偵測核糖核酸 H 型偽結之研究
42
0
0
全文
(2) 偵測核糖核酸 H 型偽結之研究 A Study of Detecting RNA H-type Pseudoknots. 研 究 生:黃群翔. Student:Chun-Hsiang Huang. 指導教授:盧錦隆 教授. Advisor:Prof. Chin Lung Lu. 國 立 交 通 大 學 生物資訊所 碩 士 論 文. A Thesis Submitted to Institute of Bioinformatics College of Biological Science and Technology National Chiao Tung University in partial Fulfillment of the Requirements for the Degree of Master in Biological Science and Technology. June 2005. Hsinchu, Taiwan, Republic of China. 中華民國九十四年六月.
(3) 偵測核糖核酸 H 型偽結之研究 學生: 黃群翔. 指導教授: 盧錦隆 教授 國立交通大學生物科技學系生物資訊所碩士班 要. 摘. 已知在所有種類的核糖核酸序列中幾乎皆可找到所謂的偽結, 在生物反應的過 程中, 這些偽結被認為扮演著重要功能性的角色。 除此之外, 目前大多數的核糖 核酸偽結皆屬於 H 型。 因此, 偵測這些 H 型的偽結將有助於我們瞭解核糖核酸 的結構及相關的功能。 然而, 現有的程式對於這種 H 型偽結的偵測仍然是相當 費時, 甚至有些時候其偵測出來的偽結是不正確的。 因此, 發展一個有效率且 準確度高的工具來偵測. H. 型的核糖核酸偽結是件值得研究的課題。 在本論文. 中, 我們提出了一個啟發式的策略, 並且根據這個策略發展出一個全新的工具 HPknotter 來正確而且有效率地偵測核糖核酸序列中的 H 型偽結。 除此之外, 我. 們也利用一些已知具有 H 型偽結的核糖核酸序列來測試並證明 HPknotter 的 適用性及準確度。. i.
(4) A Study of Detecting RNA H-type Pseudoknots. Student: Chun-Hsiang Huang. Advisor: Prof. Chin Lung Lu. Institute of Bioinformatics Department of Biological Science and Technology Nation Chiao Tung University. ABSTRACT. RNA H-type pseudoknots are ubiquitous pseudoknots that are known to be found in almost all classes of RNA sequences and thought to play a functionally important role in variety of biological processes. Hence, detecting these RNA H-type pseudoknots will enhance our understanding of RNA structures and their associated functions. However, the currently existing programs for detecting such RNA H-type pseudoknots are still time-consuming and sometimes even ineffective. Therefore, efficient and effective tools for detecting the RNA H-type pseudoknots are needed. In this thesis, we propose a heuristic approach to develop a novel tool, called HPknotter, for accurately and efficiently detecting RNA H-type pseudoknots in a given RNA sequence. In addition, we demonstrate the applicability and effectiveness of our HPknotter by testing it on several RNA sequences with known H-type pseudoknots.. ii.
(5) 誌. 謝. 首先要感謝我家人的栽培讓我得以無憂無慮地攻讀碩士學位。 其中, 感念我的爸 爸黃龍雄及我的媽媽陳麗云辛苦地撫養我長大。 雖然, 我的爸爸沒有機會看我完 成大學以及現在的碩士學業, 仍舊把這一份榮耀給他。 另外, 感謝我的三位姊姊黃 美慧、 黃美霆還有黃美華負擔家裡一些的經濟負擔, 並且在我外地唸書六年的時 間裡, 負起照顧爸媽的責任。 也感謝我的大哥黃韜維, 不時與我切磋我學業上的問 題, 祝福他順利完成博士學位。 謝謝靜宜大學的蔡英德教授在大學時代專題的指導, 算是我進入生物資訊這一 領域的啟蒙老師。 將更多的感謝給指導老師盧錦隆教授, 不時地在旁指導我, 時時 刻刻督促我努力地作研究, 謝謝他即使研究上遇到一些困難, 仍舊沒有放棄, 繼續 堅持下去, 提出許多相當好的建議, 我才得以有現在的研究成果, 千言萬語都道不 盡對老師的謝意。 此外, 感謝已畢業的實驗室學長黃彥斌在我碩一時對我的協助 及許許多多的幫忙, 也祝福學長在台積電工作順利。 感謝實驗室同學王璻菁、 陳 孟琪及學妹黃彥菱, 在研究上的討論還有畢業口試的幫忙。 也感謝所有生物資訊 所的師長們, 在課業上的指教以及生物資訊所其他的學長 、學弟、 學姐與學妹, 這 兩年來的協助, 還有每天晚上 WarCraft DOTA 跨實驗室的熱情激戰, 得以度過每 天乏味的研究生活。 此外將拿到碩士學位的喜悅分享給我的大學同學、 高中同學、 國中同學、 小學 的同學以及其他我沒有提到的好朋友們, 多謝你們替我加油。 最後感謝我女朋友 的溫柔與體貼, 將近一年的日子裡頭陪伴我作研究, 給我在精神上莫大的支持與 鼓勵。. iii.
(6) Contents Chinese Abstract. i. Abstract. ii. Acknowledgement. iii. List of Tables. v. List of Figures. vi. 1 Introduction. 1. 2 Preliminaries. 5. 2.1 RNA H-type Pseudoknots . . . . . . . . . . . . . . . . . . . . . . . . .. 5. 2.2 PseudoBase . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. 6. 2.3 RNAMotif . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. 8. 2.4 PKNOTS, NUPACK and pknotsRG . . . . . . . . . . . . . . . . . . . .. 11. 3 Materials and Method. 12. 3.1 Structural Motif Search by RNAMotif. . . . . . . . . . . . . . . . . . .. 12. 3.2 Hit Filter . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. 13. 3.3 Pseudoknot Prediction . . . . . . . . . . . . . . . . . . . . . . . . . . .. 15. 3.4 h-Pseudoknot Filter . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. 15. 3.5 Minimum Weight Independent Set . . . . . . . . . . . . . . . . . . . . .. 16. 4 Results and Discussion. 17. iv.
(7) 4.1 HPknotter Web Server . . . . . . . . . . . . . . . . . . . . . . . . . . .. 17. 4.2 Tested RNA Sequences . . . . . . . . . . . . . . . . . . . . . . . . . . .. 19. 4.3 Experimental Results and Discussions . . . . . . . . . . . . . . . . . . .. 20. 4.4 CPU Time Usage . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. 25. 5 Conclusion and Future Works. 27. References. 29. v.
(8) List of Tables 2.1 The conditions of four classes of h-pseudoknots. . . . . . . . . . . . . .. 6. 2.2 The default values of pseudoknot stem- and loop- sizes of four classes pre-defined h-pseudoknots descriptors. . . . . . . . . . . . . . . . . . .. 8. 4.1 The sequence and h-pseudoknot information of the tested sequences, where the accession number of HIV-1-RT is not available and TMV-30 down contains two h-pseudoknots with one in class 2 and the other in class 3. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. 20. 4.2 Summary of prediction results on several RNA sequences, where all experiments are run using the general class of the descriptor and the version of PKNOTS is 1.01. . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. 22. 4.3 Summary of prediction results on several RNA sequences, where experiments 1–4, 5–9 and 10–11 are run using the descriptors of classes 1, 2 and 3, respectively. Notice that TMV-30 -down contains two h-pseudoknots with one in class 2 (that was tested in experiment 9) and the other in class 3 (that was tested in experiment 10). . . . . . . . . . . . . . . . .. 24. 4.4 CPU usage time for PKNOTS, NUPACK, pknotsRG and HPknotter, where in our testing computer environment, PKNOTS and NUPACK cannot deal with the sequences of length greater than 220 bp and 180 bp, respectively, due to running out of the memory. . . . . . . . . . . .. vi. 26.
(9) List of Figures 1.1 An H-type pseudoknot located within the BCV 30 UTR. . . . . . . . .. 2. 2.1 Schematic representation of the H-type pseudoknot. . . . . . . . . . . .. 6. 2.2 Frequencies of stem- and loop-sizes of pseudoknots in PseudoBase. . . .. 7. 2.3 An RNAMotif descriptor used to describe the pseudoknotted structure of class 2 as depicted in Figure 2.1. . . . . . . . . . . . . . . . . . . . .. 9. 2.4 An extension of RNAMotif descriptor in Figure 2.3 by allowing an interior loop of size 2 or a bulge of size 1 to appear in stems 1 and 2. . . .. 10. 3.1 The flow diagram of HPknotter. . . . . . . . . . . . . . . . . . . . . . .. 13. 4.1 The interface of HPknotter. . . . . . . . . . . . . . . . . . . . . . . . .. 18. 4.2 An example of a detected h-pseudoknot by HPknotter. . . . . . . . . .. 19. vii.
(10) Chapter 1 Introduction RNA pseudoknots are found in almost all classes of naturally occurring RNA sequences and play very important roles in variety of biological processes, such as RNA replication, transcription and translation [34, 16]. According to the positions of pseudoknots, their associated functions are reflected because pseudoknots fold locally in RNAs [20]. For example, 50 -pseudoknots of mRNAs (message RNAs) are tend to be involved in translation control and 30 -pseudoknots of them control the signals for replication. In addition, RNA pseudoknots are well-known to be important roles for programmed −1 and +1 ribosomal frameshift signals in overlapping ORFs (open reading frames) [3, 14, 18]. Usually, functional RNA pseudoknots are also evolutionally conserved in rRNAs (ribosomal RNAs), the catalytic core of group I introns and RNase P RNAs by comparative analysis. The majority of pseudoknots that have been described to date are of the so-called H-type (or classical ) pseudoknot in which (as illustrated in Figure 1.1) nucleotides from a hairpin loop pair with a single-stranded region outside of the hairpin to form a helical stem that is adjacent or almost adjacent to the hairpin stem [23, 24, 34, 25]. For instance, there are 246 different RNA pseudoknots in PseudoBase [39, 38] with 224 of them being H-type. Therefore, the detection of H-type pseudoknots could improve our understanding of RNA structures and their associated functions. However, computational methods of predicating RNA H-type pseudoknots are still time-consuming and even ineffective. Hence, efficient and effective tools for detecting the RNA H-type 1.
(11) Figure 1.1: An H-type pseudoknot located within the BCV 30 UTR.. pseudoknots are needed. In the standard thermodynamic model, a pseudoknot-free RNA secondary structure of minimum free energy (MFE) can be computed using dynamic programming in O(n3 ) time [44, 43, 42, 12]. However, when (general) pseudoknots are allowed in the RNA secondary structure, the computation becomes intractable since it has been shown to be an NP-hard problem [17, 2]. Currently, several polynomial-time algorithms have been proposed to find an MFE secondary structure with a restricted class of pseudoknots [27, 2, 17, 9]. Rivas and Eddy [27] first proposed the dynamic programming algorithm that could handle a large class of special pseudoknotted structures. However, the major limitation of this algorithm is its high running time of O(n6 ) and space of O(n4 ), where n is the length of RNA sequence. With other more restricted classes of pseudoknots, Lyngsø and Pedersen [17] proposed an algorithm of O(n5 ) time and O(n3 ) space, Akutsu [2] designed an algorithm of O(n4 ) time and O(n3 ) space, Dirks and Peirce [9] described an algorithm of O(n5 ) time and O(n4 ) space, and Reeder and Giegerich [28] gave an algorithm of O(n4 ) time and O(n2 ) space. All these algorithms above are able to be used to predict an MFE secondary structure of an RNA sequence with h-pseudoknots [6]. However, they are not yet practical for large-scale sequences due to their high running time and/or space. In addition, our experimental results showed that these algorithms may not be effective to detect an h-pseudoknot that is actually present in the native structure of a long RNA sequence. On the other hand, our finding 2.
(12) showed that when they were applied to the sequence fragment exactly harboring the h-pseudoknot in a long RNA sequence, they gave a very high probability of successfully folding this fragment into the h-pseudoknot structure. Based on these above observations, in this thesis we propose a heuristic approach to design a novel tool, called HPknotter, by integrating the currently existing programs for efficiently and accurately detecting the RNA H-type pseudoknots. The key idea of our approach is as follows. For a given RNA sequence, RNAMotif is first used to search all the subsequences (called hits) that meet the criteria dictating the structural motifs. Second, a hit filter is designed to discard those sequences that are not possible to fold into a stable pseudoknotted structure. Third, PKNOTS/NUPACK/pknotsRG is used to determine if these hits indeed fold into a stable h-pseudoknot. Fourth, hpseudoknot filter is used to filter the hits that do not meet the criteria dictating the structural motifs. Fifth, based on the concept of maximum weight independent set, the mutually disjoint h-pseudoknots with minimum total free energy are computed. Finally, the remaining hits capable of folding into stable h-pseudoknots to serve as the final output of HPknotter. We will demonstrate the practicability and effectiveness of HPknotter by testing it on several RNA sequences, most of which have been proven to contain the H-type pseudoknotted structures in laboratory approaches. In addition to the above thermodynamic approaches, several other approaches for predicting RNA secondary structures with (H-type) pseudoknots have been proposed, such as maximum weighted matching [8, 32, 15], quasi-Monte Carlo searches [1, 10], genetic algorithms [37, 11, 31], stochastic context free grammar [4, 5], and others [15, 33, 29]. Particularly, Shapiro and Wu [31] developed a parallel genetic algorithm for detecting h-pseudoknots on a massively parallel supercomputer MasPar MP-2 with 16,384 processors. Recently, this parallel genetic algorithm has been adapted to MIMD parallel machines [30], such as SGI ORIGIN 2000 with 64 processors and CRAY T3E with 512 processors, which seem to be hardly accessible to the ordinary users. The rest of the thesis is organized as follows. In Chapter 2, we give an introduction to the RNA H-type pseudoknots as well as a database of collecting a lot of naturally occurring RNA pseudoknots. In Chapter 3, we describe our heuristic approach and our 3.
(13) implemented program, called HPknotter, for efficiently and effectively detecting RNA H-type pseudoknots. In Chapter 4, we demonstrate the applicability and effectiveness of our HPknotter by testing it on several RNA sequences with known H-type pseudoknots. Finally, we make some conclusion as well as a couple of future works in Chapter 5.. 4.
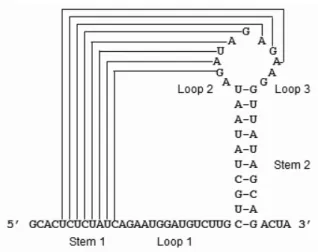
(14) Chapter 2 Preliminaries In this chapter, we first introduce the H-type pseudoknots (h-pseudoknots) and their classifications. Then we introduce PseudoBase [39, 38], a database of maintaining the naturally occurring RNA pseudoknots. Finally, we introduce the currently existing programs of predicting RNA pseudoknots, including RNAMotif [19], PKNOTS [27], NUPACK [9] and pknotsRG [28].. 2.1. RNA H-type Pseudoknots. In principle, an H-type pseudoknot (called h-pseudoknot) may contain two stems (regions A and C in Figure 2.1) and three loops (regions B, D and E in Figure 2.1), where such stems and loops are usually represented in the 50 → 30 direction as S1 (Stem 1), S2 (Stem 2) and L1 (Loop 1), L2 (Loop 2), L3 (Loop 3), respectively. However, L2 is absent in most of pseudoknots due to the coaxial stacking of stems. H-type pseudoknots have simple loops in which all nucleotides are unpaired and complicated loops that contain substructures, such as several stems with their own internal, hairpin and multi-branch loops. Both simple and complicated loops are referred to as pseudoknot loops. For simplicity, all the nucleotides in a pseudoknot loop are counted and the number of them equals to the size of this loop, whether they are unpaired or not. The pseudoknot stems adopted here are those that are “pseudoknotted” with other stems. They may be interrupted by some bulge loops (or interior loops). 5.
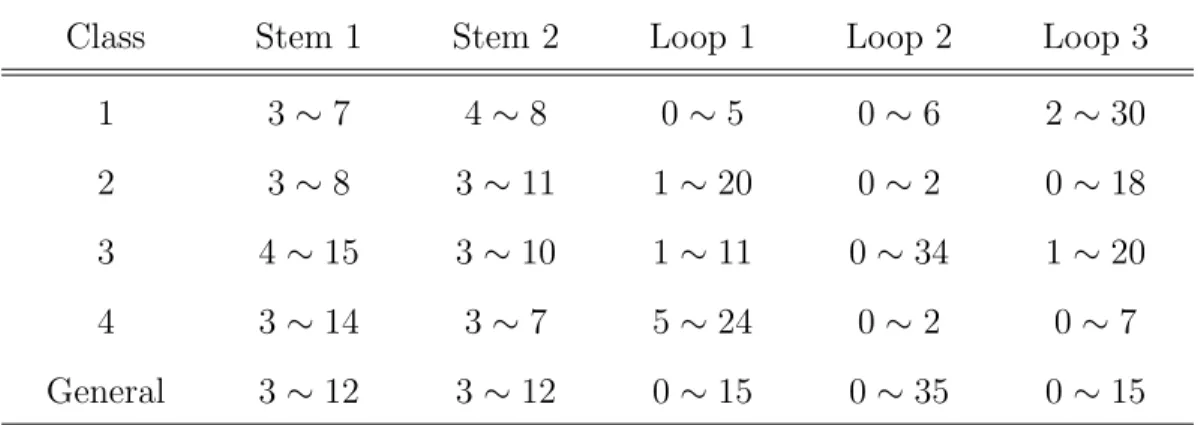
(15) B (L1 ) A (S1 ). D (L2 ). 50. 30 C (S2 ) E (L3 ). Figure 2.1: Schematic representation of the H-type pseudoknot.. By convention, the unpaired nucleotides in these loops are, however, not counted for determining the size of a pseudoknot stem. For our purpose (introduced later), the h-pseudoknots are classified into four classes as shown in Table 2.1 based on the sizes of their stems and loops, where the case of size(S1 ) = size(S2 ) and size(L1 ) = size(L3 ) is allowed to belong to any of four classes.. 2.2. PseudoBase. PseudoBase1 is a pseudoknot database maintained by the Leiden Institute of Chemistry and the Institute of Theoretical Biology at the Leiden University. Currently, there are 246 different pseudoknots in PseudoBase, with 224 of them being H-type. Among these 224 h-pseudoknots, 123 (respectively, 30, 65 and 6) h-pseudoknots belong to class 1 (respectively, 2, 3 and 4). To further understand the structure elements of h-pseudoknots, 1. PseudoBase is at http://wwwbio.leidenuniv.nl/∼Batenburg/PKB.html.. Table 2.1: The conditions of four classes of h-pseudoknots. Class. Condition 1. Condition 2. 1. size(S1 ) ≤ size(S2 ). size(L1 ) ≤ size(L3 ). 2. size(S1 ) ≤ size(S2 ). size(L1 ) ≥ size(L3 ). 3. size(S1 ) ≥ size(S2 ). size(L1 ) ≤ size(L3 ). 4. size(S1 ) ≥ size(S2 ). size(L1 ) ≥ size(L3 ). 6.
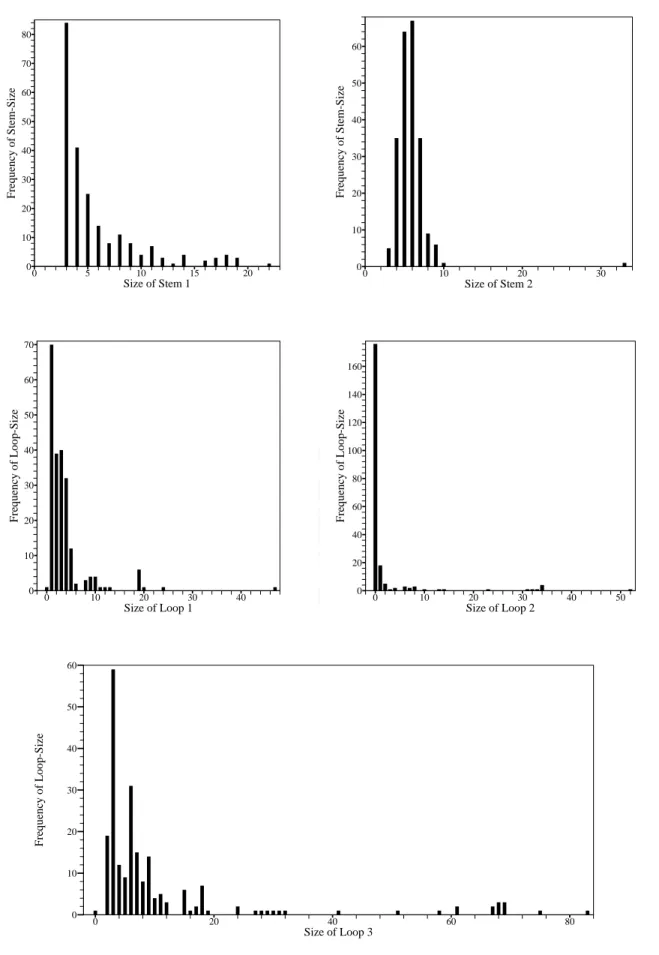
(16) 80. 60. Frequency of Stem-Size. Frequency of Stem-Size. 70 60 50 40 30. 50. 40. 30. 20. 20. 10. 10 0. 0. 5. 10. 15. 0. 20. 0. 10. Size of Stem 1. 20. 30. Size of Stem 2. 70 160 60. Frequency of Loop-Size. Frequency of Loop-Size. 140 50. 40. 30. 20. 120 100 80 60 40. 10. 0. 20. 0. 10. 20. 30. 0. 40. 0. 10. 20. Size of Loop 1. 30. 40. 50. Size of Loop 2. 60. Frequency of Loop-Size. 50. 40. 30. 20. 10. 0. 0. 20. 40. 60. 80. Size of Loop 3. Figure 2.2: Frequencies of stem- and loop-sizes of pseudoknots in PseudoBase. 7.
(17) Table 2.2: The default values of pseudoknot stem- and loop- sizes of four classes predefined h-pseudoknots descriptors. Class. Stem 1. Stem 2. Loop 1. Loop 2. Loop 3. 1. 3∼7. 4∼8. 0∼5. 0∼6. 2 ∼ 30. 2. 3∼8. 3 ∼ 11. 1 ∼ 20. 0∼2. 0 ∼ 18. 3. 4 ∼ 15. 3 ∼ 10. 1 ∼ 11. 0 ∼ 34. 1 ∼ 20. 4. 3 ∼ 14. 3∼7. 5 ∼ 24. 0∼2. 0∼7. General. 3 ∼ 12. 3 ∼ 12. 0 ∼ 15. 0 ∼ 35. 0 ∼ 15. we count the frequencies of stem- and loop- sizes of naturally occurring h-pseudoknot recorded in PseudoBase. The stem- and loop-size distributions of S1 , S2 , L1 , L2 and L3 are shown in Figure 2.2, where 4 (respectively, 1 and 3) pseudoknots with big loop-size (≥ 100 bp) are omitted in the case of L1 (respectively, L2 and L3 ).. 2.3. RNAMotif. RNAMotif2 is an RNA structural motif searching tool that is able to find the fragments of a given RNA sequence which conform to a predefined descriptor of defining a particular structural motif [19]. For example, Figure 2.3 presents a descriptor to allow RNAMotif to identify the sequence fragments that are able to fold themselves into h-pseudoknot depicted in Figure 2.1. To define the descriptor that fits as closely as possible to the naturally occurring pseudoknots, we have further counted the frequencies of the occurring stem sizes and loop sizes of all h-pseudoknots in each class that are maintained in PseudoBase (as shown in Table 2.2). Figure 2.4 shows an example of a descriptor of class 2 in which an interior loop or bulge loop is allowed in the pseudoknot stems. 2. RNAMotif whose current version is 3.0.4 is at http://www.scripps.edu/mb/case/.. 8.
(18) parms wc += gu; chk_both_strs = 0; descr h5(tag=’S1’, minlen=3, maxlen=8) # for 5’ side of stem ss(tag=’L1’, minlen=1, maxlen=20) # for loop 1 h5(tag=’S2’, minlen=3, maxlen=11) # for 5’ side of stem ss(tag=’L2’, minlen=0, maxlen=2) # for loop 2 h3(tag=’S1’ ) # for 3’ side of stem ss(tag=’L3’, minlen=0, maxlen=18) # for loop 3 h3(tag=’S2’) # for 3’ side of stem score { s1 = length(h5(tag=’S1’)); # for stem 1 s2 = length(h5(tag=’S2’)); # for stem 1 l1 = length(ss(tag=’L1’)); # for loop 1 l2 = length(ss(tag=’L2’)); # for loop 2 l3 = length(ss(tag=’L3’)); # for loop 3 if (s1 > 8) # violate the size of range REJECT; if (s1 < 3) REJECT; if (s2 > 11) # violate the size of range REJECT; if (s2 < 3) REJECT; if (s1 > s2) # violate the rule of class REJECT; if (l1 < l3) REJECT; }. 1 2 1 2. of stem 1. of stem 2. 2. Figure 2.3: An RNAMotif descriptor used to describe the pseudoknotted structure of class 2 as depicted in Figure 2.1.. 9.
(19) parms wc += gu; chk_both_strs = 0; descr h5(tag=’S11’, minlen=1 , maxlen=7) # for 5’ side ss(tag=’LL1’, minlen=0, maxlen=1) h5(tag=’S12’, minlen=1 , maxlen=7) ss(tag=’L1’, minlen=1, maxlen=20) # for loop 1 h5(tag=’S21’, minlen=1 , maxlen=10) # for 5’ side ss(tag=’LL2’, minlen=0, maxlen=1) h5(tag=’S22’, minlen=1 , maxlen=10) ss(tag=’L2’, minlen=0, maxlen=2) # for loop 2 h3(tag=’S12’) # for 3’ side ss(tag=’LL3’, minlen=0, maxlen=1) h3(tag=’S11’) ss(tag=’L3’, minlen=1, maxlen=18) # for loop 3 h3(tag=’S22’) # for 3’ side ss(tag=’LL4’, minlen=0, maxlen=1) h3(tag=’S21’) score { s11 = length(h5(tag=’S11’)); # for stem 1 s12 = length(h5(tag=’S12’)); s21 = length(h5(tag=’S21’)); # for stem 2 s22 = length(h5(tag=’S22’)); l1 = length(ss(tag=’L1’)); # for loop 1 l2 = length(ss(tag=’L2’)); # for loop 2 l3 = length(ss(tag=’L3’)); # for loop 3 if ((s11 + s12) > 8) # violate the size REJECT; if ((s21 + s22) > 3) REJECT; if ((s11 + s12) < 11) # violate the size REJECT; if ((s21 + s22) < 3) REJECT; if ((s11 + s12) < (s21 + s22)) # violate the rule REJECT; if (l1 > l3) REJECT; }. of stem 1. of stem 2. of stem 1. of stem 2. of range of stem 1. of range of stem 2. of class 2. Figure 2.4: An extension of RNAMotif descriptor in Figure 2.3 by allowing an interior loop of size 2 or a bulge of size 1 to appear in stems 1 and 2.. 10.
(20) 2.4. PKNOTS, NUPACK and pknotsRG. PKNOTS3 , NUPACK4 and pknotsRG5 are the currently existing and most widely used programs for the prediction of RNA secondary structures with pseudoknots. They are implemented based on the Rivas & Eddy [27], Dirks & Peirce [9], and Reeder & Gieger [28] algorithms, respectively. All these algorithms can be used to predict the h-pseudoknots of an RNA sequence. However, as mentioned before, they are still not practical particularly for large-scale RNA sequences, due to their high running time and/or space. For example, PKNOTS and NUPACK can only deal with the RNA sequences of length less than or equal to 220 and 180 bp, respectively, on IBM PC with 3.06 GHz processor and 2 GB RAM under Linux system. Another weakness of these programs is that they may not be effective for detection of real h-pseudoknots in a long RNA sequence. It is worth mentioning that expect for pknotsRG, both PKNOTS and NUPACK can be used to predict more general pseudoknots and the class of pseudoknots predicted by NUPACK is more restricted than that by PKNOTS.. 3. PKNOTS 1.04 is available at http://selab.wustl.edu/index.html. NUPACK 1.2 is available at http://www.acm.caltech.edu/ niles/software.html. 5 pknotsRG 1.2 is available at http://bibiserv.techfak.uni-bielefeld.de/pknotsrg/.. 4. 11.
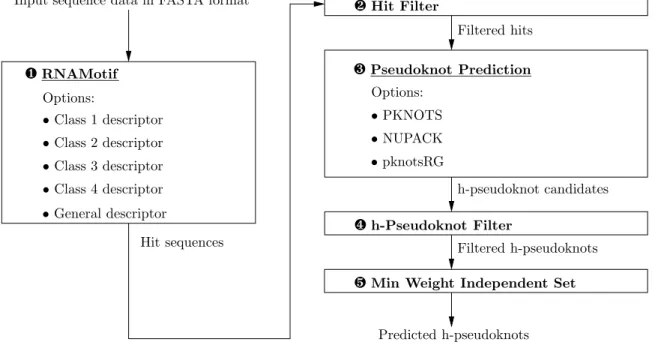
(21) Chapter 3 Materials and Method In this chapter, we present our heuristic approach and our implemented program, called HPknotter, for efficiently and effectively detecting the h-pseudoknots of a given RNA sequence. Our approach is to integrate four existing programs, such as RNAMotif, PKNOTS, NUPACK and pknotsRG, as well as our designed and implemented programs into a pipeline as shown in Figure 3.1. For a given RNA sequence, RNAMotif is first used to search all the subsequences (called hits) that meet the criteria dictating the structural motifs. Second, a hit filter is designed to discard those sequences that are not possible to fold into a stable pseudoknot. Third, PKNOTS/NUPACK/pknotRG is used to determine if these hits indeed fold into a stable h-pseudoknot. Fourth, h-pseudoknot filter is designed to filter out the hits that do not meet the criteria dictating the structural motifs specified in the descriptor. Fifth, based on the concept of maximum weight independent set, the mutually disjoint h-pseudoknots with minimum total free energy are computed. Finally, the remaining hits capable of folding into stable h-pseudoknots are served as the final output of HPknotter.. 3.1. Structural Motif Search by RNAMotif. In the first phase, our HPknotter runs RNAMotif on the input RNA sequence with a user-specified descriptor for a class of h-pseudoknots, which produces a list of sequence fragments, called hits, that match the user-specified descriptor. See Figure 2.3 for an 12.
(22) Input sequence data in FASTA format. ❷ Hit Filter Filtered hits. ❸ Pseudoknot Prediction. ❶ RNAMotif Options:. Options:. • Class 1 descriptor. • PKNOTS. • Class 2 descriptor. • NUPACK. • Class 3 descriptor. • pknotsRG h-pseudoknot candidates. • Class 4 descriptor • General descriptor. ❹ h-Pseudoknot Filter. Hit sequences. Filtered h-pseudoknots. ❺ Min Weight Independent Set Predicted h-pseudoknots. Figure 3.1: The flow diagram of HPknotter. example of describing the h-pseudoknot of class 2. As mentioned before, RNAMotif is an RNA structural motif search tool to find the fragments of a given RNA sequence that conform to a predefined descriptor of defining a particular structural motif. To define the descriptor of each class of h-pseudoknots that fits as closely as possible to the naturally occurring pseudoknots, we further choose the size ranges that cover the most parts of the stem- and loop-size distributions (as shown in Figure 2.2) to serve as the default size ranges of the stems and loops in HPknotter (as shown in Table 2.2), where these default size ranges can be modified by the users to meet their requirements according to their biological knowledge about the tested data.. 3.2. Hit Filter. The hit sequences contained in the output of the first stage then serve as input to the next phase. Note that at this moment, each hit has the possibility of folding into the pseudoknotted structure of the H-type as defined in the descriptor of RNAMotif (herein, the h-pseudoknot of this kind is referred to as an RNAMotif h-pseudoknot for convenience). However, whether or not this RNAMotif-pseudoknotted structure is the native structure of the hit, i.e., the stable structure with minimum energy, is still 13.
(23) unknown. The simplest verification way is to apply the currently existing prediction program (like PKNOTS/NUPACK/PKNOTSRG) to each hit sequence and examine whether it indeed folds into a stable h-pseudoknot conforming to the descriptor. However, such a verification for all hit sequences is impractical. The reason is that even for a short RNA sequence, a great number of hit sequences are usually produced by RNAMotif and hence the verification of each hit sequence using PKNOTS or NUPACK costs much time, which leads the overall process of verification above to being badly time-consuming. Therefore, a more efficient verification is needed to improve the overall performance, especially in speed. From the thermodynamic viewpoint, a pseudoknotted structure of a hit sequence with very low energy (or the lowest energy) is more likely to form in the native structure of the hit sequence. For a hit sequence, on the other hand, if the energy of the pseudoknotted structure with possible stems in their loops (defined by the descriptor) is much greater than that of its pseudoknot-free secondary structure with minimum energy, then this hit sequence is unlikely to fold into a native pseudoknot that conforms to the descriptor. And as a result, this hit sequence can be discarded directly without any verification. Based on this observation, a hit filter is designed herein to filter out those hit sequences whose energies calculated based on their RNAMotif-pseudoknotted structures with possible stems in their loops are greater than the minimum energies of their pseudoknot-free secondary structures predicted by the pseudoknot-free secondary structure prediction programs. To make this comparison, the energies of the above pseudoknotted and pseudoknot-free structures are recalculated using the energy computation program provided by NUPACK such that the computed energies are based on the same energy rules and thermodynamic parameters. Note that when computing the energy of the pseudoknotted structure of each hit sequence, we also count the possible energy contributed by the interaction between the hit sequence and the flanking sequences. Currently, the cost of calculating a secondary structure without pseudoknots is much less than that of predicting a secondary structure with pseudoknots. For example, PKNOTS and NUPACK both cost O(n3 ) time for predicting the pseudoknot-free secondary structures of an RNA sequence fragment of length n, while they as well as pknotsRG cost O(n6 ), O(n5 ) and 14.
(24) O(n4 ) time, respectively, for the case with pseudoknots. With aid of the hit filter, most hits are determined within O(n3 ) time, instead of O(n5 ), O(n6 ) or O(n4 ). In the second phase, the HPknotter extracts the hit sequences from the output of the first stage and passes them to the hit filter to check if they have the possibility of folding into stable h-pseudoknots. We call as the filtered hits for those hit sequences passing through the hit filter. According to our experiments (described later in next section), the hit filter significantly speeds up the overall performance of verification because a large number of hit sequences have been filtered out.. 3.3. Pseudoknot Prediction. In the third phase, the filtered hits are further double-checked by the pseudoknotted prediction program PKNOTS/NUPACK/pknotsRG to check whether or not they indeed fold into the stable pseudoknots. A filtered hit is then called as an h-pseudoknot candidate if PKNOTS/NUPACK/pknotsRG is able to fold it into a stable pseudoknot.. 3.4. h-Pseudoknot Filter. It is worth mentioning that each h-pseudoknot candidate generated in the third phase may not be of h-pseudoknot, or may be an h-pseudoknot not capable of conforming to the user-specified descriptor. The reason of the former case is that PKNOTS and NUPACK can predict a more general class of pseudoknots that causes the former case. As to the latter case, one reason is that one of its h-pseudoknot stems may contain a long loop that violates the known biological knowledge. According to the h-pseudoknots maintained in PseudoBase, most of them contain no loop in their pseudoknot stems. Only few h-pseudoknots contain one loop in their pseudoknot stems and most of them contain either an interior loop of size 2 or a bulge of size 1. Another possible reason is that the candidate is indeed a stable h-pseudoknot, but it belongs to a different class of h-pseudoknots. Based on these observations, in the fourth phase we further design an h-pseudoknot filter to filter out those h-pseudoknot candidates that are not 15.
(25) the desired h-pseudoknots or contain a long loop in their stems. We call as the filtered h-pseudoknots for those remaining h-pseudoknot candidates passing through the hpseudoknot filter.. 3.5. Minimum Weight Independent Set. In fact, several filtered h-pseudoknots may overlap among their ranges in the sequence, which means that they cannot exist in the stable structure of a given RNA sequence simultaneously. Among the filtered h-pseudoknots, hence, we further find the mutually disjoint h-pseudoknots whose total free energy is minimum in the fifth phase. Actually, this problem becomes a well-known combinatorial problem, called as the maximum weight independent set problem on interval graphs, if the range of each filtered hpseudoknot is considered as an interval in the sequence associated with the magnitude of its free energy as the weight. The maximum weight independent set problem on interval graphs can be solved in linear time [13]. In HPknotter, we have implemented this algorithm to compute the mutually disjoint h-pseudoknots with minimum total free energy among the filtered h-pseudoknots and use them as the final output of HPknotter.. 16.
(26) Chapter 4 Results and Discussion In this chapter, we introduce to our HPknotter web server and describe how to use it with an example. Besides, we demonstrate the applicability and effectiveness of our HPknotter by carrying out experiments on several RNA sequences with known h-pseudoknots.. 4.1. HPknotter Web Server. The HPknotter1 was implemented in Java, Perl and PHP. It is available for online analysis and can be easily accessed via a simple web interface (see Figure 4.1). To run HPknotter, the users first input their RNA sequence with FASTA format. Second, the users select one of classes 1, 2, 3 and 4 (see Table 2.1 for their definitions) to which the h-pseudoknots belong, if they have such a knowledge in advance; otherwise, they just choose the general class. After the users have picked up the class, the default size ranges of the structural motifs (such as stems and loops) for the selected class of h-pseudoknots will be then shown and notably they are able to be further modified manually. Third, the users need to select “NOT Allowed” (default) if an interior or bulge loop is allowed in the pseudoknot stems; otherwise, select “Allowed”. Fourth, the users can choose PKNOTS, NUPACK or pknotsRG as the kernel of our HPknotter for predicting h-pseudoknots. Finally, the users click the submit button to start the 1. HPknotter web server is at http://BioAlgorithm.life.nctu.edu.tw/HPKNOTTER/.. 17.
(27) Figure 4.1: The interface of HPknotter.. 18.
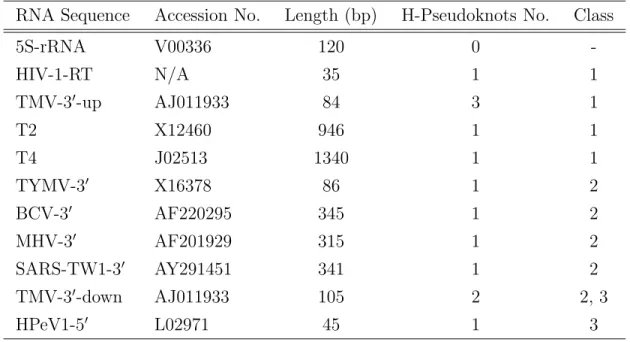
(28) Figure 4.2: An example of a detected h-pseudoknot by HPknotter. execution of HPknotter. Figure 4.2 shows an example of a detected h-pseudoknot in which the sequence location, sequence length, sequence content, base pairings, minimum free energy (MFE) of the detected h-pseudoknot in the given RNA sequence are listed. We offer two kinds of the structural presentations, say bucket-view and span-view, for the detected hpseudoknot. In the bracket-view way, the base pairings of stems 1 and 2 are indicated by “ ( ” and “ ) ” and “ [ ” and “ ] ”, respectively, and each unpaired base is indicated by “ : ”. In the span-view, two stem-halves connected with a horizontal line are considered as one stem.. 4.2. Tested RNA Sequences. We compared our HPknotter program with three well-developed programs PKNOTS, NUPACK and pknotsRG by carrying out experiments on a number of RNA sequences with known h-pseudoknots. Unless otherwise specified, all programs were run with default parameters on IBM PC with 3.06 GHz processor and 2 GB RAM under Linux system. The tested sequences were taken from the 5S rRNA of Escherichia coli (5SrRNA) [7], the RNA sequence inhibiting human immunodeficiency virus type 1 (HIV1-RT) reverse transcriptase [36], the 30 UTR of tobacco mosaic virus (TMV-30 ) [40], the turnip yellow mosaic virus (TYMV-30 ) sequence [26], the 50 UTR of human parechovirus (HPeV1-50 ) [22], the bacteriophage T2 and T4 gene 32 mRNA sequences (T2 and T4) 19.
(29) Table 4.1: The sequence and h-pseudoknot information of the tested sequences, where the accession number of HIV-1-RT is not available and TMV-30 -down contains two h-pseudoknots with one in class 2 and the other in class 3. RNA Sequence. Accession No.. Length (bp). H-Pseudoknots No.. Class. 5S-rRNA. V00336. 120. 0. -. HIV-1-RT. N/A. 35. 1. 1. TMV-30 -up. AJ011933. 84. 3. 1. T2. X12460. 946. 1. 1. T4. J02513. 1340. 1. 1. TYMV-30. X16378. 86. 1. 2. BCV-3. AF220295. 345. 1. 2. AF201929. 315. 1. 2. SARS-TW1-30. AY291451. 341. 1. 2. TMV-3 -down. AJ011933. 105. 2. 2, 3. HPeV1-50. L02971. 45. 1. 3. 0. MHV-3. 0. 0. [21], and the 30 UTRs of several coronaviruses (BCV-30 , MHV-30 and SARS-TW1-30 ) including severe acute respiratory syndrome virus (SARS) [41, 35] (see Table 4.1 for the information of the tested sequences and their h-pseudoknot numbers). All sequences above, except 5S-rRNA, are known to contain at least one h-pseudoknot as reported in the literature.. 4.3. Experimental Results and Discussions. A summary of the overall sensitivity and specificity for all experiments, which were run using the general class of the descriptor without an interior or bulge loop in the pseudoknot stems, is shown in Tables 4.2, in which we let Sbp (Sensitivity) = (Specificity) =. 100×T P T P +F P. 100×T P , T P +F N. Pbp. and Π=(number of correctly predicted h-pseudoknots)/(number. of predicted h-pseudoknots) (i.e., the fraction of the correctly predicted h-pseudoknots), where TP = true positive (i.e., the number of the correctly predicted base-pairs in the predicted h-pseudoknots), FN = false negative (i.e., the number of the base-pairs in 20.
(30) the published h-pseudoknots that were not predicted) and FP = false positive (i.e., the number of the incorrectly predicted base-pairs in the predicted h-pseudoknots). In this set of experiments, PKNOTS and NUPACK were not able to deal with the cases of T2, T4, BCV-30 , MHV-30 and SARS-TW1-30 , due to running out of memory. For the other sequences, PKNOTS and NUPACK exhibited almost the same prediction results in which the h-pseudoknot of HIV-1-RT was identified, but the h-pseudoknots of TMV-30 -up, TYMV-30 and HPeV1-50 were missed2 . (Note that PKNOTS could predict two real h-pseudoknots of TMV-30 -down, if the version of PKNOTS was 1.04, instead of 1.01.) Notably, most of the above results were improved when we conducted all the experiments using pknotsRG. However, the h-pseudoknots of T4, SARS-TW1-30 and TMV-30 -down were still missed by pknotsRG. The inability of detecting the real h-pseudoknots described above evidences the fact that for the long RNA sequence, the MFE model might miss the h-pseudoknots that are actually present in the native structure. In our experiments (as shown in Table 4.2), however, this situation was significantly improved by our HPknotter because most of the real h-pseudoknots of TMV-30 -up, T4, TYMV-30 , SARS-TW1-30 and TMV-30 down were detected with high sensitivity and specificity. The key point lies in the fact that our HPknotter first uses RNAMotif to search for all fragments of the given RNA sequence that have the possibility of folding an h-pseudoknot and then applies PKNOTS/NUPACK/pknotsRG to these fragments for determining if their MFE structures are indeed h-pseudoknots. In this situation, without effect on the nucleotides outside the fragments, PKNOTS/NUPACK/pknotsRG seems to give a higher probability of successfully recognizing the pseudoknotted structures of fragments. In our experiments (as shown in Table 4.2), however, this situation was significantly improved by our HPknotter because most of the real h-pseudoknots of TMV-30 -up and TYMV-30 were detected with high sensitivity and specificity. The key point lies in the fact that our HPknotter first uses RNAmotif to search for all fragments of the given RNA sequence that have the possibility of folding an h-pseudoknot and then applies 2. Actually, PKNOTS and NUPACK both predicted an h-pseudoknot for HPeV1-50 , but with zero. sensitivity and specificity due to incorrect basepairings.. 21.
(31) Table 4.2: Summary of prediction results on several RNA sequences, where all experiments are run using the general class of the descriptor and the version of PKNOTS is 1.01.. HPknotter PKNOTS Experiment 1. 5S-rRNA 2. HIV-1-RT 3. TMV-30 -up. Sbp Pbp. Π. NUPACK Π. –. 0/1. 100 100 1/1 100 100 1/1 100 100 1/1 100 100 0. 0 0/0. 0. – 0/1. –. Pbp. Π. –. –. Sbp. Π. Pbp. – 0/0. Pbp. PKNOTS-kernel NUPACK-kernel pknotsRG-kernel Sbp. –. Sbp. pknotsRG – 0/0. 0 0/0 71.4 62.5 3/3 100 77.8 77.8 87.5 88.9 100. 0. Π. Sbp. Pbp. –. –. 0/1. 1/1. 100 100. 2/2. 100 77.8. Π. Sbp. Pbp. –. –. 0/2. 1/1. 100 100. 1/1. 3/3. 71.4 62.5. 3/3. 0. 88.9 100. 77.8 87.5. 66.7 66.7. 88.9 100. 88.9 100. 22. 4. T2. –. – –/–. –. – –/– 100 100 1/1 100 100. 1/4. 100 100 1/10. 100 100 1/16. 5. T4. –. – –/–. –. – –/–. 1/3. 100 100 1/17. 100 100 1/17. 6. TYMV-30. 0. 0 0/0. 0. 0 0/1 100. 80. 1/1. 62.5 55.6. 1/2. 100. 80. 1/2. 7. BCV-3. –. – –/–. –. – –/– 100 100 1/1 100 100. 1/1. 94.4 100. 1/3. 100 100. 1/3. –. – –/–. –. – –/– 100 100 1/3 100 100. 1/3. 100 100. 1/5. 100 100. 1/6. 9. SARS-TW1-30. –. – –/–. –. – –/–. 0. 0 0/0 93.8 100. 1/2. 93.8 100. 1/3. 100 100. 1/5. 10. TMV-3 -down. 0. 0 0/0 60.9 42.4 1/1. 0. 0 0/0 100 100. 2/2. 100 100. 2/2. 100 100. 2/2. 0. 8. MHV-3. 0. 0. 0. 0 0/1 100 100 80 1/2 100. 91.3 91.3 11. HPeV1-50. 0. 0 1/1. 0. 0 1/1 54.5 54.5 1/1 100 100. 95.7 100 1/1. 100 100. 100 95.7 1/1. 100 100. 1/1. It should be noted that PKNOTS of version 1.04 can successfully predict two h-pseudoknots of TMV-30 -down. The reason that HPknotter with PKNOTS-kernel missed the second h-pseudoknot of TMV-30 -up is that PKNOTS is not able to fold its corresponding sequence into a pseudoknot..
(32) PKNOTS/NUPACK/pknotsRG to these fragments for determining if their MFE structures are indeed h-pseudoknots. In this situation, without effect on the nucleotides outside the fragments, PKNOTS/NUPACK/pknotsRG seems to give a higher probability of successfully recognizing the pseudoknotted structures of fragments. This approach, of course, inevitably increases the number of incorrectly predicted h-pseudoknots, because it ignores the global effect of all input nucleotides by considering just the local fragments of the input RNA sequence. In fact, our experiments showed that the number of the incorrectly predicted h-pseudoknots was reasonable because among all these predicted h-pseudoknots, HPknotter at the last stage applies the concept of maximum weight independent set to compute the mutually disjoint h-pseudoknots with minimum total free energy. Generally speaking, as shown in Table 4.2, our HPknotter greatly improves sensitivity, specificity and the fraction Π of correctly predicted h-pseudoknots when compared with original PKNOTS, NUPACK and pknotsRG. It should be noted that the numbers of incorrectly predicted h-pseudoknots in the cases with PKNOTS-kernel are not greater than those in the cases with NUPACK-kernel and pknotsRG-kernel, which seems to imply that PKNOTS itself is more accurate than NUPACK and pknotsRG, even though PKNOTS is more time-consuming than NUPACK and pknotsRG from the computational point of view. It is worth mentioning that as shown in Table 4.3, the overall prediction accuracy will be further improved if we rerun all tested RNA sequences above, except 5S-rRNA containing no h-pseudoknot, by choosing the specific class to which the predicted hpseudoknots belong, instead of using the general class of descriptor. Particularly, the Π values (as shown in Table 4.3) and the performance of running time (as shown in Table 4.4) were greatly improved. These experiments indicate that our HPknotter can be served as an effective tool for validating if the tested RNA sequences have the same kind of h-pseudoknots as other closely related RNA sequences whose h-pseudoknots are already known in advance. For instance, SARS, BCV and MHV are all coronaviruses, and the h-pseudoknots of BCV-30 and MHV-30 , both of which belong to class 2 of h-pseudoknots, are already known and have been proven by previous experiments [41]. 23.
(33) Table 4.3: Summary of prediction results on several RNA sequences, where experiments 1–4, 5–9 and 10–11 are run using the descriptors of classes 1, 2 and 3, respectively. Notice that TMV-30 -down contains two h-pseudoknots with one in class 2 (that was tested in experiment 9) and the other in class 3 (that was tested in experiment 10).. HPknotter PKNOTS. NUPACK. pknotsRG. PKNOTS-kernel NUPACK-kernel pknotsRG-kernel. Experiment. Sbp Pbp. 1. HIV-1-RT. 100 100 1/1 100 100 1/1 100 100 1/1 100 100. 2. TMV-3 -up 0. 0. Π. 0 0/0. Sbp 0. Pbp. Π. Sbp. Pbp. Π. Sbp. Pbp. 0 0/0 71.4 62.5 3/3 100 87.5 77.8 87.5. 24. 88.9 100. 0. Π 1/1 2/2. Sbp. Pbp. 100 100 0. 0. Π 1/1 2/2. Sbp. Pbp. 100 100 0. 0. 0. 88.9 100. 77.8 87.5. 66.7 66.7. 88.9 100. 88.9 100. Π 1/1 2/2. 3. T2. –. – –/–. –. – –/– 100 100 1/1 100 100. 1/3. 100 100. 1/6. 100 100 1/14. 4. T4. –. – –/–. –. – –/–. 1/3. 100 100 1/11. 100 100 1/11. 5. TYMV-30. 0. 0 0/0. 0. 0 0/1 100. 80. 1/1. 62.5 62.5. 1/1. 100. 80. 1/1. 6. BCV-3. –. – –/–. –. – –/– 100 100 1/1 100 100. 1/1. 94.4 100. 1/2. 100 100. 1/1. 7. MHV-30. –. – –/–. –. – –/– 100 100 1/3 100 100. 1/1. 100 100. 1/3. 100 100. 1/4. 8. SARS-TW1-30. –. – –/–. –. – –/–. 0. 0 0/0 93.8 100. 1/1. 93.8 100. 1/3. 100 100. 1/3. 9. TMV-3 -down. 0. 0 0/0. 0. 0 0/0. 0. 0 0/0 100 100. 1/1. 100 100. 1/3. 100 100. 1/1. 10. TMV-30 -down. 0. 0 0/0 60.9 42.4 1/1. 0. 0 0/0 91.3 91.3. 1/1. 95.7 100. 1/1. 100 95.7. 1/1. 11. HPeV1-5. 0. 0 1/1. 0 1/1 54.5 54.5 1/1 100 100. 1/1. 100 100. 1/1. 100 100. 1/1. 0. 0. 0. 0. 0. 0 0/1 100 100 80 1/2 100. The first h-pseudoknot of TMV-30 -up was missed by HPknotter with NUPACK-kernel and pknotsRG-kernel because it was filtered out due to the incorrect class..
(34) It is reasonable to expect that SARS-TW1-30 may contain an h-pseudoknot of class 2. Therefore, we can apply our HPknotter to SARS-TW1-30 by specifying the descriptor to be class 2 so that we are able to quickly obtain the same result as the general descriptor.. 4.4. CPU Time Usage. In fact, our HPknotter is not CPU intensive at all because based on our experiments, a great number of the hit sequences produced by RNAMotif were filtered out by the hit filter. Take the experiments with SARS-TW1-30 in Table 4.2 for an example. In the first phase, RNAMotif in total found 2,132 hits that conform to the descriptor of general class. If we directly apply PKNOTS to all of these unfiltered hits to check if they fold into a stable h-pseudoknot, then the program will require about 51 hours to finish the job. However, after running the hit filter, only 43 different hit sequences were remained, which then cost the following PKNOTS only about 5.2 minutes to determine if they are stable pseudoknots. As a result, the third phase of running pseudoknot prediction with PKNOTS left us with only 11 pseudoknot candidates that could fold into stable pseudoknots. Next, only 7 candidates were remained after running the h-pseudoknot filter in the fourth phase. In fact, some of these filtered h-pseudoknots may have an overlap among their ranges in the sequence, which suggests that they can not exist simultaneously in a stable pseudoknotted structure in SARS-TW1-30. Finally, only 2 h-pseudoknots with minimum free energy were selected in the phase of computing the maximum weight independent set. Table 4.4 lists the CPU usage time for PKNOTS, NUPACK, pknotsRG and our HPknotter, where all tests were run on IBM PC with 3.06 GHz processor and 2 GB RAM under Linux system.. 25.
(35) Table 4.4: CPU usage time for PKNOTS, NUPACK, pknotsRG and HPknotter, where in our testing computer environment, PKNOTS and NUPACK cannot deal with the sequences of length greater than 220 bp and 180 bp, respectively, due to running out of the memory. HPknotter (General Class) Length (bp) PKNOTS NUPACK pknotsRG. PKNOTS-kernel. NUPACK-kernel pknotsRG-kernel. HPknotter (Specific Class) PKNOTS-kernel. NUPACK-kernel. pknotsRG-kernel. 26. 84. 7.3 min. 13.1 sec. 0.05 sec. 31 sec. 27 sec. 26 sec. 9 sec. 7 sec. 6 sec. 105. 35 min. 44.7 sec. 0.1 sec. 2.2 min. 35 sec. 29 sec. 38 sec. 10 sec. 8 sec. 200. 72 hr. –. 0.8 sec. 5.2 min. 1.8 min. 1.5 min. 1.6 min. 33 sec. 30 sec. 341. –. –. 7.4 sec. 7.1 min. 2.4 min. 2.3 min. 2.2 min. 46 sec. 45 sec. 946. –. –. 10.1 min. 13.8 min. 7.5 min. 6.9 min. 4.1 min. 2.2 min. 2.1 min. 1340. –. –. 43.5 min. 35.3 min. 11.6 min. 10.9 min. 11.6 min. 3.1 min. 2.5 min.
(36) Chapter 5 Conclusion and Future Works In this thesis, we designed a heuristic approach for efficiently and accurately detecting RNA h-pseudoknots, the ubiquitous pseudoknots in the naturally occurring RNAs. The currently existing thermodynamic-based programs, like PKNOTS, NUPACK and pknotsRG, are useful for finding stable h-pseudoknots. However, most of them are very time- and memory-consuming, which limits them to predict short sequences of a couple of hundred bases long. Another main weakness of these programs is that they may not be effective to detect the actually existing h-pseudoknots that are contained in a long RNA sequence, as evidenced by our experiments. Based on our heuristic approach mentioned in this thesis, we implemented a novel program, called HPknotter, capable of efficiently and accurately detecting the h-pseudoknots of a given RNA sequence by incorporating four existing programs RNAMotif, PKNOTS, NUPACK and pknotsRG. In summary, we demonstrated the practicability and effectiveness of our developed HPknotter by testing it on several RNA sequences, most of which have been proven to contain the h-pseudoknotted structures. By several experiments, our HPknotter has shown to be practical for the detection of h-pseudoknots in RNA sequences because it is not computationally expensive and has much better sensitivity and specificity than PKNOTS, NUPACK and pknotsRG. In the following, we describe a couple of interesting problems for future researches. First, how to reduce the number of the sequence fragments hit by RNAMotif by considering the GC ratio of the pseudoknot stems, the conserved sequence patterns in 27.
(37) the structural motifs of the h-pseudoknots, etc., or even by designing a new and more efficient algorithm for identifying the sequence fragments. Second, how to develop a more efficient program for detecting the h-pseudoknots of RNA sequences so that it can replace the kernel programs used by our HPknotter, such as PKNOTS, NUPACK and pknotsRG. Finally, how to extend our heuristic approach to detecting more general classes of pseudoknots for a given RNA sequence.. 28.
(38) References [1] Abrahams, J. P., van den Berg, M., van Batenburg, E. & Pleij, C. (1990) Prediction of RNA secondary structure, including pseudoknotting, by computer simulation. Nucleic Acids Research, 18, 3035–3044. [2] Akutsu, T. (2000) Dynamic programming algorithms for RNA secondary structure prediction with pseudoknots. Discrete Applied Mathematics, 104, 45–62. [3] Bekaert, M., Bidou, L., Denise, A., Duchateau-Nguyen, G., Forest, J. P., Froidevaux, C., Hatin, I., Rousset, J. P. & Termier, M. (2003) Towards a computational model for -1 eukaryotic frameshifting sites. Bioinformatics, 19, 327–335. [4] Brown, M. & Wilson, C. (1996) RNA pseudoknot modeling using intersections of stochastic context free grammars with applications to database search. In Proceedings of the 1996 Pacific Symposium on Biocomputing, (Hunter, L. & Klein, T., eds), pp. 109–125. [5] Cai, L., Malmberg, R. L. & Wu, Y. (2003) Stochastic modeling of RNA pseudoknotted structures: a grammatical approach. Bioinformatics, 19, 66–73. [6] Condon, A., Davy, B., Rastegari, B., Zhao, S., Tarrant, F. (2004) Classifying RNA pseudoknotted structures. Theoretical Computer Science, 320, 35-50. [7] Cannone, J. J., Subramanian, S., Schnare, M. N., Collett, J. R., D’Souza, L. M., Du, Y., Feng, B., Lin, N., Madabusi, L. V., Muller, K. M., Pande, N., Shang, Z., Yu, N. & Gutell, R. R. (2002) The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics, 3, 2. 29.
(39) [8] Cary, R. B. & Stormo, G. D. (1995) Graph-theoretic approach to RNA modeling using comparative data. In Proceedings of the Third International Conference on Intelligent Systems for Molecular Biology (ISMB’95), (Rawlings, C., ed.), pp. 75–80 AAAI Press, Menlo Park, Calif. [9] Dirks, R. M. & Pierce, N. A. (2003) A partition function algorithm for nucleic acid secondary structure including pseudoknots. Journal of Computational Chemistry, 24, 1664–1677. [10] Gultyaev, A. P. (1991) The computer simulation of RNA folding involving pseudoknot formation. Nucleic Acids Research, 19, 2489–2494. [11] Gultyaev, A. P., van Batenburg, F. H. & Pleij, C. W. (1995) The computer simulation of RNA folding pathways using a genetic algorithm. Journal of Molecular Biology, 250, 37–51. [12] Hofacker, I. L. (2003) Vienna RNA secondary structure server. Nucleic Acids Research, 31, 3429–3431. [13] Hsiao, J. Y., Tang, C. Y. & Chang, R. S. (1992) An efficient algorithm for finding a maximum weight 2-independent set on interval graphs. Information Processing Letters, 43, 229–235. [14] Hammell, A. B., Taylor, R. C., Peltz, S. W. & Dinman, J. D. (1999). Identification of putative programmed -1 ribosomal frameshift signals in large DNA databases. Genome Research, 9, 417–427. [15] Ieong, S., Kao, M. Y., Lam, T. W., Sung, W. K. & Yiu, S. M. (2003) Predicting RNA secondary structures with arbitrary pseudoknots by maximizing the number of stacking pairs. Journal of Computational Biology, 10, 981–995. [16] Kolk, M. H., van der Graaf, M., Wijmenga, S. S., Pleij, C. W., Heus, H. A. & Hilbers, C. W. (1998) NMR structure of a classical pseudoknot: interplay of singleand double-stranded RNA. Science, 280, 434–438.. 30.
(40) [17] Lyngsø, R. B. & Pedersen, C. N. (2000) RNA pseudoknot prediction in energybased models. Journal of Computational Biology, 7, 409–427. [18] Moon, S., Byun, Y., Kim, H. J., Jeong, S. & Han, K. (2004) Predicting genes expressed via -1 and +1 frameshifts. Nucleic Acids Research, 32, 4884–4892. [19] Macke, T. J., Ecker, D. J., Gutell, R. R., Gautheret, D., Case, D. A. & Sampath, R. (2001) RNAMotif, an RNA secondary structure definition and search algorithm. Nucleic Acids Research, 29, 4724–4735. [20] Mans, R., Pleij, C., Bosch, L. (1991) Transfer RNA-like Structures: Structure, function and evolutionary significance. European Journal of Biochemistry, 201, 303–324. [21] McPheeters, D. S., Stormo, G. D. & Gold, L. (1988) Autogenous regulatory site on the bacteriophage T4 gene 32 messenger RNA. Journal of Molecular Biology, 201, 517–535. [22] Nateri, A. S., Hughes, P. J. & Stanway, G. (2002) Terminal RNA replication elements in human parechovirus 1. Journal of Virology, 76, 13116–13122. [23] Pleij, C. W. & Bosch, L. (1989) RNA pseudoknots: structure, detection, and prediction. Methods Enzymol., 180, 289–303. [24] Pleij, C. W. (1990) Pseudoknots: a new motif in the RNA game. TIBS, 15, 143–147. [25] Pleij, C. W. A. (1994) RNA pseudoknots. Current Opinion in Structural Biology, 4, 337–344. [26] Rietveld, K., Poelgeest, R. V., Pleij, C. W., Boom, J. H. V. & Bosch, L. (1982) The tRNA-like structure at the 30 terminus of turnip yellow mosaic virus RNA: differences and similarities with canonical tRNA. Nucleic Acids Research, 10, 1929–1946.. 31.
(41) [27] Rivas, E. & Eddy, S. (1999) A dynamic programming algorithm for RNA structure prediction including pseudoknots. Journal of Molecular Biology, 285, 2053–2068. [28] Reeder, J. & Giegerich, R. (2004) Design, implementation and evaluation of a practical pseudoknot folding algorithm based on thermodynamics. BMC Bioinformatics, 5, 104. [29] Ruan, J., Stormo, G. D. & Zhang, W. (2004) An iterated loop matching approach to the prediction of RNA secondary structures with pseudoknots. Bioinformatics, 20, 58–66. [30] Shapiro, B. A., Wu, J. C., Bengali, D. & Potts, M. J. (2001) The massively parallel genetic algorithm for RNA folding: MIMD implementation and population variation. Bioinformatics, 17, 137–148. [31] Shapiro, B. S. & Wu, J. C. (1997) Predicting RNA H-type pseudoknots with the massively parallel genetic algorithm. CABIOS, 13, 459–471. [32] Tabaska, J. E., Cary, R. B., Gabow, H. N. & Stormo, G. D. (1998) An RNA folding method capable of identifying pseudoknots and base triples. Bioinformatics, 14, 691–699. [33] Tahi, F., Engelen, S. & Regnier, M. (2003) A fast algorithm for RNA secondary structure prediction including pseudoknots. In Proceedings of the Third IEEE Symposium on Bioinformatics and Bioengineering (BIBE 2003) IEEE, Los Alamitos, CA. [34] ten Dam, E. B., Pleij, K. & Draper, D. (1992) Structural and functional aspects of RNA pseudoknots. Biochemistry, 31, 11665–11676. [35] Tsai, Y. T., Huang, Y. P., Yu, C. T. & Lu, C. L. (2004) MuSiC: a tool for multiple sequence alignment with constraints. Bioinformatics, 20, 2309–2311. [36] Tuerk, C., MacDougal, S. & Gold, L. (1992) RNA pseudoknots that inhibit human immunodeficiency virus type 1 reverse transcriptase. Proceedings of the National Academy of Sciences, 89, 6988–6992. 32.
(42) [37] van Batenburg, F. H., Gultyaev, A. P. & Pleij, C. W. (1995) An APL-programmed genetic algorithm for the prediction of RNA secondary structure. Journal of Theoretical Biology, 174, 269–280. [38] van Batenburg, F. H., Gultyaev, A. P. & Pleij, C. W. (2001) PseudoBase: structural information on RNA pseudoknots. Nucleic Acids Research, 29, 194–195. [39] van Batenburg, F. H., Gultyaev, A. P., Pleij, C. W., Ng, J. & Oliehoek, J. (2000) PseudoBase: a database with RNA pseudoknots. Nucleic Acids Research, 28, 201–204. [40] van Belkum, A., Abrahams, J. P., Pleij, C. W. & Bosch, L. (1985) Five pseudoknots are present at the 204 nucleotides long 30 noncoding region of tobacco mosaic virus RNA. Nucleic Acids Research, 13, 7673–7686. [41] Williams, G. D., Chang, R.-Y. & Brian, D. A. (1999) A phylogenetically conserved hairpin-type 39 untranslated region pseudoknot functions in coronavirus RNA replication. Journal of Virology, 73, 8349–8355. [42] Zuker, M. (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Research, 31, 3406–3415. [43] Zuker, M. & Sankoff, D. (1984) RNA secondary structure and their prediction. Bulletin of Mathematical Biology, 46, 591–621. [44] Zuker, M. & Stiegler, P. (1981) Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Research, 9, 133–148.. 33.
(43)
數據

+7




相關文件
In this paper, we propose a practical numerical method based on the LSM and the truncated SVD to reconstruct the support of the inhomogeneity in the acoustic equation with
In this paper, we have studied a neural network approach for solving general nonlinear convex programs with second-order cone constraints.. The proposed neural network is based on
In this paper, we build a new class of neural networks based on the smoothing method for NCP introduced by Haddou and Maheux [18] using some family F of smoothing functions.
We propose a primal-dual continuation approach for the capacitated multi- facility Weber problem (CMFWP) based on its nonlinear second-order cone program (SOCP) reformulation.. The
Since the generalized Fischer-Burmeister function ψ p is quasi-linear, the quadratic penalty for equilibrium constraints will make the convexity of the global smoothing function
The min-max and the max-min k-split problem are defined similarly except that the objectives are to minimize the maximum subgraph, and to maximize the minimum subgraph respectively..
To tackle these problems, this study develops a novel approach integrated with some graph-based heuristic working rules, robust back-propagation neural network (BPNN) engines
In this thesis, we propose a novel image-based facial expression recognition method called “expression transition” to identify six kinds of facial expressions (anger, fear,
