預測 transfer RNA 二級結構使用互補式基因演算法
莊麗月
a, 林昱達
b, 楊正宏
b,c*Li-Yeh Chuang
a,
Yu-Da Lin
b,
Cheng-Hong Yang
b,c*a
義守大學化學工程系
b國立高雄應用科技大學電子工程系
c稻江科技暨管理學院網路系統學系
*通訊作者: 楊正宏, chyang@cc.kuas.edu.tw
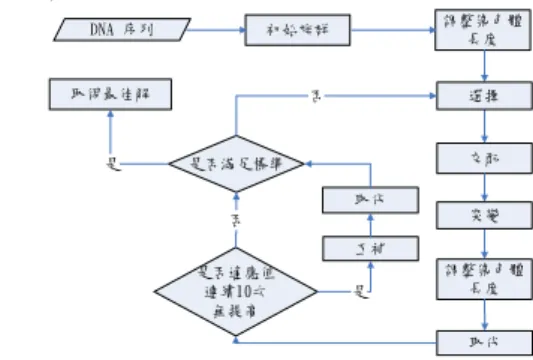
摘要 tRNA 為蛋白質合成時,擔任轉譯作用環節之轉運分 子,每條序列各自擁有反密碼子以能相對應 mRNA 密 碼子,並組織蛋白質序列延續生物生命。要如何精確 預測 tRNA 序列中之反密碼子為研究 tRNA 二級結構 的目標。在本研究中,提供一個敏感度高達 99.6%之 預測 tRNA 二級結構方法。利用互補式基因演算法模 擬生物演化,逐步組合最佳折疊途徑及預測結構,並 找出反密碼子訊息。在實驗中一般基因演算法因太早 收斂而落入區域最佳解,導致結果不理想。故本文使 用互補觀念來改善基因演算法,實驗結果證明能有效 跳脫區域最佳解。本研究採用資料庫是經實驗證明過 之 Sprinzl tRNA database,並分成 4 種類型(archaeal, eubacterial, eukaryotic, chloroplasts genes)測詴敏感度。 關鍵字:tRNA、tRNA 二級結構預測、互補式基因演 算法 1、前言 tRNA 是合成蛋白質時,轉譯作用(translation) 環節中 擔任轉運的分子,在 1958 年由 Crick 推測有一分子介 於基因及多胜肽之間,也就是介於核苷酸序列及氨基 酸 (amino acid) 序列間之連繫,此分子被稱為 tRNA (transfer RNA),且已經由 Crick 進行蛋白質合成實驗證 實。每個 tRNA 皆有反密碼子(anticodon)。其作用是為 精確對應由讀取 DNA 訊息所轉錄成的 mRNA 序列, 該序列由5’端往 3’端方向以三個核苷酸組成的密碼子 與 tRNA 反密碼子做互補動作。當成功互補時,接附 在tRNA3’端 OH 基的胺基酸會被帶往核糖體進行結合 胺基酸序列,進而成為蛋白質來維持生物活性。研究 tRNA 常應用於蛋白質工程,生物學家能藉由分析 tRNA 反密碼子來改變其相對應的胺基酸,進而結合至 胺基酸序列,嘗詴創造具有全新屬性或功能的蛋白質。 第一條 tRNA 序列的二級結構已在 1965 年由 Holley 所證明 [1, 2],至今已超過 4000 條不同 tRNA 被驗證, 由這增加數量的曲線證明,Holley 所提出的三葉草結 構已被普遍採用[3, 4]。一條典型 tRNA 序列大約由 70 至 95 個核苷酸(nucleotide)所組成,其中核苷酸包含胞 嘧 啶 (Cytosine) 、 鳥 糞 嘌 呤 (Guanine) 、 腺 嘌 呤 (Adenine)、尿嘧啶(Uracil)、及稀有鹼基。在序列中核 苷酸以反平行互補的方式並滿足標準 Watson - Crick base pairing (為 C-G、A-U 配對)或 wobble base pair (為 G-U 配對)的條件配對形成螺旋區。其中每個核苷酸最 多只能與一個核苷酸配對,此螺旋區稱 tRNA 的莖 (stem),連續且不滿足配對部份則形成環(loop)。而環 加上莖的結構稱為臂(arm),整條序列總共摺疊成四個 螺旋區( acceptor stem、D stem、anticodon stem、T stem ) 與四個環( D Loop、anticodon Loop、variable Loop、T Loop)的基本結構,此結構稱為三葉草結構(clover leaf structure)。一個標準 tRNA 結構及結構中各螺旋區及環 的特徵如 Figure 1 所示, 其中 acceptor stem 在 3’端通 常含有 CCA-OH 序列,亦是胺基酸所接附的地方。 anticodon Loop 中位於中間 3 個核苷酸則稱之為反密碼 子 (anticodon),也是預測 tRNA 的重點。 現今能預測 tRNA 的工具或方法有 tRNAscan-SE 系統 [5]、運用啟發式演算法 ARAGORN 系統 [7] 或運用圖 形理論來分析反密碼子[6]。tRNAscan-SE 是於 1997 年 被提出,是目前生物學者多數選擇的系統。此系統結 合 快 速 heuristic 演 算 法 及 擁 有 高 敏 感 度 、 選 擇 性 covariance model 及 tRNAscan 和 EufindtRNA 演算法。 在 2003 年一個新的預測 tRNA 程序由 Vickie tsui 等人提出,此方法優勢在於能適用於預測多種 tRNA 種類 且不需選擇種類,其作法根據計算 tRNA 結構折疊的 自由能變化[8]。但 Dean Laslett 等人認為 Vickie tsui 等 人提出的方法並非完善的,之後發表 ARAGORN 系 統。ARAGORN 系統是採用啟發式演算法任意組合 tRNA 結構,再依 A-Box(由 D stem 的 upstream 及 D loop 組成)及 B-Box(由 T arm 組成)各位置的規則來調整並 組織出二級結構,其優點在於能快速完成預測。 本研究則提出一個利用進化式演算法來預測 tRNA 二 級結構的方法,並且能適用於 4 種類的 tRNA,分別為 archaeal, eubacterial, eukaryotic, chloroplasts。在研究中 我們參考 Christian Marck 等人對超過 4000 條 tRNA 序 列 進 行 分 析 [4] , 利 用 分 析 其 三 域 種 類 (archaeal, eubacterial, eukaryotic)序列可能共同擁有的結構及不 可能的結構,以及當要組成 tRNA 三級結構時必要的 核苷酸位置做為判斷條件,並組合出 tRNA 二級結構。 在本文中使用基因演算法 (Genetic Algorithms) 以自 然演變的方式,逐步組合最佳折疊途徑來預測 tRNA 二級結構。但一般基因演算法會有落入區域最佳解問 題,本研究即加入互補式觀念來提升染色體多樣性, 實驗結果顯示此方法能有效克服落入區域最佳解。 Figure 1 tRNA 二級結構圖 2、方法 2.1、基因演算法 達爾文的進化論提及『適者生存,不適者淘汰』,解 釋自然界基本現象。即生物在惡劣環境中為了生存及 適應環境而不斷的進化,並產生適應力更強的下一 代。生物在繁殖過程中染色體會進行交配及突變來改 變基因其組成,使得子代和母代間產生差異性。而基 因演算法是由此理論基礎下所建構,由 John Holland 於 1975 年所提出[10]。本研究針對 tRNA 二級結構預 測所提出之互補式基因演算法,對於一般基因演算法 容易落入區域最佳解問題,藉由產生補數染色體改善 其搜索範圍並提升敏感度,該方法流程如 Figure 2 所 示。步驟將於各小節詳細介紹。 初始族群 調整染色體 長度 交配 取代 是否滿足標準 取得最佳解 是否適應值 連續10次 無提高 DNA 序列 是 調整染色體 長度 突變 選擇 否 互補 否 取代 是 Figure 2 演算法流程圖 2.2、編碼表示法 本研究採用實數型編碼,如 Figure 1 所示,其中 Base0 (位置 0)、D Loop(位置 14~21)、V Loop(位置 44~48)、 CCA-OH(位置 74~76)這三種非固定數量的結構分別以 Gene1、Gene2、Gene3、Gene4表示。如 Figure 3 所示。 Figure 3 編碼示意圖 2.3、初始族群 初始族群以各結構之限制範圍,利用亂數隨機產生各 基因值來進行初始化,其限制範圍為:Base0 為亂數 0 到 1、D Loop 為亂數 7 到 11、V Loop 為亂數 3 到 22、 CCA-OH 為亂數 0 到 1,其中 CCA-OH 結構中是以 3 個核苷酸為一組別,若為 0 則同時不存在,若為 1 則 同時存在,當亂數和大於序列長度則進行微調。微調 方式為隨機選取一種結構來做遞減動作至最小值,並 重新選擇其他結構繼續遞減動作,如 Figure 4 所示。 Figure 4 初始族群示意圖
2.4、適應函數(fitness function) 計算適應函數是以各染色體亂數所產生 tRNA 結構, 再依配對成功數量給予加分動作,但若符合 Table 1 及 Table 2 不可能結構則進行扣分動作,如公式(1)所示, 其中加分權重值為 1 分,而扣分權重值則為 4 分,扣 分值比加分值高其目的是為引導各染色體至最佳解進 一步提升族群品質。換言之,若其中某染色體出現不 可能結構則被其他染色體取代機率將會提升,最後將 所有的分數加總,即為此染色體之適應值。如 Figure 5 所示,有一染色體,首先依照此組合建構出 tRNA 模 型,再將其序列放入對應位置,此時假設 acceptor stem 結構裡,5’端的 7 個核苷酸為 UGACUCG 與依照組合 而放入3’端的 7 個核苷酸 GCAGACC 依序相互配對, 只要達成配對就給予分數 1,如 G-C、C-G、U-A、G-C, 若兩端皆為 A 時因無法形成配對就給予分數 0,如 A-A、C-C,而不符合結構則給予-4 分,如 Table 1 中 1-72 的 UG。適應函數公式如下: 適應函數 = 加分權重值 × 符合結構數量 – 扣分權重值 × 不符合結構數量 ( 1 ) Figure 5 加分適應函數示意圖 2.5、選擇 在本研究中,選擇兩條母體染色體的方式乃採用輪盤 法(Roulette wheel),依據每條染色體適應值計算挑選機 率,其公式如(2)(3),首先計算所有染色體適應值的總 和,並利用公式(2)計算每一個染色體個體的機率,再 利用公式(1)計算每一個染色體累計的機率,最後隨機 產生介於 0 到 1 之間的亂數,若此亂數介於某染色體 機率範圍,則將其染色體挑出,此動作執行 2 次。
i j iP(
j
)
,
,...,P
P
i
11
( 2 )sum
fit
fit( i )
P( j )
( 3 ) 其中 P 為染色體的數量,fit(i)為族群中第 i 個染色體的 適應值,fitsum為族群所有染色體的適應值加總。 Table 1 不可能出現的配對 註:mm 為不配對 核苷酸配對 不符合的配對 1 – 72 UG 8 – 14 GC、CG、AU、GU 10 – 25 UG 11 – 24 GU 15 – 48 CG 18 – 55 CG、AU、UA、UG、mm 19 – 56 CG、UA、UG 32 – 38 GC、GU 53 – 61 CG、UA、UG、mm 54 – 58 GC、CG、AU、GU、UG Table 2 位置上不可能出現的核苷酸 位置 不可能出現 位置 不可能出現 8 A、G 14 G 53 U、C 54 G 55 A、G 57 C 58 G、U、C 61 A、G 2.6、交配及突變 交配方面,採用兩點交配(Two-Point Crossover)方式將 兩條選擇出來之母體染色體,隨機挑選兩交配點,並 於兩交配點間進行基因互換產生子代,如 Figure6(A) 所示。再將所產生的兩條子代染色體進行突變,突變 點為各基因。若該點所得機率小於所設定的突變率, 則進行突變動作,如 Figure6(B)所示。交配目的是讓染 色體保持或增加多樣性,突變則是為了尋找未知染色 體。 Figure 6 (A) 雙點交配示意圖, (B) 突變示意圖 2.7、取代 本研究採用保留菁英方式,將舊族群中的適應函數值最高者保留下來,並淘汰掉最差之 2 條母代染色體並 以新的子代染色體取代,成為新的族群。 2.8、判斷是否進行互補 新的子代經排序後,若最佳適應值連續 10 次沒有改 變,即判斷落入區域最佳解。為有效克服此狀況,本 研究選擇適應值較佳之前 50%染色體進行互補來取代 較差 50%染色體,藉由互補來提升族群多樣性,跳脫 區域最佳解,如 Figure7 所示。實數互補公式如下: selected id min max Complement id
(G
G
)
-
g
g
( 4 ) 在公式中,g
selectedid 為所選擇的基因, 經由公式轉換為補數
g
idComplement,而 Gmax與 Gmin分別為該基因的上限值與下限值。 Figure 7 判斷互補示意圖 3、實驗結果與討論 3.1、資料集來源 1997 年Lowe和Eddy建構的tRNAscan-SE[5]測詴採用 589 條細胞質tRNA的序列,其資料庫是 1995 年發表的 sprinzl database ( 來 源 :
Hftp://ftp.ebi.ac.uk/pub/databases/trna/H)。Sprinzl database 是經實驗證實其正確性且具有註釋的tRNA序列資料 庫,其細分為病毒(VIRUS) 、古細菌(Archaebacteria)、 細 菌 (Eubacteria) 、 Cyanelle 、 葉 綠 粒 (Chloroplast) 、 Cytoplasm、粒線體(Mitochondria)類型。2004 年Dean Laslett和Bjorn Canback發表ARAGORN,其演算法使用 1290 條細胞質的tRNA序列,資料庫則為current Sprinzl
database( 來 源 :
Hhttp://www.old.uni-bayreuth.de/departments/biochemie/s prinzl/trna/H)。本實驗亦採用此current Sprinzl database測 詴以利比較,目前資料庫更新為 2004 年,且新增至 3279 條實驗證明過的tRNA序列。 欲將資料以三域分類來劃分三大類,預先資料庫先分 為原核生物(Prokaryotic)、真核生物(Eukaryota),其中 原核生物包含兩大類群,分別為真細菌(Eubacteria)和 古細菌(Archaebacteria),現今稱細菌(Bacteria)和古菌 (Archaea)。其中,細菌包含真細菌共 686 條序列,古 菌 則 包 含 古 細 菌 共 161 條 序 列 。 真 核 生 物 包 含 Cytoplasm、Cyanelle 共 443 條序列。此外本文多增加 葉綠粒(Chloroplast)種類來預測其敏感度,共 376 條序 列,故本次實驗預測之 tRNA 序列共 1666 條序列。 3.2、參數設定 基因演算法之參數部份,迭代次數 G=300,母體族群 數量 P=30,基因池數量 P’=4,交配率 X-rate=1.0,突 變率 M-rate=1/P[11]。tRNAscan-SE 系統設定部份,預 測 Archaeal 及 Bacteria 使用-A 及-B,Eukaryota 使用-E, chloroplast 使用-M/C。
3.2、實驗結果
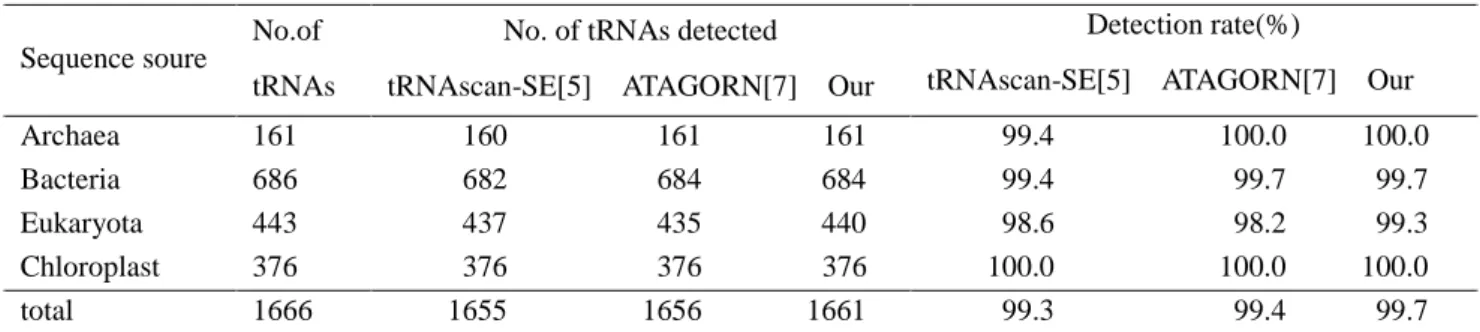
本次實驗中經由 Sprinzl 所建構之 database 來測詴敏感 度,如 Table 3 所示,其結果顯示本方法在 Archaea 種 類稍微較 tRNAscan-SE 和 ATAGORN 系統的敏感度高 (100 versus 99.4 versus 100%)。Bacteria 種類稍微較 tRNAscan-SE 和 ATAGORN 系統敏感度高(99.7 versus 99.4 versus 99.7%)。Eukaryota 種類比 tRNAscan-SE 和 ATAGORN 系統的敏感度高(99.3 versus 98.6 versus 98.2%)。在 Chloroplast 種類則皆為 100%,整體敏感度 皆較 tRNAscan-SE 和 ATAGORN 系統高(99.7 versus 99.3 versus 99.4%) 。 在 錯 誤 預 測 方 面 , 我 們 利 用
tRNAdb[9] 資 料 庫
(http://trnadb.bioinf.uni-leipzig.de/Search) 所 儲 存 的 Sprinzl database 來觀察預測錯誤之序列結構,發現 Bacteria 種類中 Sprinzl ID:DZ1430,11 與 24 號核苷 酸配對出現 GU 配對,以及 8 號位置出現鹼基 A 特例, Sprinzl ID:DZ2000,11 與 24 號核苷酸配對出現 GU 配對,在 Eukaryota 種類中 Sprinzl ID:DQ8510、 DA9360、DG9300 皆在 D Loop 結構出現非標準結構, 這 3 條序列並沒有 D Loop 的結構。
3.3、討論
序列結構,整理出 3 域分類中共同可能的結構及不可 能的結構,來建構出三葉草結構並預測反密碼子,並 利用此方法測詴 Chloroplast 種類。結果顯示能有效的 預測 Chloroplast,整體方面獲得 99.7%敏感度,而錯誤 的預測方面,出現極少數的例外結構,尤其以 Eukaryota 的 3 條缺乏 D Loop 結構,在其他預測軟體皆無法預 測。但根據文獻[4]所知,D Loop 中 18 及 19 號位置分 別與 T Loop 的 55 及 56 位置互補折疊並形成三級結構 關鍵,故屬不正規結構。而在比對其他預測正確的 tRNA 序列,發現有著其他非典型結構存在,例如不該 變動的 T Loop 及 acceptor stem 皆出現非典型結構,但 反密碼子則為正確。於對這些非典型結構進行分析, 發現許多不應該存在之結構出現,但本方法所預測亦 之結構較為完整(例如各 stem 的 base pair 數量),然而 利用其他預測系統[5][7],其視覺化結構發現大多與本 方法結構類似,有些預測其結構差異度很大,但皆為 正確預測。而對某序列進行重複預測,則發現會有結 構不同但反密碼子正確的情形且皆遵守規則。總而言 之,雖然架構皆不同,但其預測皆正確,故此 tRNA 模型及規則為正確之架構,另外在此次實驗選擇之資 料庫皆無包含 intron 序列,故預測包含 intron 的序列並 不在本次實驗列入考慮。儘管如此,此方法優點為適 用多種類型的 tRNA,使用者不需要選擇序列種類,皆 較其他方法有效達到正確預測及擴充彈性佳,若未來 有文獻指出本文固定結構為可調整之結構,只需再針 對其結構,擴充染色體之基因體個數並限制結構範圍 即可。 本方法之關鍵在於調整序列長度的步驟,其原因為 fitness 是以染色體基因值及其他固定結構所組成的 tRNA 模型為基底,再放入 tRNA 序列到各自位置。以 此評估分數。若發生模型長度大於需預測之 tRNA 序 列長度,則會發生模型中某些位置呈現無鹼基狀態, 導致 fitness 無法正確評估,故必須調整可變動結構長 度。此外,模型最短長度為 69 個鹼基,故序列長度必 須大於 68 個鹼基長度,為極重要之需要。由上述得知 模型長度必須小於 tRNA 序列長度,故在初始化步驟 隨機給予各長度總和,若超出序列長度,即依照多出 的序列長度之隨機刪除長度,其靈感來自於背包問題 [12],要預測的序列長度為背包大小,而各結構長度為 重量,當放入後所計算出的適應值為總價值。 而在測詴過程中發現對某序列重複預測時,會產生適 應值不同的解。其原因為一般基因演算法中是以交配 運算來進行演化,故在經過多次迭代後,會產生染色 體的編碼越來越相似的情形(如 Figure 8 左邊),導致後 來大多數的迭代會產生相同的最佳解,而有可能落入 區域最佳解,且無法找出其最佳的結構排列,造成反 密碼子預測失誤。為避免染色體編碼相似度提高,本 方法利用互補式觀念增加染色體的多樣性,以跳脫區 域最佳解,若適應值多次無法改善(即使目前已經是最 佳解),即假設已落入區域最佳解,並藉由實數互補方 式跳脫。如 Figure 8,假如有四條染色體,此時最佳的 染色體為 0、1、4、11、0、5,適應值為 20,在連續 10 次迭代後皆沒有改善,就對前 50%染色體進行互補 動作並做排序動作,最後經由隨機選擇交配,來達到 多樣性(如 Figure 8 右邊),其結果不但能有效克服區域 最佳解問題,並可增強求解效能。 Figure 8 補數跳脫區域最佳解示意圖 4、結論 本研究提出一個以互補式基因演算法於預測 tRNA 二 級結構之方法,以二級及三級結構可能出現的結構為 關鍵完成預測。在實驗中因一般基因演算法求解過程 中容易落入區域最佳解,故藉由取補數方式產生編碼 具多樣性的初始染色體群族及求解過程中的母體染色 體族群。實驗結果顯示能有效產生所期望的排列組 合,並找出正確反密碼子,倘若 tRNA 屬於非典型之 結構,則會導致錯誤率提升。在未來研究方面期望增 加預測包含 intron 的序列及由 genome 序列中辨識出
tRNA 序列,進而達到更完善之預測系統。
參考文獻
[1] Holley R.W. “Structure of an alanine transfer ribonucleic acid,” JAMA 194, pp.868-871, 1965. [2] Ernard S. D., Gerson K, Ellen K. T, Robert W.
Holley, "Primary structure of wheat germ phenylalanine transfer RNA*," Biochemistry, Vol.62, January 21, pp.941-945, 1969.
[3] Dirheimer G., Keith G., Dumas P., Westhof E. “Primary secondary and tertiary structures of tRNAs,” ln: Söll D, RajBhandary U, eds.tRNA: Structure, biosynthesis, and function. Washington DC:ASM Press. pp.93-126, 1995.
[4] Christian M. and Henri G. “tRNomics:Analysis of tRNA genes from 50 genomes of Eukarya, Archaea, and Bacteria reveals anticodon-sparing strategies and domain-specific features,” Bioinformatics, Vol.8, pp.1189-1232, 2002.
[5] Todd M.L. and Sean R.E.* “tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence,” Nucleic Acids Research, Vol.25, No.5, pp.955-964, 1997.
[6] Johan F.G., Clara I.B., Edgar E.D.. “tRNA structure from a graph and quantum theoretical perspective,”
Journal of Theoretical Biology 240, pp.574-582, 2006.
[7] Dean L. and Bjorn C. “ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequence,” Nucleic Acids Research, Vol. 32, No.1 pp.11-16, 2004.
[8] Vickie T., Tom M. and David A. “A novel method for finding tRNA genes,” RNA 2003 9: pp.507-517, 2003.
[9] Frank J., Mario M., Roland K.H., Mathias S., Peter F.S. and Joern P.* “tRNAdb 2009: compilation of tRNA sequences and tRNA genes,” Nucleic Acids Research, Vol. 37, pp.D159–D162, 2009.
[10] Holland, J.H. “Adaptation in Nature and Artificial Systems,” MIT Press, 1992.
[11] Erol O.K., Eksin I. “New optimization method: Big Bang-Big Crunch?,” Advances in Engineering Software, Vol. 37, pp.106-111, 2006.
[12] Kuk-Hyun H., Kui-Hong P., Chi-Ho L., Jong-Hwan K. “Parallel Quantum-inspired Genetic Algorithm for Combinatorial Optimization Problem,” IEEE Congress on Evolutionary Computation Seoul, pp.1422–1429, 2001.
Table 3 tRNA 預測正確率 註:tRNAscan-SE version 1.23. Sequence soure
No.of No. of tRNAs detected Detection rate(%)
tRNAs tRNAscan-SE[5] ATAGORN[7] Our tRNAscan-SE[5] ATAGORN[7] Our Archaea 161 160 161 161 99.4 100.0 100.0 Bacteria 686 682 684 684 99.4 99.7 99.7 Eukaryota 443 437 435 440 98.6 98.2 99.3 Chloroplast 376 376 376 376 100.0 100.0 100.0 total 1666 1655 1656 1661 99.3 99.4 99.7