Development of a feasible assay for the detection of GAA mutations in
patients with Pompe disease
Tze-Kiong Er
a,b,1, Chih-Chieh Chen
c,d,e,1, Yin-Hsiu Chien
f, Wen-Chen Liang
g,h,
Tzu-Min Kan
g, Yuh-Jyh Jong
a,g,i,j,⁎
a
Division of Molecular Diagnostics, Department of Laboratory Medicine, Kaohsiung Medical University Hospital, Kaohsiung Medical University, 100 Shih-Chuan 1st Rd., Kaohsiung 80708, Taiwan
b
Translational Research Center, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung, Taiwan
c
Institute of Bioinformatics and Systems Biology, College of Biological Science and Technology, National Chiao Tung University, 75 Bo-Ai Street, Hsinchu 30068, Taiwan
d
Center for Lipid Biosciences, Kaohsiung Medical University Hospital, 100 Shih-Chuan 1st Rd., Kaohsiung 80708, Taiwan
eBiomedical Technology and Device Research Laboratories, Industrial Technology Research Institute, 195, Sec. 4, Chung Hsing Rd., Hsinchu 31040, Taiwan f
Department of Medical Genetics and Pediatrics, National Taiwan University Hospital, Taipei 10041, Taiwan
g
Department of Pediatrics, Kaohsiung Medical University Hospital, 100 Shih-Chuan 1st Rd., Kaohsiung 80708, Taiwan
h
Department of Pediatrics, School of Medicine, College of Medicine, Kaohsiung Medical University, 100 Shih-Chuan 1st Rd., Kaohsiung 80708, Taiwan
i
Department of Biological Science and Technology, College of Biological Science and Technology, National Chiao Tung University, Hsinchu, Taiwan
j
Graduate Institute of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
a b s t r a c t
a r t i c l e i n f o
Article history: Received 24 July 2013
Received in revised form 30 September 2013 Accepted 16 October 2013
Available online 24 October 2013
Keywords:
Acidα-glucosidase (GAA) gene High resolution melting Mutation analysis Pompe disease
Background: Pompe disease is an inherited autosomal recessive deficiency of acid α-glucosidase (GAA) and is due to pathogenic sequence variants in the corresponding GAA gene. While the analysis of enzyme activity remains the diagnostic test of choice for individuals with Pompe disease, mutation analysis remains for establishing a de-finitive diagnosis.
Methods: High resolution melting (HRM) analysis was performed to screen GAA mutations. Genomic DNA was extracted from peripheral blood samples of the two patients with Pompe disease and 250 normal controls. Exons 2 through 20 of the GAA gene were screened by the HRM analysis. The results were subsequently con-firmed by direct sequencing.
Results: This assay proved to be feasible in detecting seven known (c.2TNC, c.1726GNA, c.1845GNA, c.1935CNA, c.1958CNA, c.2238GNC, and c.2815_2816del) GAA mutations. Each mutation could be readily and accurately iden-tified in the difference plot curves. We estimated the carrier frequency of the most common mutation, c.1935GNA (p.D645E), in the Taiwanese population to be 0.2%.
Conclusions: In clinical practice, we suggest that HRM analysis is assumed as a fast and reliable method for screen-ing GAA gene mutations especially the most common mutations which are responsible for Pompe disease among the Taiwanese populations.
© 2013 Elsevier B.V. All rights reserved.
1. Introduction
Pompe disease (glycogen storage disease type II, acid maltase de-ficiency, OMIM 232300) is an autosomal recessive progressive mus-cular disorder caused by the deficiency of acid α-glucosidase (GAA; EC.3.2.10.20) that results in impaired lysosomal degradation and the
accumulation of glycogen. The lysosomal glycogen accumulated in many tissues such as skeletal, cardiac, and smooth muscles[1]. Pompe disease is classified into infantile, juvenile, and adult types based on clinical presentation and the onset of the disorder. The clinical manifes-tations of the infantile form are cardiomyopathy with cardiomegaly and rapidly progressive myopathy[1]. Late-onset Pompe disease is the re-sult of a partial deficiency of GAA. The primary symptom is muscle weakness progressing to respiratory weakness. They may develop re-spiratory failure at a later stage during the course of the disease[2,3]. In general, GAA activity correlates with the age at onset and severity of disease. Infantile patients have less than 1% GAA activity, and late-onset patients have 2–40% of normal GAA activity in cultured skin fibro-blasts[4]. The estimated frequency of Pompe disease in southern China and Taiwan newborns is 1 in 20,000–40,000[5]. The combined inci-dence of all forms of Pompe disease is approximately 1:40,000[6].
Measurement of GAA activity in skinfibroblasts is the current gold standard for the diagnosis of Pompe disease. Recently, isolation of GAA
Abbreviations: GAA, acidα-glucosidase; HRM, high resolution melting; DBS, dried blood spot; PCR, polymerase chain reaction; WCN, weighted contact number; DHPLC, de-naturing high performance liquid chromatography.
⁎ Corresponding author at: Department and Institute of Biological Science and Technology, College of Biological Science and Technology, National Chiao Tung University, 75 Bo-Ai Street, Hsinchu, Taiwan. Graduate Institute of Medicine, College of Medicine, Kaohsiung Medical University, 100, Shih-Chuan 1st Road, Kaohsiung, 80708, Taiwan. Tel.: +886 3 5712121 56900; fax: +886 3 5729288.
E-mail addresses:tzekiong@totalbb.net.tw(T.-K. Er),chieh.bi91g@nctu.edu.tw
(C.-C. Chen),chienyh@ntu.edu.tw(Y.-H. Chien),peggieliang@yahoo.com.tw(W.-C. Liang),
nekokan1983@yahoo.com.tw(T.-M. Kan),yjjong@gap.kmu.edu.tw(Y.-J. Jong).
1These authors contribute equally to this article.
0009-8981/$– see front matter © 2013 Elsevier B.V. All rights reserved.
http://dx.doi.org/10.1016/j.cca.2013.10.013
Contents lists available atScienceDirect
Clinica Chimica Acta
from the dried blood spot (DBS) extracts has been developed for GAA activity assay. These novel DBS methods are not only suitable for new-born screening of Pompe disease, but also provide a quick and noninva-sive diagnostic test for Pompe disease [1]. While enzyme activity analysis remains the diagnostic test of choice for individuals with Pompe disease, mutation analysis remains important. It is particularly useful in identifying carriers when a familial mutation is known. In ad-dition, there are also some common mutations with useful genotype– phenotype correlations[3,7]. On the other hand, the availability of en-zyme replacement therapy has increased the demand for rapid and more sophisticated diagnosis at both enzymatic and molecular level[8]. The human structural gene encoding GAA is located at chromosome 17q25.2–q25.3 and contains 20 exons, the first of which is non-coding
[9]. The GAA cDNA is over 3.6 kb in length and with 2856 nucleotides of coding sequence. The resulting product is a protein of 952 amino acids[10,11]. The start codon begins at position 33 of exon 2[9]. Muta-tions are randomly spread over the whole gene and some mutaMuta-tions ap-pear with considerable frequency in distinct ethnic groups[12].
High-resolution melting (HRM) analysis is a mutation-scanning technology and offers considerable time and cost savings compared with screening methods. HRM analysis has recently been tested in a va-riety of clinical mutation-scanning and genotyping applications and is shown to be sensitive, cost-effective, and economical. Unlike other scan-ning method (such as sequencing), it is time-consuming and labor in-tensive. HRM analysis offers a faster and more convenient closed-tube method of assessing the presence of mutations and gives a result that can be further investigated if it is of interest. Besides, the closed-tube system reduces the risk of contamination. van der Stoep N et al.[13]
compared HRM analysis with sequence analysis, which is the current gold standard for mutation detection in most diagnostic laboratories. They reported that HRM analysis is a highly sensitive method with a sensitivity of 100% in detecting all heterozygous mutations. Similarly, a meta-analysis[14]indicated that HRM analysis is a highly sensitive, simple and low-cost test to detect human disease-associated mutations, especially for samples with mutations of low incidence. Recently, our re-view article[15]also discussed about the effectiveness and limitations of HRM analysis in the diagnosis of genetic disorders including autoso-mal recessive, dominant, and X-linked disorders.
Identification of mutations in the GAA coding sequence is advan-tageous that it can provide a noninvasive method for the con firma-tion of the clinical and biochemical diagnosis. In addifirma-tion, it is useful in identifying rare or novel mutations and facilitates genotype– phenotype correlations. Therefore, this present study aimed to as-sess the value of the HRM analysis using real-time polymerase chain reaction (PCR) (LightCycler® 480; Roche Applied Science) for scanning GAA gene mutations in patients with Pompe disease. 2. Materials and methods
2.1. Diagnosis of patients
Confirmatory diagnosis included both biochemical and molecu-lar tests. GAA activity in DBS was measured using artificial substrate 4-methylumbelliferyl-α-D-glucopyrosidase (Fluka Chemical Corp, Ronkonkoma, NY, USA) and the nature substrate glycogen. Genomic DNA was isolated using the proteinase K and QIAamp® mini DNA ex-traction kit (QIAGEN) according to the manufacturer's protocol. The quality and quantity of the extracted DNA samples were measured using the Nano-200 Nucleic Acid Analyzer (MEDCLUB, Tainan, Taiwan). This study was approved by the Institutional Review Board (IRB) of Kaohsiung Medical University Hospital (KMUHIRB-20130027). 2.2. Optimization of HRM curve analysis
Seven DNA samples with previously identified GAA mutations were used for the optimization and validation of HRM. Optimized HRM
conditions were applied for further screening of GAA mutations in two additional Pompe patients from our medical center. Meanwhile, we also recruited 250 healthy subjects in order to estimate the allele fre-quency of the most common GAA gene mutation, c.1935CNA (p.D645E). 2.3. Assay design and PCR conditions
Good amplicon design is essential to obtain a robust and reproduc-ible HRM analysis. The difference between wild-type and heterozygote curves became smaller and more difficult to differentiate when the product length was increased[16]. Besides, extra care is needed to de-sign PCR conditions to avoid primer dimers and non-specific amplifica-tion in HRM analysis. In this study, all 25 pairs of primers for HRM analysis were newly designed using Primer3 software (Supplementary data 1). For exon 2 and exon 4, six sets and two sets of primers were used to amplify the exons in overlapping segments, respectively. All synthesized primers were of standard quality in molecular biology quality (Protech Technology Enterprise Co., Ltd, Taiwan).
2.4. The HRM technique
PCR reactions were carried out in duplicate in 10μL final volume using the LightCycler® 480 High-Resolution Melting Master (reference 04909631001, Roche Diagnostics) 1× buffer, containing Taq polymerase, nucleotides and the dye ResoLight, and 50ng DNA. The primers and MgCl2 were used at a concentration of 0.25μM and 2.5 mM, respectively for de-tecting the GAA gene mutations. The HRM assays were conducted using the LightCycler® 480 Instrument (Roche Diagnostics) provided with the software LightCycler® 480 Gene-Scanning Software Version 1.5 (Roche Diagnostics). The PCR program required a SYBR Green Ifilter (533 nm), and it consisted of an initial denaturation–activation step at 95 °C for 10min, followed by a 45-cycle program (denaturation at 95°C for 15s, an-nealing at 58 °C or 60 °C for 15 s and elongation at 72 °C for 15 s with the reading of thefluorescence; acquisition mode: single). The melting pro-gram included three steps: (a) denaturalization at 95 °C for 1 min, (b) re-naturation at 40°C for 1min, and (c) subsequent melting that consists of a continuousfluorescent reading from 60 to 90°C at the rate of 25 acquisi-tions per degree centigrade. The difference plot curves of the duplicate for each DNA sample must be reproducible both in shape and peak height. 2.5. Sequencing of GAA exons 2–20
Exons 2 through 20 of the GAA gene were amplified by PCR as pre-viously described with some modified reaction conditions[17]. Each locus was amplified in a 20 μL volume including buffer [60 mM Tris– SO4(pH 8.9), 18 mM (NH4)2SO4, 2 mM MgSO4], 0.2 mM dNTP, 0.2μM forward PCR primer, 0.2μM reverse PCR primer, 50–120 ng genomic DNA and 1 U of AccuPrime Taq DNA polymerase (Invitrogen Canada Inc., Ont.). The DNA was amplified using the following amplification conditions: 94 °C for 3 min; 35 cycles of 92 °C for 15 s, 50 °C for 15 s, and 68 °C for 1 min. After PCR, the samples were purified using the Geneaid Gel/PCR DNA fragments extraction kit. The sequence reac-tion was performed in afinal volume of 10 μL, comprising 1 μL of the purified PCR product, 2.5 μM of each PCR primer and 2 μL of ABI PRISM terminator cycle sequencing kit v3.1 (Applied Biosystems). The sequencing program is a 25-cycle PCR program (denaturation at 96 °C for 10 s; annealing at 50 °C for 5 s and elongation at 60 °C for 4 min). The sequence detection was performed in the ABI Prism 3130 Genetic Analyzer (Applied Biosystems).
2.6. Weighted contact number (WCN) and the sequence conservation profiles
Protein packing is described using weighted contact number[18]. The weighted contact number (WCN) of an atom is the sum of the inverse square of the distances between it and other atoms. The sequence-specific
conservation scores were computed following the protocol of CONSURF
[19], based on the phylogeny of the sequences. The conservation scores were normalized to the corresponding z-scores in such a way that the average conservation score is zero and the standard deviation is one. The series of the conservation scores of a sequence is referred to as its conservation profile. Notably, in the conservation profile of a protein, the residue of a lower conservation score is more conserved than that of a higher conservation score[20].
2.7. Structure prediction of human GAA protein
Three-dimensional (3D) structures of the human GAA were predict-ed using the (PS)2server[21]. (PS)2automatically uses an effective con-sensus strategy. The above mentioned strategy combines structural- and profile-based[22]comparison methods for both template selection and target-template alignment. (PS)2server selected the Human intestinal maltase-glucoamylase (PDB entry: 2QMJ)[23]as the model template. 3. Results
3.1. Diagnosis and clinical manifestations in patients 3.1.1. Patient 1
A 5 year-old boy was presented with a gross motor developmental delay since infantile. He started to head control when he was 5 months old. Then, he could sit alone at the age of 9 months and be able to walk independently at the age of 1year and 9months. He could not jump high even when he was 3-years-old. He also had difficulty in climbing up
stairs and running. At hisfirst visit to our clinic at the age of 3 years, the patient showed positive Gowers' sign and deep tendon reflexes in his lower extremities were absent. Creatine kinase levels were mildly elevated. Muscle biopsy revealed intracytoplasmic huge au-tophagic vacuoles with high acid phosphatase activity. Glycogen storage disease type II was thus suspected; DBS were collected for GAA activity assay and it was confirmed by Pompe disease due to the deficient activity of acid α-glucosidase (GAA) in net GAA (with inhibitor), 0.29μmol/L/h (reference range: 12.22 ± 5.88 μmol/L/h). Mutational analysis showed that he harbored compound heterozy-gous mutations (p.R594H/p.D645E) (Fig. 1). His muscle biopsy re-sults were shown in Fig. S1(C) & (D).
3.1.2. Patient 2
An 11 year-old girl presented with slowly progressive lower extrem-ity weakness since the age of 10years. She had mild difficulty in climbing upstairs and could not do sit-ups at all. On herfirst visit to our clinic, marked abdominal and tongue weakness were observed. Proximal mus-cle weakness was relatively mild but Gowers' sign was positive. Creatine kinase levels were mildly increased to 300 IU/L and muscle biopsy was thus performed. Cytoplasmic bodies with high acid phosphatase activity were found in some musclefibers. Late-onset glycogen storage disease type II was therefore highly suspected. DBS were collected for GAA activ-ity assay. Pompe disease was confirmed by deficient activity of acid α-glucosidase (GAA) in net GAA (with inhibitor), 0.26μmol/L/h (reference range: 12.22 ± 5.88μmol/L/h). Mutational analysis showed that she har-bored compound heterozygous mutations (p.R190H/p.D645E) (Fig. 2). Her muscle biopsy results were shown in Fig. S1(A) & (B).
Fig. 1. The melting profiles of the GAA mutations in patient 1. (A) Normalized and temperature-shifted difference plots, the melting profile of c.1935CNA; (B) normalized plots, and nor-malized and temperature-shifted difference plots, the melting profile of c.569GNA; electropherogram of mutation: (C) c.1935CNA; Electropherogram of mutation: (D) c.569GNA.
3.2. Optimization of HRM curve analysis
Seven known GAA mutations (c.2TNC, c.1726GNA, c.1845GNA, c.1935CNA, c.1958CNA, c.2238GNC, and c.2815_2816del) were used to assess the sensitivity of the HRM method for mutation scanning. Complete GAA mutational screening required investigation of exon 2 through exon 20. Twentyfive PCR amplicons (169–377 bp) were evaluated with an average of 214 bp to cover exon 2 through exon 20 using LightCycler® system.
The normalized and temperature-shifted difference plots, the melt-ing profile of GAA mutations c.2TNC, c.1845GNA, and c.2815_2816del are shown in Figs. S1, S2, and S3, respectively. The results were con-firmed by direct sequencing.Fig. 3shows that c.1726GNA (heterozy-gous, homozy(heterozy-gous, and wild-type) are clearly described in the normalized temperature-shifted difference plots. Fig. 4 shows that c.1935CNA (heterozygous, homozygous, and wild-type) are clearly de-scribed in the normalized temperature-shifted difference plots.Fig. 5
shows that c.2238GNA (heterozygous, homozygous, and wild-type) are clearly described in the normalized temperature-shifted difference plots. All results were confirmed by direct sequencing. Intriguingly, we identified a non-pathogenic mutation among the Taiwanese population, c.1920TNG (p.P640P)[24]in normal populations (Fig. S4).
3.3. Allele frequency of the most common mutation, c.1935CNA (p.D645E), among the Taiwanese population
In the current study, we recruited 250 healthy Taiwanese sub-jects. We screened the subjects for the most common mutation,
c.1935CNA (p.D645E), using HRM analysis and computed the allele frequency for this mutant site. The results were confirmed by direct sequencing. We found that only one subject harbors a heterozygous mutation, c.1935CNA (p.D645E). The estimated allele frequency was 0.002 for c.1935CNA (p.D645E).
3.4. Structure packing density and sequence conservation analyses of the GAA protein
The protein packing density is described using weighted contact number (WCN)[25]. The larger the WCN of a residue, the more packed its environment. The reciprocal of the WCN (rWCN) profile is used to be compared with the sequence conservation profile. The conservation scores of a protein were calculated using ConSurf[19]. These scores are a relative measure of evolutionary conservation at each sequence site of target chain. The lowest score represents the most conserved position in a protein. Qualitatively, the residues of larger WCNs (or in more packed environments) are more con-served than those of smaller contact WCNs (or in less packed envi-ronments). Indeed,Fig. 6demonstrates that rWCN profile (dotted line) and the conservation profile (solid line) overlap extremely well in the GAA protein. The recent study shows that the catalytic site residues tend to be located in regions of high packing density
[26]. Our results demonstrate that these mutations (p.R190H, p.G576S, p.R594H, p.D645E, and p.W746C) are conserved residues and also located in the more packed environments. Therefore, it is reasonable to assume that these residues are highly associated with the functionality of the GAA protein.
Fig. 2. The melting profiles of the GAA mutations in patient 2. (A) Normalized and temperature-shifted difference plots, the melting profile of c.1935CNA; (B) normalized plots, and nor-malized and temperature-shifted difference plots, the melting profile of c.1781GNA; electropherogram of mutation: (C) c.1935CNA; electropherogram of mutation: (D) c.1781GNA.
4. Discussion
In this study, we have successfully established a diagnostic strategy by HRM analysis for identifying seven known (c.2TNC, c.1726GNA, c.1845GNA, c.1935CNA, c.1958CNA, c.2238GNC, and c.2815_2816del) GAA mutations. We have also identified two previously described
mutations, c.569GNA (p.R190H)[27]and c.1781GNA (p.R594H) in our patients[28].
To date, approximately 453 different sequence variants in the GAA gene are listed in the Pompe disease mutation database
(www.pompecenter.nl). Numerous sequence variants have been
demonstrated to be pathogenic. The most frequent mutation
Fig. 4. Optimization of HRM curve analysis specific for c.1935CNA. (A) Normalized and temperature-shifted difference plots, the melting profile of c.1935CNA (homozygous mutation, het-erozygous mutation, and wild-type) and c.1935CNA + c.1958CNA; electropherogram of mutation: (B) c.1935GNA (hethet-erozygous mutation); (C) c.1935GNA (hethet-erozygous mutation); (D) c.1958CNA (heterozygous mutation).
Fig. 3. Optimization of HRM curve analysis specific for c.1726GNA. (A) Normalized and temperature-shifted difference plots, the melting profile of c.1726GNA (homozygous mutation, het-erozygous mutation, and wild-type); electropherogram of mutation: (B) c.1726GNA (hethet-erozygous mutation); (C) c.1726GNA (hethet-erozygous mutation).
among late-onset Pompe disease is the leaky c.-32-13TNG, which gives rise to alternatively spliced transcripts, including a deletion of thefirst coding exon, but still allows the production of a low amount of normally processed mRNA[1]. The c.-32-13TNG muta-tion was found over 40% patients of Caucasian origin[29–33]. In contrast, the c.-32-13TNG mutation has not been found among Asian population and this may indicate that the mutation shows ethnic specificity. Among the Taiwanese population, Yang et al.
[34] revealed that two dual mutations in the GAA gene c.1935CNA; c.1726GNA and c.2238GNC; c.1726GNA represented 66.5% of the mutated chromosomes. Besides, Wan et al. [24]
demonstrated that, the c.1935CNA (p.D645E) mutation was iden-tified in 57.7% of both infantile and juvenile-onset GSD II. The au-thor suggested that the c.1935CNA (p.D645E) mutation should be detectedfirst when one suspects a GSD II patient. The mutation, c.1935CNA (p.D645E) has also been observed in up to 80% of infantile
cases. It is associated with a haplotype and suggested as a founder ef-fect[35]. Meanwhile, Amarinthnukrowh et al.[36]revealed that the mutation, c.1935CNA (p.D645E) accounts for 80% of the GAA muta-tions in Thai patients with infantile-onset Pompe disease. In the cur-rent study, both of the patients were compound heterozygous for this mutation. Notably, the reported mutation, c.1781GNA (p.R594H), is probably pathogenic because it involves the conserved residue during the evolution suggesting its essential functional role
(Fig. 6). However, larger scale control experiment or further
func-tional experiment may be necessary.
It should be noted that, the pseudodeficiency mutation, c.1726GNA (p.G576S), was found to be associated with severe mutation. The allele frequency of pseudodeficiency mutation among Taiwanese population is 14.5%[37]. It is far higher than that of European and sub-Saharan/African populations[38]. Also, the pseudodeficiency mutation may decrease the residual GAA activity of another mutation
Fig. 5. Optimization of HRM curve analysis specific for c.2238GNC. (A) Normalized and temperature-shifted difference plots, the melting profile of c.2238GNC (homozygous mutation, het-erozygous mutation, and wild-type); electropherogram of mutation: (B) c.2238GNC (hethet-erozygous mutation); (C) c.2238GNC (hethet-erozygous mutation).
Fig. 6. The rWCN profile (dotted line) and ConSurf profile (solid line) of the GAA protein. The GAA mutations are marked in red circles. Both the rWCN and the conservation scores are normalized to their respective z-scores.
[39]. This may be the reason that Chinese patients with later-onset Pompe disease have earlier symptom onset than white patients. Indeed, both patients harbored the pseudodeficiency mutation having earlier symptom in the present study (Fig. S5). Aforementioned, two dual mutations in the GAA gene c.1935CNA; c.1726GNA and c.2238GNC; c.1726GNA were represented in 66.5% of the mutated chromosomes in the Taiwanese population[34]. On the other hand, the c.2238GNC (p.W746C) was shown to be associated with juvenile-onset GSD II and this mutation leads to a moderate disease phenotype. In this study, we designed three of primer-specific pairs for identifying c.1726GNA (p.G576S), c.1935CNA (p.D645E), and c.2238GNC (p.W746S) using HRM analysis (Supplementary data-1). The melting curve data showed the difference in normalized temperature-shifted data between the homozygous mutation, heterozygous mutation, and wild-type samples (Figs. 3–5). Once Pompe disease is suspected, physicians can screen these GAA mutations using HRM analysis.
Recently, Lobato et al.[32]developed the real-time PCR only for the detection of c.-32TNG (IVS1-13TNG) mutation of Pompe disease from DBS specimen. Besides, Pittis et al.[31]revealed 28 polymorphisms spread over the GAA gene using denaturing high performance liquid chromatography (DHPLC) analysis. The authors indicated that the c.-32-13TNG was the most frequent mutation, present as compound heterozygote in Italian population. Besides, the RT-PCR and long range-RT-PCR have also been used for the identification of GAA mu-tation in Pompe disease[34,40]. Notably, Chou et al.[41] demon-strated that HRM analysis had better sensitivity and specificity than DHPLC with the added advantages that some homozygous sequence alterations could be identified. Indeed, we could differentiate the heterozygous, homozygous, and wild-type DNA samples in the cur-rent study (Figs. 3–5). Till very recently, most screening approaches are now utilizing the more sensitive techniques of massively paral-lel sequencing. It has revolutionized genomics, providing a wide range of novel application on a high throughput, genome wide level. Although massively parallel sequencing has become the promising tool in genetic and genomic analysis, this approach is more expensive and the large amount of bioinformatics data creates informatics challenges. Meanwhile, it is not affordable for a small to the middle scale laboratory. In contrast, equipment, labor, reagent, and supply costs for analysis using the HRM analysis are substan-tially lower than the costs associated with analysis using massively parallel sequencing[42]. The HRM analysis is faster and less expen-sive than next-generation sequencing, facilitating rapid analysis of disease-causing mutations especially the high frequency of hotspot mutations.
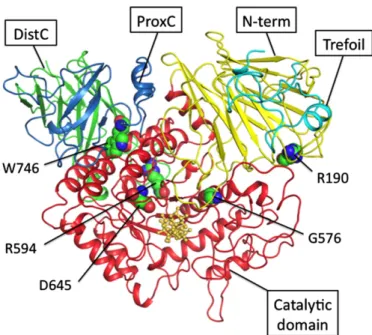
In the current study, we also intended to model the proteins encoded by GAA to understand the structural implications of the mu-tations identified in this study. The atomic coordinates of the model structure were predicted using maltase-glucoamylase (PDB entry: 2QMJ)[23]as the template. The model of human GAA protein is shown inFig. 7. The mutations of GAA are spread over the protein structure and distributed from the core to the surface of the enzyme molecule. A study showed that the substitution of G576 with S causes a small conformational change of side chains and does not af-fect the active site of the enzyme. The expressed protein may be un-stable but exhibits considerable residual activity and normal affinity for substrates[39]. The structure analyses also reveal that both resi-dues R190 and R594 have the salt bridge interactions with resiresi-dues D253 and D860, respectively. A salt bridge is actually a combination of two non-covalent interactions: hydrogen bonding and electrostat-ic interactions. This is commonly observed to contribute stability to the proteins. Therefore, the substitution R190 with H or substitution D594 with H may reduce the salt bridge interaction and then desta-bilize the structure of GAA. The D645 is a conserved residue and is located at the catalytic domain. As previously described, the D645E substitution results in a decrease in enzyme activity and ac-counts for the observed defects in transport, phosphorylation and,
proteolytic processing of the newly synthesizedα-glucosidase pre-cursor of the patient[43]. The W746 residue is located at the proxi-mal C terminal. Tryptophan (W) is unique in terms of chemistry and size. The substitution of W746 to S, a polar uncharged size change, may lead to a deleterious effect on protein function and structure.
In summary, we have demonstrated that HRM for mutation scan-ning of the GAA in patients with Pompe disease is a feasible and ef fi-cient method. This method differentiated all 7 known mutations (c.2TNC, c.1726GNA, c.1845GNA, c.1935CNA, c.1958CNA, c.2238GNC, and c.2815_2816del). Accordingly, HRM analysis is proved to be a workable, sensitive and in-tube methodology to scan GAA mutations in clinical practice. As aforementioned, the mutational analysis serves as an important role in establishing a definitive diagnosis especially in genotype–phenotype correlations in patients with Pompe disease. Therefore, we suggest that patients who are suspected of having Pompe disease should receive GAA gene analysis via HRM and direct se-quencing for further diagnosis and appropriate treatments.
Supplementary data to this article can be found online athttp://dx. doi.org/10.1016/j.cca.2013.10.013.
Competing interests
The authors report no conflict of interest.
Authors' contributions
Concept and design of the experiments: TKE, CCC, YHC, WCL, and YJJ. Perform the experiments: TKE and CCC. Data analysis and discussion: TKE, CCC, WCL, YHC, and YJJ. Contribution of reagents/materials/analy-sis tools: CCC, TMK and YJJ. Clinical information: WCL, YHC and YJJ. Man-uscript preparation: TKE, CCC, WCL, YHC, and YJJ. All authors read and approved thefinal manuscript.
Fig. 7. Molecular modeling of Human GAA. The predicted wild type of human GAA model. The predicted structure comprisesfive domains: trefoil type P domain (light blue), N-terminal (yellow), catalytic domain (red), proximal C (blue), N-terminal and distal C termi-nal (green). Thefive residues R190, G576, R594, D645, and W746 are shown in a sphere mode. The 3D molecular graphs are displayed using PyMOL.
Acknowledgement
We are grateful for both hardware and software support from the Structural Bioinformatics Core Facility at National Chiao Tung Universi-ty. This study was supported by a grant from Kaohsiung Medical Univer-sity Hospital (KMUH101-1M64).
References
[1]Kishnani PS, Howell RR. Pompe disease in infants and children. J Pediatr 2004;144: S35–43.
[2]Taglia A, Picillo E, D'Ambrosio P, Cecio MR, Viggiano E, Politano L. Genetic counseling in Pompe disease. Acta Myol 2011;30:179–81.
[3]Laforet P, Nicolino M, Eymard PB, et al. Juvenile and adult-onset acid maltase defi-ciency in France: genotype–phenotype correlation. Neurology 2000;55:1122–8.
[4]Disease AWGoMoPKishnani PS, Steiner RD, et al. Pompe disease diagnosis and man-agement guideline. Genet Med 2006;8:267–88.
[5]Lin CY, Shieh JJ. Molecular study on the infantile form of Pompe disease in Chinese in Taiwan. Zhonghua Min Guo Xiao Er Ke Yi Xue Hui Za Zhi 1996;37:115–21.
[6]Martiniuk F, Chen A, Mack A, et al. Carrier frequency for glycogen storage disease type II in New York and estimates of affected individuals born with the disease. Am J Med Genet 1998;79:69–72.
[7]Raben N, Plotz P, Byrne BJ. Acid alpha-glucosidase deficiency (glycogenosis type II, Pompe disease). Curr Mol Med 2002;2:145–66.
[8]Kishnani PS, Beckemeyer AA, Mendelsohn NJ. The new era of Pompe disease: ad-vances in the detection, understanding of the phenotypic spectrum, pathophysiolo-gy, and management. Am J Med Genet C Semin Med Genet 2012;160:1–7.
[9]Hoefsloot LH, Hoogeveen-Westerveld M, Reuser AJ, Oostra BA. Characterization of the human lysosomal alpha-glucosidase gene. Biochem J 1990;272:493–7.
[10]Martiniuk F, Bodkin M, Tzall S, Hirschhorn R. Isolation and partial characteriza-tion of the structural gene for human acid alpha glucosidase. DNA Cell Biol 1991;10:283–92.
[11]Hoefsloot LH, Hoogeveen-Westerveld M, Kroos MA, van Beeumen J, Reuser AJ, Oostra BA. Primary structure and processing of lysosomal alpha-glucosidase; homol-ogy with the intestinal sucrase-isomaltase complex. EMBO J 1988;7:1697–704.
[12]Herzog A, Hartung R, Reuser AJ, et al. A cross-sectional single-centre study on the spectrum of Pompe disease, German patients: molecular analysis of the GAA gene, manifestation and genotype–phenotype correlations. Orphanet J Rare Dis 2012;7:35.
[13]van der Stoep N, van Paridon CD, Janssens T, et al. Diagnostic guidelines for high-resolution melting curve (HRM) analysis: an interlaboratory validation of BRCA1 mutation scanning using the 96-well LightScanner. Hum Mutat 2009;30:899–909.
[14]Li BS, Wang XY, Ma FL, Jiang B, Song XX, Xu AG. Is high resolution melting analysis (HRMA) accurate for detection of human disease-associated mutations? A meta analysis. PLoS One 2011;6:e28078.
[15]Chang JG, Er TK. High-resolution melting: applications in genetic disorders. Clin Chim Acta 2012;414:197–201.
[16]Reed GH, Wittwer CT. Sensitivity and specificity of single-nucleotide polymorphism scanning by high-resolution melting analysis. Clin Chem 2004;50:1748–54.
[17]Ko TM, Hwu WL, Lin YW, et al. Molecular genetic study of Pompe disease in Chinese patients in Taiwan. Hum Mutat 1999;13:380–4.
[18]Lu CH, Huang SW, Lai YL, et al. On the relationship between the protein structure and protein dynamics. Proteins 2008;72:625–34.
[19]Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: calculating evolu-tionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res 2010;38:W529–33.
[20]Chang CM, Huang YW, Shih CH, Hwang JK. On the relationship between the se-quence conservation and the packing density profiles of the protein complexes. Pro-teins 2013;81:1192–9.
[21]Chen CC, Hwang JK, Yang JM. (PS)2-v2: template-based protein structure prediction server. BMC Bioinformatics 2009;10:366.
[22]Schaffer AA, Wolf YI, Ponting CP, Koonin EV, Aravind L, Altschul SF. IMPALA: matching a protein sequence against a collection of PSI-BLAST-constructed position-specific score matrices. Bioinformatics 1999;15:1000–11.
[23]Sim L, Quezada-Calvillo R, Sterchi EE, Nichols BL, Rose DR. Human intestinal maltase-glucoamylase: crystal structure of the N-terminal catalytic subunit and basis of inhi-bition and substrate specificity. J Mol Biol 2008;375:782–92.
[24]Wan L, Lee CC, Hsu CM, et al. Identification of eight novel mutations of the acid alpha-glucosidase gene causing the infantile or juvenile form of glycogen storage disease type II. J Neurol 2008;255:831–8.
[25]Lin CP, Huang SW, Lai YL, et al. Deriving protein dynamical properties from weighted protein contact number. Proteins 2008;72:929–35.
[26]Huang SW, Yu SH, Shih CH, Guan HW, Huang TT, Hwang JK. On the relationship be-tween catalytic residues and their protein contact number. Curr Protein Pept Sci 2011;12:574–9.
[27]Kroos M, Pomponio RJ, van Vliet L, et al. Update of the Pompe disease mutation da-tabase with 107 sequence variants and a format for severity rating. Hum Mutat 2008;29:E13–26.
[28]Kroos M, Hoogeveen-Westerveld M, Michelakakis H, et al. Update of the pompe disease mutation database with 60 novel GAA sequence variants and additional studies on the functional effect of 34 previously reported variants. Hum Mutat 2012;33:1161–5.
[29]Montalvo AL, Bembi B, Donnarumma M, et al. Mutation profile of the GAA gene in 40 Italian patients with late onset glycogen storage disease type II. Hum Mutat 2006;27:999–1006.
[30]Gort L, Coll MJ, Chabas A. Glycogen storage disease type II in Spanish patients: high frequency of c.1076-1GNC mutation. Mol Genet Metab 2007;92:183–7.
[31]Pittis MG, Filocamo M. Molecular genetics of late onset glycogen storage disease II in Italy. Acta Myol 2007;26:67–71.
[32]Bobillo Lobato J, Sanchez Peral BA, Duran Parejo P, Jimenez Jimenez LM. Detection of c.-32TNG (IVS1-13TNG) mutation of Pompe disease by real-time PCR in dried blood spot specimen. Clin Chim Acta 2013;418:107–8.
[33]Joshi PR, Glaser D, Schmidt S, et al. Molecular diagnosis of German patients with late-onset glycogen storage disease type II. J Inherit Metab Dis 2008;31(Suppl. 2): S261–5.
[34]Yang CC, Chien YH, Lee NC, et al. Rapid progressive course of later-onset Pompe dis-ease in Chinese patients. Mol Genet Metab 2011;104:284–8.
[35]Shieh JJ, Lin CY. Frequent mutation in Chinese patients with infantile type of GSD II in Taiwan: evidence for a founder effect. Hum Mutat 1998;11:306–12.
[36]Amarinthnukrowh P, Tongkobpetch S, Kongpatanayothin A, Shotelersuk V. p.D645E of acid alpha-glucosidase is the most common mutation in thai pa-tients with infantile-onset pompe disease. Genet Test Mol Biomarkers 2010;14:835–7.
[37]Chiang SC, Hwu WL, Lee NC, Hsu LW, Chien YH. Algorithm for Pompe disease newborn screening: results from the Taiwan screening program. Mol Genet Metab 2012;106:281–6.
[38]Kroos MA, Mullaart RA, Van Vliet L, et al. p.[G576S; E689K]: pathogenic combination or polymorphism in Pompe disease? Eur J Hum Genet 2008;16:875–9.
[39]Tajima Y, Matsuzawa F, Aikawa S, et al. Structural and biochemical studies on Pompe disease and a“pseudodeficiency of acid alpha-glucosidase”. J Hum Genet 2007;52:898–906.
[40]Hermans MM, van Leenen D, Kroos MA, Reuser AJ. Mutation detection in glycogen storage-disease type II by RT-PCR and automated sequencing. Biochem Biophys Res Commun 1997;241:414–8.
[41]Chou LS, Lyon E, Wittwer CT. A comparison of high-resolution melting analysis with denaturing high-performance liquid chromatography for mutation scanning: cystic fibrosis transmembrane conductance regulator gene as a model. Am J Clin Pathol 2005;124:330–8.
[42]Cousins MM, Ou SS, Wawer MJ, et al. Comparison of a high-resolution melting assay to next-generation sequencing for analysis of HIV diversity. J Clin Microbiol 2012;50:3054–9.
[43]Hermans MM, de Graaff E, Kroos MA, et al. The conservative substitution Asp-645–NGlu in lysosomal alpha-glucosidase affects transport and phosphorylation of the enzyme in an adult patient with glycogen-storage disease type II. Biochem J 1993;289(Pt 3):687–93.