2H), 10.02 (s, 1H).
Synthesis of p-PBBBP-OC8(8). A 0.78 g (33.0 mmol) sample of NaH was slowly added into a solution containing 5.0 g (24.0 mmol) of compound m and 6.59 g (11.0 mmol) of compound k in 50 mL of anhydrous THF at 0 °C under nitrogen. The reaction mixture was slowly warmed to refluxed for 20 h. Upon cooling to room temperature, the solvent was removed under reduced pressure, and the residue was washed with ethyl acetate and ethyl alcohol. The residue was collected and recrystallized with THF/MeOH (1: 1) to get yellow solids. Yield: 1.48 g (18%).1H NMR (300 MHz,
CDCl3, ppm): δ 0.86 (t, J ) 6.6 Hz, 6H), 1.26-1.57 (m, 20H), 1.80-2.04 (m, 4H), 4.08 (s, J ) 6.6 Hz, 4H), 7.03 (d, J ) 16.2 Hz, 2H), 7.14 (s, 2H), 7.16 (d, J ) 16.5 Hz, 2H), 7.32 (d, J ) 16.0 Hz, 2H), 7.38 (d, J ) 6.0 Hz, 4H), 7.53 (d, J ) 12.6 Hz, 2H), 7.55 (s, 8H), 8.58 (d, J ) 6.0 Hz, 4H).13C NMR (CDCl 3, δ): 14.12, 22.68, 26.32, 29.32, 29.43, 29.51, 31.84, 69.60, 110.67, 120.78, 124.15, 125.60, 126.94, 127.40, 128.21, 132.79, 135.21, 138.54, 144.66, 150.19. Anal. Calcd for C52H60N2O2: C, 83.83; H, 8.12,
N, 4.29. Found: C, 83.89; H, 8.12, N, 4.19.
Synthesis of p-PBBBP-OC12(9). This compound was obtained from compound m 5.0 g (24.0 mmol) and compound l 8.22 g (11.0 mmol), following a procedure similar to that described for compound p-PBBBP-OC8(8). This crude product was isolated as
a yellow solid. Yield: 3.21 g (34%).1H NMR (300 MHz, CDCl 3, ppm): δ 0.87 (t, J ) 6.6 Hz, 6H), 1.26-1.56 (m, 36H), 1.86-1.89 (m, 4H), 4.08 (t, J ) 6.4 Hz, 4H), 7.04 (d, J ) 16.2 Hz, 2H), 7.14 (s, 2H), 7.16 (d, J ) 16.5 Hz, 2H), 7.32 (d, J ) 16.2 Hz, 2H), 7.37 (d, J ) 6.1 Hz, 4H), 7.53 (d, J ) 12.6 Hz, 2H), 7.55 (s, 8H), 8.57 (d, J ) 6.0 Hz, 4H).13C NMR (CDCl 3, δ): 14.08, 22.67, 26.31, 29.38, 29.47, 29.50, 29.63, 29.68, 29.73, 31.92, 69.57, 110.66, 120.76, 124.13, 125.58, 126.93, 127.38, 128.20, 132.77, 135.19, 138.52, 144.62, 150.19, 151.22. Anal. Calcd for C60H76N2O2:
C, 84.06; H, 8.94, N, 3.27. Found: C, 83.71; H, 9.11, N, 3.20. Proton donors (10-16) were identified as the required materials by 1H,13C NMR spectroscopy and elementary analyses. Proton
donors (10-12) were reported in our previous results.14 Donor
monomers p-MC10BA (13) and m-MC10BA (15) were prepared
according to the procedure reported by Portugal et al.49The NMR
results of the monomers are as follows.
4-((6-(Acryloyloxy)decyl)oxy)benzoic Acid Monomer, p-MC10
-BA (13). Yield: 35%.1H NMR (300 MHz, CDCl 3, δ): 1.32-1.46 (m, 12H), 1.65-1.69 (m, 2H), 1.76-1.83 (m, 2H), 4.02 (t, 2H, J ) 6.4 Hz), 4.15 (t, 2H, J ) 6.6 Hz), 5.83 (d, J ) 9.0 Hz, 1H), 6.14 (m, 1H), 6.38 (d, J ) 16.1 Hz, 1H), 6.94 (d, J ) 8.8 Hz, 2H), 8.06 (d, J ) 8.8 Hz, 2H). 13C NMR (ppm, CDCl 3): 23.5, 25.9, 27.5, 28.6, 29.1, 29.2, 29.3, 29.4, 64.7, 68.2, 114.2, 121.4, 128.6, 130.5, 132.3, 163.6, 166.4, 171.7. Anal. Calcd for C20H28O5: C, 68.91; H, 8.1. Found: C, 68.8; H, 7.96.
3-((6-(Acryloyloxy)decyl)oxy)benzoic Acid Monomer, m-MC10
-BA (15). Yield: 25%.1H NMR (300 MHz, CDCl 3, δ): 1.25-1.99 (m, 16H), 4.01 (t, J ) 6.6 Hz, 2H), 4.15 (t, J ) 6.9 Hz, 2H), 5.80 (d, J ) 9.0 Hz), 6.07-6.16 (m, 1H), 6.34 (d, J ) 15.9 Hz, 1H), 7.13 (d, J ) 8.4 Hz, 1H), 7.28 (t, J ) 8.1 Hz, 1H), 7.59 (s, 1H), 7.68 (d, J ) 7.5 Hz, 1H).13C NMR (300 MHz, CDCl 3,δ): 25.90, 25.98, 28.59, 29.17, 29.31, 39.40, 52.98, 54.97, 55.95, 64.71, 68.23, 91.52, 115.07, 120.86, 122.45, 128.60, 129.46, 130.44, 159.16, 186.63. Anal. Calcd for C20H28O5: C, 68.94; H, 8.10.
Found: C, 68.88; H, 8.18.
Polymerization. The synthetic procedures of polymers p-PC10 -BA (14) and m-PC10BA (16) are shown as follows: A solution of
1 g of monomers 13 and 15 in 5 mL of anhydrous THF was prepared, and then 1 mol % of benzoyl peroxide was added as an initiator and reacted at 70°C for 40 h under nitrogen atmosphere. The reacted mixture was poured into a large amount of petroleum ether. The polymers 14 and 16 were purified by repeated dissolution in THF and precipitation from petroleum ether (40-65°C).
Poly{4-((6-(acryloyloxy)decyl)oxy)benzoic acid}, p-PC10BA
(14). Yield: 58%.1H NMR (300 MHz, CDCl
3,δ): 1.22-1.65 (m,
16H), 3.94 (t, 4H), 6.93 (d, 2H), 7.83 (d, 2H).
Poly{3-((6-(Acryloyloxy)decyl)oxy)benzoic acid}, m-PC10BA
(16). Yield: 55%.1H NMR (300 MHz, CDCl
3,δ): 1.27-2.25 (m,
16H), 3.97 (t, 4H), 7.10 (m, 1H), 7.32-7.35 (t, 1H), 7.54-7.69 (m, 2H).
The number-average molecular weight (Mn) and polydispersity
index (PDI) of polymers are as follows. p-PC10BA (14): Mn) 5800 and PDI ) 1.21. m-PC10BA (16): Mn) 4400 and PDI ) 1.06. Results and Discussion
Thermal Properties. Phase transition temperatures and thermal behavior of bonded acceptor emitters (1-9), H-bonded donors (10-16), and their H-H-bonded complexes char-acterized by differential scanning calorimetry (DSC) and polarizing optical microscopy (POM) are summarized in Tables 1-4. The phase transition temperatures of bis(pyridyl) acceptor emitters (1-9), i.e., the melting and crystallization temperatures (Tmand Tcr), are demonstrated in the following order: p-PBP (1) > p-PBP-OMe (4) > o-PBP (3) > o-PBP-OMe (6) > m-PBP (2) > m-PBP-OMe (5) > p-PBP-OC8 (7), and p-PBBBP-OC8(8) > p-PBBBP-OC12(9) > p-PBP-OC8(7) (shown in Table 1). In general, the phase transition temperatures (i.e., Tmand Tcr) of bis(pyridyl) emitters (1-9) decrease with increasing the length of side chains. This phenomenon may be as a result of the long alkoxy side chains which hinder the molecular packing and result in the decrease of phase transition
temperatures. For instance, the phase transition temperatures of p-PBBBP-OC8(8) > p-PBBBP-OC12(9) and p-PBP (1) > p-PBP-OMe (4) > p-PBP-OC8(7) are observed with the same rigid cores. Besides, longer rigid cores with the same side chains have higher transition temperatures, for example, p-PBBBP-OC8 (8) > p-PBP-OC8 (7). As for the phase transition temperatures of p-PBP-OMe (4) > o-PBP-OMe (6) > m-PBP-OMe (5) in Tmand Tcr, it can be explained by that stronger dipole-dipole interaction occurred between para-pyridyl groups in the linear conjugated direction. Similar to
methoxy substituted compounds (4-6), the phase transition temperatures of compounds (1-3) without methoxy side groups have the same tendency (i.e., p-PBP (1) > o-PBP (3) > m-PBP (2)).
For H-bonded donors C10ONA (10), C10OBA (11), and C10 -COOTHA (12), their phase transition temperatures (i.e., Tmand Tcr) are in the following order: C10COOTHA (12) < C10OBA (11) < C10ONA (10) (shown in Table 2). In general, the phase transition temperatures will increase while the central cores are more linear and rigid. As for the phase transition temperatures of donor monomers p-MC10BA (13) > m-MC10BA (15) and donor polymers p-PC10BA (14) > m-PC10BA (16), it can be explained by a more linear H-bonded dimeric architecture in para-acid structure and therefore results in higher transition temperatures. In addition, comparing donor monomers (p-MC10 -BA (13) and m-MC10BA (15)) with donor polymers (p-PC10 -BA (14) and m-PC10BA (16)), polymers have larger molecular weights to produce higher transition temperatures of Tm and Tcr.
By evaporation of the mixture of acceptor/donor (molar ratio ) 1:2) in the THF solution, fully H-bonded complexes are formed. Since bis(pyridyl) acceptor emitters (1-9) are bifunc-tional acceptors, double molar values of H-bonded donors are needed to complex with central bifunctional acceptors to generate fully H-bonded trimers. The evidence of different Table 1. Thermal Properties of Bis(pyridyl) Acceptor Emitters (1-9)
aThe enthalpy (J/g) is shown in parentheses.bThe phase transition temperatures were obtained from POM.cCrystallization was not observed during the cooling process.
Table 2. Thermal Properties of Proton Donors (10-16)
aThe enthalpy (J/g) is shown in parentheses.bThe phase transition temperatures were obtained from POM.
Table 3. Thermal Properties of H-Bonded Complexes Containing Donors (10-12)
aThe enthalpy (J/g) is shown in parentheses.bThe phase transition temperatures were obtained from POM.
Table 4. Thermal Properties of H-Bonded Complexes Containing Donor Polymers (14 and 16)
extents of H-bonding in H-bonded trimers consisting of acid donors and isomeric pyridyl acceptors (with various positions of N-heterocyclic atoms), i.e., p-PBP-OMe (4), m-PBP-OMe (5), and o-PBP-OMe (6), can be confirmed by IR spectra.50 As shown in Figure 1, due to the H-bonding, the O-H bands at 2672 and 2556 cm-lin pure donor acid p-MC10BA (13) are shifted to lower energy wavenumber around 2664-2514 cm-l and 1920 cm-lin H-bonded complex p-PBP-OMe (4)/p-MC10 -BA (13) with para-N-heterocycles and around 2698-2442 cm-l and 1920 cm-lin H-bonded complex o-PBP-OMe (6)/p-MC10 -BA (13) with o-N-heterocycles, which shows that the degree of H-bonding between acids and o-N-heterocyclic emitter o-PBP-OMe (6) is similar to that between acids and p-N-heterocyclic emitter p-PBP-OMe (4). However, the O-H bands at 2672 and 2558 cm-l in H-bonded complex m-PBP-OMe (5)/p-MC10BA (13) with m-N-heterocycles are almost the same as the pure donor acid p-MC10BA (13). Therefore, the degree of H-bonding between acids and m-N-heterocyclic emitter m-PBP-OMe (5) is much less than the other isomeric emitters and may remain partially as non-H-bonded moieties in the mixture.
Because of H-bonds, the physical properties of the bispyridyl emitters can be modified by donor monomers and polymers in the H-bonded complexes, including the formation of LC phases. Some of the thermal properties of H-bonded trimers are shown in Table 3. The nematic phase is introduced in all H-bonded trimers, except p-PBP-OC8(7)/C10COOTHA (12) due to the bending tail of thiophene structure in C10COOTHA (12) and the shortest core of p-PBP-OC8(7). In general, the bending structure of thiophene donor, i.e., C10COOTHA (12), induces the lowest isotropization temperature in analogous H-bonded trimers, and the most rigid structure of naphthalene donor, i.e., C10ONA (10), causes the highest isotropization temperatures in H-bonded trimer counterparts. Moreover, the longer conju-gated cores induce the broader nematic phase in analogous H-bonded trimers. Once the proton donor polymers p-PC10BA (14) and m-PC10BA (16) are used, the bis(pyridyl) acceptor emitters will act as H-bonded cross-linkers. Some of the thermal properties of fully H-bonded polymer networks (acceptor:donor ) 1:2 by mole) containing H-bonded cross-linking emitters p-PBBBP-OC8(8) and p-PBBBP-OC12(9) are shown in Table 4. Only H-bonded complexes containing donor polymers are reported, because donor monomers of H-bonded complexes seem easier to react in the H-bonded complexes than pure donor
monomers during the heating process. The simplified schematic drawing of idealized H-bonded complexes formed among bis-(pyridyl) acceptor emitters and donor polymers are revealed in Figure 2, where the layered smectic structures are favored to form in the H-bonded polymeric networks. Table 4 reveals that lower melting temperatures are observed in analogous H-bonded polymeric networks bearing longer side chain of acceptor emitter p-PBBBP-OC12(9). However, regardless of the lower isotro-pization temperature in m-PC10BA (16), the H-bonded polymeric networks containing donor polymer m-PC10BA (16) possess a little higher isotropization temperatures. This might be possibly due to the stronger acidity of m-PC10BA (16) compared with that of p-PC10BA (14),51and thus the stronger H-bonded effect is induced in the H-bonded polymeric networks containing donor polymer m-PC10BA (16). Above all, mesomorphism is intro-duced in most H-bonded series (shown in Tables 3 and 4) by the complexation of nonmesogenic acceptor emitters with H-bonded donors.
Optical Properties. While materials possess mesomorphism, the polarized emission is anticipated for using the LC phase to be aligned in a rubbing cell. The PL emission has the highest intensity (PL|) when the polarizer is parallel to the rubbing
direction of the aligned cell, and has the lowest intensity (PL⊥) perpendicular to the rubbing direction. The polarization ratio is defined by (P|/P⊥), where P| and P⊥ are the maximum PL emission intensities as the polarizer is parallel and perpendicular to the rubbing direction, respectively. The maximum polarization ratios of H-bonded complexes (acceptor:donor ) 1:2 by mole) containing bis(pyridyl) acceptor emitters p-PBBBP-OC8(8) and p-PBBBP-OC12 (9) are shown in Table 5. The p-PBBBP-OC12(9)/C10ONA (10) complex shows the highest polarization ratio (P|/P⊥ ) 9.5) among all of these H-bonded complexes.
The polarized PL spectra (PL|and PL⊥) of H-bonded complex p-PBBBP-OC12(9)/C10ONA (10) in the LC phase (the nematic phase at 198°C) are also shown in Figure 3. In general, the polarization values of polymeric H-bonded complexes are lower
Figure 1. FTIR spectra of donor acid p-MC10BA (13) and its H-bonded
trimers containing isomeric pyridyl acceptors, i.e., p-PBP-OMe (4),
m-PBP-OMe (5), and o-PBP-OMe (6).
Figure 2. Simplified schematic drawing of idealized H-bonded polymer
networks formed among bis(pyridyl) acceptor emitters and donor polymers.
Table 5. Maximum Polarization Ratio of H-Bonded Complexes
H-bonded complexes (molar ratio ) 1:2)
maximum polarization ratio p-PBBBP-OC8(8)/C10ONA (10) 6.8 p-PBBBP-OC12(9)/C10ONA (10) 9.5 p-PBBBP-OC8(8)/C10OBA (11) 4.7 p-PBBBP-OC12(9)/C10OBA (11) 4.1 p-PBBBP-OC8(8)/C10COOTHA (12) 1.8 p-PBBBP-OC12(9)/C10COOTHA (12) 2.2 p-PBBBP-OC8(8)/p-MC10BA (13) 3.2 p-PBBBP-OC8(8)/p-PC10BA (14) 2.1 p-PBBBP-OC8(8)/m-MC10BA (15) 2.1 p-PBBBP-OC8(8)/m-PC10BA (16) 1.7 p-PBBBP-OC12(9)/p-MC10BA (13) 3.7 p-PBBBP-OC12(9)/p-PC10BA (14) 1.6 p-PBBBP-OC12(9)/m-MC10BA (15) 2.1 p-PBBBP-OC12(9)/m-PC10BA (16) 1.6
than those of monomeric H-bonded complexes, because polymer chains are more difficultly aligned in the rubbing direction, which may be due to higher viscosity and entanglement of polymer chains in the H-bonded polymer networks.
The UV-vis absorption and PL spectra of bis(pyridyl) emitters in solutions (THF as solvent) are shown in Figure 4 andλmaxvalues of UV-vis, PL, and EL spectra are listed in Table 6. The peaks of UV-vis spectra are about 357-359 nm for PBP derivatives, 397-403 nm for PBP-OMe derivatives,
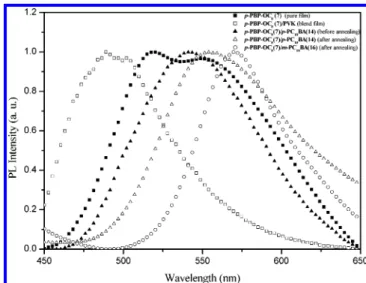
408 nm for p-PBP-OC8(7), and 434-435 nm for p-PBBBP derivatives, respectively. Compared with p-PBP-OC8 (7), p-PBBBP-OR (-OR ) -OC8and -OC12) derivatives have largerλmaxvalues of UV-vis and PL spectra in solutions, i.e., red-shifted wavelengths, due to the smaller energy band gaps of longer conjugations in five-conjugated rings of p-PBBBP-OR derivatives. Owing to the aggregation in solid states, the λmaxvalues of PL in pure films are red-shifted to different extent in contrast to those of solution states. Theπ-π stacking effect appears in pure films to form excimers, so the red-shifted phenomena were observed. However, compared with pure films, the blend films (dopant emitter:PVK ) 5:100 wt %) have less red-shifted λmax values of PL, where the polymer matrixes (PVK) act as solvents in the blend films. Similar results in PL spectra of bis(pyridyl) acceptor emitter p-PBP-OC8(7) in pure film and blend film are observed in Figure 5. This indicates dopant emitters in pure films have stronger aggregation them-selves than in PVK matrix. The blend films are similar to solid solutions which show concentration effects on the aggregation of dopant emitters as well as theλmaxvalues of PL emissions. Hence, compared with pure films, the dilution effect of the solid solvent (PVK) induce blue-shift of PL in the blend films.
In H-bonded complexes, the acids play both roles as solid solvents and H-bonded donors at the same time. Thus, the dilution effect of the acids (as solid solvents) to induce blue-shift of PL and the H-bonded effect of the acid donors to cause red-shift of PL are competitive. Table 7 shows both of the dilution effect and the H-bonded effect onλmax(PL) values of fully H-bonded polymer networks (acceptor:donor ) 1:2 by mole) containing various bis(pyridyl) acceptor emitters (1-9) and polymer donors, i.e., p-PC10BA (14) and m-PC10BA (16). PL spectra of fully H-bonded complexes of bis(pyridyl) acceptor Table 6. UV-Vis, PL, and EL Spectra of Bis(pyridyl) Acceptor Emitters
λmax(nm)
compd UV-vis (abs)a PL (solution)a PL (pure film) PL (blend film)b EL (blend film)b
p-PBP (1) 357 414 (392)c 444 (463) 458 454 m-PBP (2) 357 412 (392) 474 (456) 441 450 o-PBP (3) 359 415 462 428 428 p-PBP-OMe (4) 403 463 558 464 468 m-PBP-OMe (5) 397 450 531 462 464 o-PBP-OMe (6) 403 452 548 456 456 p-PBP-OC8(7) 408 464 519 (555) 490 494 p-PBBBP-OC8(8) 434 494 549 502 502 p-PBBBP-OC12(9) 435 494 553 503 504
aThe solvent is THF.bThe matrix is PVK (dopant emitter: PVK ) 5:100 wt %).cSecond peaks are shown in the parentheses.
Figure 3. PL spectra (PL|and PL⊥) of H-bonded complex p-PBBBP-OC12(9)/C10ONA (10) measured at 198°C (in the nematic phase),
where PL|and PL⊥are PL spectra measured as the polarizer parallel and perpendicular to the rubbing direction of LC cells, respectively.
Figure 4. UV-vis spectra of bis(pyridyl) acceptor emitters in solutions
(THF as solvents).
Figure 5. PL spectra of bis(pyridyl) acceptor emitter p-PBP-OC8(7)
emitters (1-9) with m-PC10BA (16) (molar ratio ) 1:2) are even widely distributed from blue to red colors (from 390 to 642 nm). Compared with pure bis(pyridyl) acceptor emitters, red-shifts of PL emission wavelengths have occurred, i.e., the H-bonded effect is dominant, in most of the H-bonded plexes. Some exceptions occur in polymeric H-bonded com-plexes containing bis(pyridyl) acceptor emitters (1-3), where bis(pyridyl) acceptor emitters (1-3) are highly aggregated in pure films owing to the lack of side chains (-OR ) -OMe, -OC8, and -OC12) in PBP derivatives (1-3). In the meanwhile, the lack of side groups (-OR) in PBP derivatives (1-3) are also short of electron donor groups and thus to reduce the possible H-bonded effect. Thus, the dilution effect of the acid donors to induce blue-shift of PL is more dominant than the H-bonded effect (red-shift of PL) in polymeric H-bonded complexes containing bis(pyridyl) acceptor emitters (1-3).
Among emitters 1-3 and 4-6 (see Table 6),λmax(PL) values in solution do not change substantially with regard to their heteroatom position of nitrogen, i.e., para-, meta-, and ortho-positions. Besides, the trend of heteroatom position effect is not consistent in pure and blend films. However, comparing analogous emitters PBP-OMe (4-6) with side alkoxy groups, p-heteroatom emitter, i.e., p-PBP-OMe (4), has the largestλmax (PL) values in various states of Table 6. As for H-bonded polymer networks (in Table 7), the heteroatom position effect is more obvious in H-bonded polymer networks containing emitters PBP-OMe (4-6) with side alkoxy groups, and polymer donors have induced the largest λmax(PL) values in p-heteroatom emitter, i.e., p-PBP-OMe (4). Whereas H-bonded polymer networks contain emitters PBP (1-3) without side chains, λmax (PL) values of H-bonded complexes are almost equivalent and regardless of p-, m-, and o-heteroatoms in emitters PBP (1-3).
Comparing H-bonded complexes of various bis(pyridyl) acceptor emitters (1-9) H-bonded with p-PC10BA (14) and m-PC10BA (16), respectively, due to the higher acidity value of m-PC10BA (16), stronger H-bonded effect occurs in H-bonded complexes containing m-PC10BA (16), so largerλmaxvalues of PL are observed in H-bonded complexes than those containing p-PC10BA (14) (shown in Table 7). This is possibly because of the dissimilar acidity values of donor polymers p-PC10BA (14) and m-PC10BA (16), i.e., pKa) 4.36 for p-PC10BA (14) and pKa ) 4.19 for m-PC10BA (16),51which may affect the red-shift ofλmaxvalue in PL as described in our previous work.14 For instance, the pKa values of C10ONA (10), C10OBA (11), and C10COOTHA (12) are 4.17, 4.21, and 3.49, respectively, so H-bonded complexes of p-PBP-OC8(7) with C10ONA (10), C10OBA (11), and C10COOTHA (12) have different red-shifts in λmax values of PL, i.e., 4, 16, and 94 nm, respectively, compared with bis(pyridyl) acceptor emitter p-PBP-OC8 (7) in pure films withλmax(PL) ) 516 nm. In addition, H-bonded
complexes of p-PBP (1) with C10ONA (10), C10OBA (11), and C10COOTHA (12) have 14, -5, and 24 nm of red-shifts inλmax values of PL, respectively, compared with bis(pyridyl) acceptor emitter p-PBP (1) in pure films withλmax(PL) ) 443 nm.14 Similar results in PL spectra of p-PBP-OC8(7)/p-PC10BA (14) and p-PBP-OC8(7)/m-PC10BA (16) after annealing, i.e.,λmax (PL) ) 554 and 570 nm, correspondingly, are observed in Figure 5. Besides, a more complete H-bonded polymer network p-PBP-OC8(7)/p-PC10BA (14) can be further established after annealing at 150°C (in the LC phase) compared with its pristine film by spin-coating (before annealing), so an additional red-shift of 15 nm (from 539 to 554 nm) in PL has been detected by annealing (see Figure 5).
On the other hand, the varition ofλmaxvalues (PL) fluctuates differently in each pair of H-bonded polymer networks as acceptor emitters (1-9) are H-bonded to donor polymers p-PC10 -BA (14) and m-PC10BA (16), respectively. For instance, The largest difference of λmax values (PL), i.e., 63 nm, happens between H-bonded complexes p-PBBBP-OC8 (8)/p-PC10BA (14) and p-PBBBP-OC8 (8)/m-PC10BA (16) in Table 7. Moreover, the largest red-shift of 93 nm inλmaxvalue of PL can be obtained from p-PBBBP-OC8 (8)/m-PC10BA (16) in contrast to bis(pyridyl) acceptor emitter p-PBBBP-OC8(8) in pure films. Interestingly, very small variations ofλmaxvalues (PL), i.e., 1-3 nm, occur in all H-bonded polymer networks contain emitters PBP (1-3) without side chains, e.g. the smallest variation of 1 nm take places between H-bonded complexes p-PBB (1)/p-PC10BA (14) and p-PBB (1)/m-PC10BA (16). Thus, it depends on the types of acceptor emitters, i.e., PBP derivatives (1-3) without side chains or the other derivatives (4-9) with Table 7. PL Spectra of Bis(pyridyl) Acceptor Emitters and Their H-Bonded Polymer Networks in Solid Films
λmax(nm) compd pure emitter (without acid) H-bonded with p-PC10BA (14)a H-bonded with m-PC10BA (16)a p-PBP (1) 444 (463)b 394 396 m-PBP (2) 474 (456)b 392 395 o-PBP (3) 462 389 390 p-PBP-OMe (4) 558 586 597 m-PBP-OMe (5) 531 539 551 o-PBP-OMe (6) 548 570 582 p-PBP-OC8(7) 519 (555)b 542 577 p-PBBBP-OC8(8) 549 579 642 p-PBBBP-OC12(9) 573 589 612
aAcceptor:donor ) 1:2 by mole to form fully H-bonded polymer networks.bSecond peaks are shown in parentheses.
Figure 6. EL spectra of devices PVK:dopant(100:5 by
weight)/TPBI-(40 nm)/Mg:Ag containing bis(pyridyl) emitters, i.e., p-PBP (1), m-PBP (2), o-PBP (3), p-PBP-OMe (4), p-PBP-OC8(7), and
side chains, to distinguish the donor polymers p-PC10BA (14) and m-PC10BA (16). Therefore, the acidity of p-PC10BA (14) and m-PC10BA (16) may not be the only controlling factor in the red-shift ofλmaxvalues (PL) for H-bonded polymer networks. Furthermore, the small difference of acidity between p- and m-alkoxybenzoic acid may not be the only feature to introduce such a large red-shift of 93 nm inλmaxvalue of PL in the fully H-bonded polymer network p-PBBBP-OC8(8)/m-PC10BA (16) in contrast to pure emitter p-PBBBP-OC8(8), so some other uncertain reason, such as various H-bonded conformation or conjugation depending on the bis(pyridyl) acceptor emitters, may influence the red-shift of PL in H-bonded complexes by different ways.
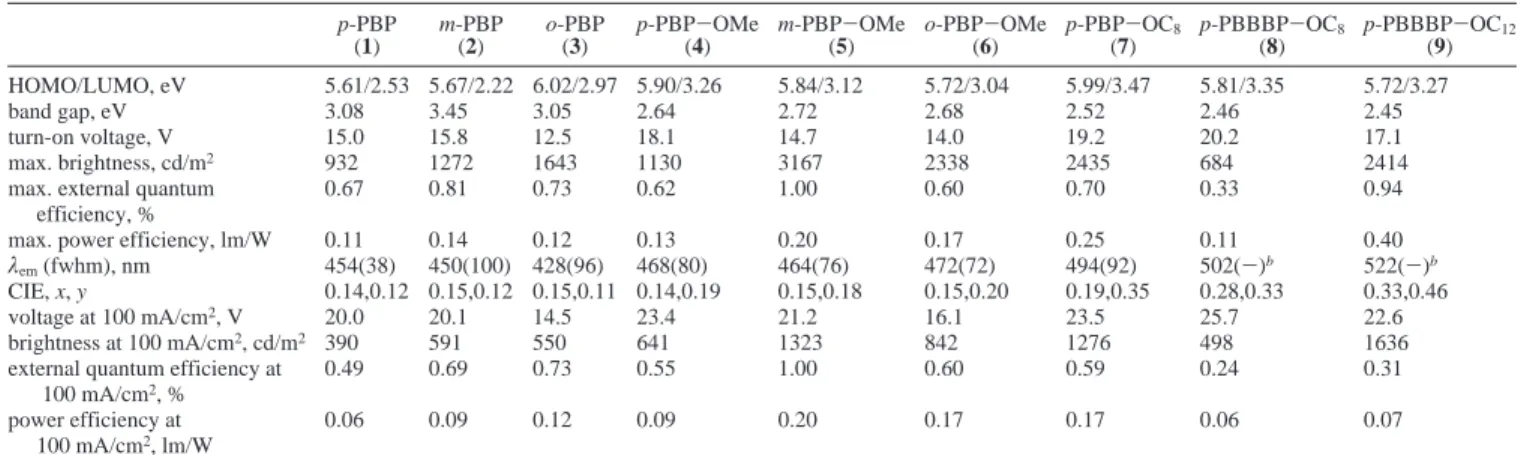
Electrooptical Properties. The λmaxvalues of PL and EL luminescence in pure PVK are usually found around 400 nm and sometimes this emission wavelength is not detectable in the PL and EL spectra of PVK doped systems due to the energy transfer from PVK to dopants. Some of the energy-transfer effects of PVK in EL were found in Figure 6, such as p-PBP (1), p-PBP-OMe (4), and p-PBP-OC8(7) in devices of PVK: dopant(100:5)/TPBI(40 nm)/Mg:Ag, and EL spectra of analo-gous derivatives are compared. Therefore, the PVK matrix acts as an energy-transfer component as well as hole transporting component in some of the blend film systems. However, a strong emission of PVK or TPBI around 425 nm was observed in of PVK doped devices containing emitting dopants m-PBP (2), o-PBP (3), and p-PBBBP-OC8 (8). The lowest occupied molecular orbital (LUMO) values are calculated by the highest occupied molecular orbital (HOMO) values (from AC-2) minus the optical band gap values (from UV-vis), which are also listed in Table 8. The band gaps of PBP and PBP-OMe derivatives are between 3.05 and 3.45 eV and between 2.64 to 2.72 eV, respectively. Compared with PBP and PBP-OMe derivatives, p-PBBBP-OR derivatives possess smaller band gaps between 2.45 and 2.46 eV. The smaller band gaps are by reason for their
longer conjugated lengths and their PL and EL spectra show greenish-blue light emissions.
Because of the poor film quality of H-bonded complexes and polymers, including polymer donors p-PC10BA (14) and m-PC10 -BA (16), PVK is utilized as the polymer matrix in further study. To analyze the H-bonding effect on the LED device performance of H-bonded complexes, acceptor emitter p-PBP-OC8 (7), which possesses the best solubility among acceptor emitters, is doped into PVK either with or without proton donor C10OBA (11). Hence, devices 1-3 containing PVK:p-PBP-OC8 (7) (100:5 by weight, i.e., without H-bonding) and PVK:p-PBP-OC8(7):C10OBA (11) (100:5:50 by weight, i.e., with H-bonding) Table 8. Electroluminescence Characteristics of PLED Devices Containing Various Bis(pyridyl) Acceptor Emitters
p-PBP (1) m-PBP (2) o-PBP (3) p-PBP-OMe (4) m-PBP-OMe (5) o-PBP-OMe (6) p-PBP-OC8 (7) p-PBBBP-OC8 (8) p-PBBBP-OC12 (9) HOMO/LUMO, eV 5.61/2.53 5.67/2.22 6.02/2.97 5.90/3.26 5.84/3.12 5.72/3.04 5.99/3.47 5.81/3.35 5.72/3.27 band gap, eV 3.08 3.45 3.05 2.64 2.72 2.68 2.52 2.46 2.45 turn-on voltage, V 15.0 15.8 12.5 18.1 14.7 14.0 19.2 20.2 17.1 max. brightness, cd/m2 932 1272 1643 1130 3167 2338 2435 684 2414
max. external quantum 0.67 0.81 0.73 0.62 1.00 0.60 0.70 0.33 0.94
efficiency, %
max. power efficiency, lm/W 0.11 0.14 0.12 0.13 0.20 0.17 0.25 0.11 0.40
λem(fwhm), nm 454(38) 450(100) 428(96) 468(80) 464(76) 472(72) 494(92) 502(-)b 522(-)b
CIE, x, y 0.14,0.12 0.15,0.12 0.15,0.11 0.14,0.19 0.15,0.18 0.15,0.20 0.19,0.35 0.28,0.33 0.33,0.46
voltage at 100 mA/cm2, V 20.0 20.1 14.5 23.4 21.2 16.1 23.5 25.7 22.6
brightness at 100 mA/cm2, cd/m2 390 591 550 641 1323 842 1276 498 1636
external quantum efficiency at 0.49 0.69 0.73 0.55 1.00 0.60 0.59 0.24 0.31
100 mA/cm2, %
power efficiency at 0.06 0.09 0.12 0.09 0.20 0.17 0.17 0.06 0.07
100 mA/cm2, lm/W
aThe thickness of TPBI is 40 nm and PVK: chromophore ) 100:5.bThe fwhm cannot be measured due to overlapping peaks.
Table 9. EL Characteristics of Devices 1-3 Containing Bis(pyridyl) Acceptor Emitter p-PBP-OC8(7), i.e., Device 1 of PVK:p-PBP-OC8(7)
(100:5 by Weight)/TPBI(40 nm)/Mg:Ag, Device 2 (with annealing at 170°C for 2 min), and Device 3 (with Annealing at 230°C for 2 min) of PVK:p-PBP-OC8(7):C10OBA(11) (100:5:50 by Weight)/TPBI(40 nm)/Mg:Ag
device 1 device 2 device 3
turn-on voltage, V 19.2 26.5 22.3
max. brightness, cd/m2 2599 1803 5184
max. external quantum efficiency, % 0.76 0.71 0.79
max. power efficiency, lm/W 0.27 0.23 0.31
λem(fwhm), nm 494(92) 510(38) 508(42)
CIE, x, y 0.19, 0.35 0.08, 0.65 0.09, 0.64
voltage at 100 mA/cm2, V 23.5 34.4 26.2
brightness at 100 mA/cm2, cd/m2 1362 1685 1777
external quantum efficiency at 100 mA/cm2, % 0.64 0.61 0.64
power efficiency at 100 mA/cm2, lm/W 0.18 0.15 0.21
Figure 7. EL spectra of devices 1-3 containing p-PBP-OC8(7), i.e.,
device 1 of PVK:p-PBP-OC8(7) (100:5)/TPBI(40 nm)/Mg:Ag, device 2 (with annealing at 170°C for 2 min) and device 3 (with annealing at 230°C for 2 min) of PVK:p-PBP-OC8(7):C10OBA (2) (100:5:50)/
blend films are investigated (with and without annealing). However, devices (with 40 nm of TPBI) of PVK:p-PBP-OC8 (7) (100:5 by weight) annealed at 170 and 230°C (below and above Tgof PVK ∼200 °C, respectively) and PVK:p-PBP-OC8(7):C10OBA (11) (100:5:50 by weight) without annealing have worse optical properties, so above device data are not demonstrated. With regard to devices 1-3 (see Figure 7 and Table 9), EL spectra obtained from devices 2 and 3 of PVK: p-PBP-OC8(7):C10OBA (11) (100:5:50 by weight, i.e., with H-bonding)/TPBI(40 nm)/Mg:Ag, which were annealed at 170 and 230°C (above and below Tgof PVK), respectively, possess more narrow full width at half-maximum (fwhm) values (38 and 42 nm) of EL spectra than that from device 1 (92 nm) of PVK:p-PBP-OC8 (7) (100:5 by weight, i.e., without H-bonding)/TPBI(40 nm)/Mg:Ag. More narrow fwhm values of EL spectra in the H-bonded LED devices may be owing to the induction of fewer electron transition states in the supramo-lecular architecture. As shown in Table 9, device 3 annealed at 230°C possesses a better brightness of 5184 cd/m2than device 2 annealed at 170°C and also better than device 1 without donor C10OBA (11). Finally, Devices with H-bonding between ac-ceptor emitters and donors can not only induce more narrow fwhm values of EL spectra but also redder shift in EL spectra in this supramolecular architecture.
Conclusions
In conclusion, the mesomorphic and photophysical properties of the H-bonded polymer networks and trimers can be easily adjusted by tuning the H-bonded donors (donor polymers) and acceptor emitters in the H-bonded complexes. Unique meso-morphic properties may be introduced into these supramolecular structures containing nonmesogenic acceptor emitters. In addi-tion, the emission properties of bis(pyridyl) acceptor emitters can be manipulated by their surrounding nonphotoluminescent proton donors. Compared with pure bis(pyridyl) acceptor emitters, red-shifts of PL emission wavelengths have occurred, i.e., the H-bonded effect is dominant, in most of the H-bonded complexes. Because of the higher acidity value of m-PC10BA (16) than that of p-PC10BA (14), stronger H-bonded effect occurs in H-bonded complexes containing m-PC10BA (16), so largerλmaxvalues of PL are observed in H-bonded complexes. The largest red-shift of 93 nm inλmaxvalue of PL has occurred in the fully H-bonded polymer network p-PBBBP-OC8 (8)/m-PC10BA (16) in contrast to pure emitter p-PBBBP-OC8(8). However, the small difference of acidity between p- and m-alkoxybenzoic acid, i.e., p-PC10BA (14) and m-PC10BA (16), may not be the only feature to introduce such large red-shifts inλmaxvalues of PL in the fully H-bonded polymer networks. PL and EL spectra of all blend films in PVK (dopant emitter: PVK ) 5:100 wt %) show blue to green light emissions. PL spectra of fully H-bonded complexes of bis(pyridyl) acceptor emitters (1-9) with m-PC10BA (16) (molar ratio ) 1:2) are even widely distributed from blue to red colors (from 396 to 642 nm). A very narrow fwhm value (38 nm) of EL spectra can be obtained in the H-bonded complexes blended with PVK, and higher brightness of EL is produced at an appropriate annealing temperature.
Acknowledgment. We are grateful for the financial support provided by the National Science Council of Taiwan (ROC) through Grant NSC 92-2113-M-009-016.
References and Notes
(1) Ikkala, O.; Brinke, G. Science 2002, 295, 2407.
(2) Brunsveld, L.; Folmer, B. J. B.; Meijer, E. W.; Sijbesma, R. P. Chem. ReV. 2001, 101, 4071.
(3) Paleos, C. M.; Tsiourvas, D. Liq. Cryst. 2001, 28, 1127.
(4) Kreuzer, M.; Benkler, E.; Paparo, D.; Casillo, G.; Marrucci, L. Phys. ReV. E 2003, 68, 011701.
(5) Huyskens, P. L.; Luck, W. A. P. Intermolecular Forces, 3rd ed.; Springer-Verlag: Berlin, 1991.
(6) Kato, T.; Kihara, H.; Ujiie, S.; Uryu, T.; Fre´chet, J. M. J. Macromol-ecules 1996, 29, 8734.
(7) Shandryuk, G. A.; Kuptsov, S. A.; Shatalova, A. M.; Plate’, N. A.; Talroze, R. V. Macromolecules 2003, 36, 3417.
(8) Cheuk, K. K. L.; Lam, J. W. Y.; Lai, L. M.; Dong, Y.; Tang, B. Z. Macromolecules 2003, 36, 9752.
(9) Lu, L.; Jenekhe, S. A. Macromolecules 2001, 34, 6249.
(10) Osuji, C.; Chao, C. Y.; Bita, I.; Ober, C. K.; Thomas, E. L. AdV. Funct. Mater. 2002, 12, 753.
(11) Barmatov, E.; Filippov, A.; Andreeva, L.; Barmatova, M.; Kremer, F.; Shibaev, V. Macromol. Rapid Commun. 1999, 20, 521. (12) Kihara, H.; Kato, T.; Uryu, T.; Fre´chet, J. M. J. Chem. Mater. 1996,
8, 961.
(13) Thote, A. J.; Gupta, R. B. Ind. Eng. Chem. Res. 2003, 42, 1129. (14) Lin, H. C.; Sheu, H. Y.; Chang, C. L.; Tsai, C. J. Mater. Chem. 2001,
11, 2958.
(15) Lin, H. C.; Ko, C. W.; Guo, K.; Cheng, T. W. Liq. Cryst. 1999, 26, 613.
(16) Lin, H. C.; Shiaw, J. M.; Wu, C. Y.; Tsai, C. Liq. Cryst. 2000, 27, 1103.
(17) Burroughes, J. H.; Bradley, D. D. C.; Brown, A. R.; Marks, R. N.; MacKay, K.; Friend, R. H.; Burn, P. L.; Holmes, A. B. Nature (London) 1990, 347, 539.
(18) Godbert, N.; Burna, P. L.; Jonathan, S. G.; Markham, P. J.; Samuel, I. D. W. Appl. Phys. Lett. 2003, 83, 5347.
(19) West, J. L.; Magyar, G. R.; Kelly, J. R. Appl. Phys. Lett. 1995, 67, 155.
(20) Lam, J. W. Y.; Kong, X.; Dong, Y.; Cheuk, K. K. L.; Xu, K.; Tang, B. Z. Macromolecules 2000, 33, 5027.
(21) Chen, J.; Xie, Z.; Lam, J. W. Y.; Law, C. C. W.; Tang, B. Z. Macromolecules 2003, 36, 1108.
(22) Neher, D. Macromol. Rapid Commun. 2001, 22, 1365.
(23) Banach, M. J.; Friend, R. H.; Sirringhaus, H. Macromolecules 2003, 36, 2838.
(24) (a) Zen, A.; Neher, D.; Bauer, C.; Asawapirom, U.; Scherf, U.; Hagen, R.; Kosttomine, S.; Mahrt, R. F. Appl. Phys. Lett. 2002, 80, 4699. (b) Sainova, D.; Zen, A.; Nothofer, H. G.; Asawapirom, U.; Scherf, U.; Hagen, R.; Bieringer, T.; Kostromine, S.; Neher, D. AdV. Funct. Mater.
2002, 12, 49.
(25) Whitehead, K. S.; Grell, M.; Bradley, D. D. C.; Jandke, M.; Strohriegi, P. Appl. Phys. Lett. 2000, 76, 2946.
(26) Miteva, T.; Meisel, A.; Knoll, W.; Nothofer, H. G.; Scherf, U.; Muller, D. C.; Meerholz, K.; Yasuda, A.; Neher, D. AdV. Mater. 2001, 13, 565.
(27) Grozema, F. C.; Savenije, T. J.; Vermeulen, M. J. W.; Siebbeles, L. D. A.; Warman, J. M.; Meisei, A.; Neher, D.; Nothofer, H. G.; Scherf, U. AdV. Mater. 2001, 13, 1627.
(28) (a) Sung, H. H.; Lin, H. C. Macromolecules 2004, 37, 7945. (b) Sung, H. H.; Lin, H. C. J. Polym. Sci. Part A: Polym. Chem. 2005, 43, 2700.
(29) Geng, Y.; Chen, A. C. A.; Ou, J. J.; Chen, S. H.; Klubek, K.; Vaeth, K. M.; Tang, C. W. Chem. Mater. 2003, 15, 4352.
(30) Hulvat, J. F.; Stupp, S. I. AdV. Mater. 2004, 16, 589.
(31) Chen, J.; Peng, H.; Law, C. C. W.; Dong, Y.; Lam, J. W. Y.; Williams, I. D.; Tang, B. Z. Macromolecules 2003, 36, 4319.
(32) Mochizuki, H.; Hasui, T.; Kawamoto, M.; Ikeda, T.; Adachi, C.; Tanighchi, Y.; Shirota, Y. Macromolecules 2003, 36, 3457. (33) Kawamoto, M.; Mochizuki, H.; Shishido, A.; Tsutsumi, O.; Ikeda,
T.; Lee, B.; Shirota, Y. Macromolecules 2003, 107, 4887. (34) Grahchev, I.; Moneva, I.; Bojinov, V.; Guittonneau, S. J. Mater. Chem.
2000, 10, 1291.
(35) Chen, A. C. A.; Culligan, S. W.; Geng, Y.; Chen, S. H.; Klubek, K. P.; Vaeth, K. M.; Tang, C. W. AdV. Mater. 2004, 16, 783. (36) Culligan, S. W.; Geng, Y.; Chen, S. H.; Klubek, K.; Vaeth, K. M.;
Tang, C. W. AdV. Mater. 2003, 15, 1176.
(37) Wang, C.; Kilitziraki, M.; Hugh MacBride, J. A.; Bryce, M. R.; Horsburgh, L. E.; Sheridan, A. K.; Monkman, A. P.; Samuel, D. W. AdV. Mater. 2000, 12, 217.
(38) Monkman, A. P.; Pa˚lsson, L. O. Higgins, R. W. T.; Wang, C.; Bryce, M. R.; Batsanov, A. S.; Howard, J. A. K. J. Am. Chem. Soc. 2002, 124, 6049.
(39) Porzio, W.; Destri, S.; Giovanella, U.; Meille, S. V.; Raos, G.; Consonni, R.; Zotti, G. Chem. Mater. 2005, 17, 242.
(40) Pei, J.; Liu, X. L.; Chen, Z. K.; Zhang, X. H.; Lai, Y. H.; Huang, H. Macromolecules 2003, 36, 323.
(41) Zhang, H.; Zhou, Z.; Liu, K.; Wang, R.; Yang, B. J. Mater. Chem.
2003, 13, 1356.
(42) Sierra, C. A.; Lahti, P. M. Chem. Mater. 2004, 16, 55.
(43) Knaapila, M.; Lkkala, O.; Torkkeli, M.; Jokela, K.; Serimaa, R.; Dolbnya, I. P.; Bras, W.; Brinke, G.; Horsburgh, L. E.; Pålsson, L. O.; Monkman, A. P. Appl. Phys. Lett. 2002, 81, 1489.
(44) Ajayaghosh, A.; George, S. J. J. Am. Chem. Soc. 2001, 123, 5148. (45) Yuan, C. H.; Hoshino, S.; Toyoda, S.; Suzuki, H.; Fujiki, M.;
Matsumoto, N. Appl. Phys. Lett. 1997, 71, 3326. (46) Liu, J.; Shi, Y.; Yang, Y. Appl. Phys. Lett. 2001, 79, 578. (47) Niu, Y. H.; Yang, W.; Cao, Y. Appl. Phys. Lett. 2002, 81, 2884.
(48) Zhang, Y.; Peng, J.; Mo, Y.; Cao, Y. Appl. Phys. Lett. 2004, 85, 5170. (49) Portugall, M.; Ringsdorf, H.; Zentel, R. Makromol. Chem. 1982, 183,
2311.
(50) Kato, T.; Fre´chet, J. M. J.; Wilson, P. G.; Saito, T.; Uryu, T.; Fujishima, A.; Jin, C.; Kaneuchi, F. Chem. Mater. 1993, 5, 1094.
(51) Tehan, B. G.; Lloyd, E. J.; Wong, M. G.; Pitt, W. R.; Montana, J. G.; Manallack, D. T.; Gancia, E. Quant. Struct.-Act. Relat. 2002, 21, 457.