Reactions of Ruthenium Cyclopropenyl Complexes
Containing Pentamethylcyclopentadienyl Ligands
Chao-Wan Chang,
†Ying-Chih Lin,*
,‡Gene-Hsiang Lee,
‡and Yu Wang
‡ National Overseas Chinese Student University Preparation School, Linkou, Taipei Hsien,Taiwan, Republic of China, and Department of Chemistry, National Taiwan University, Taipei, Taiwan 106, Republic of China
Received February 3, 2000
Deprotonation of cationic pentamethylcyclopentadienyl dppp ruthenium vinylidene com-plexes containing electron-withdrawing groups at the vinylidene ligand yielded cyclopropenyl
complexes [Ru]-CdC(Ph)CHR ([Ru] ) (η5-C
5Me5)(dppp)Ru, dppp ) Ph2PCH2CH2CH2PPh2,
R ) CN, 3a; R ) CO2CH3, 3b). Insertion of acetone into the three-membered ring of 3a gave
the neutral dihydrofuranyl complex [Ru]-CdC(Ph)CH(CN)CMe2O (4). Electrophilic addition
at Cβ of 4 afforded cationic carbene complexes [[Ru]dCC(R′)(Ph)CH(CN)C(CH3)2O]+(R′)
CH2CN, 5a; R′) CH2CO2CH3, 5b; R′) H, 7; R′) HgCl, 8). Complexes 5a and 5b transformed
to [[Ru]dCC(Ph)dC(CN)C(CH3)2O]+(6) by elimination of small organic molecules. Reactivity
of ruthenium complexes with a C5Me5ligand is different from those with a C5H5ligand and
could be ascribed to electronic and steric effects. Reaction of [Ru]N3with ICH2CN gave
[Ru]-NCH2I (11). Crystal structures of complexes 3a, 4, 6, 7, and 11 are also reported.
Introduction
Metal vinylidene complexes have attracted consider-able attention since they offer the possibility of develop-ment of new types of organometallic intermediates that
may have unusual reactivity.1-4Extensive reviews on
this subject have appeared recently.5-7The best entry
into the transition metal vinylidene complexes is the addition of electrophiles to the electron-rich carbon of
metal alkynyl complexes.8-18 A theoretical study of
vinylidene complexes indicated localization of electron
density on Cβ (HOMO) and the electron deficiency at
CR.19,20 A study of the reaction of alcohols with
ruthe-nium vinylidene complexes indicated that electron-withdrawing groups on the acetylide unit or on the
metal facilitate nucleophilic attack on CR.21
Recently we demonstrated that the presence of an
electron-withdrawing group at Cγ of the vinylidene
ligand combined with the cationic character of the ruthenium complex enhances the acidity of the proton
at Cγ. Thus deprotonation of such complexes could be
readily achieved, and subsequent intramolecular cy-cloaddition could lead to the formation of cyclopropenyl
complexes.1,2,22,23For example, [Cp(PPh
3)2
RudCdC(Ph)-CH2CN]+was found to undergo deprotonation to afford
the cyclopropenyl complex Cp(PPh3)2RuCdC(Ph)CHCN.
Unfortunately, without a methoxy substituent, the three-membered ring readily undergoes electrophilic addition to cause ring opening in the presence of acid or other electrophiles. The presence of a methoxy substituent in the cyclopropenyl ring enhances the stability of the three-membered ring, and protonation of such a ruthenium complex yields a cyclopropenylium complex by loss of methanol. In our study, we also noticed that replacement of the Cp ligand with a Tp
ligand23makes one of the phosphines labile, and a facile
substitution reaction was observed for Tp ruthenium cyclopropenyl complexes. To extend the breadth, we †National Overseas Chinese Student University Preparation School.
‡National Taiwan University.
(1) Ting, P. C.; Lin, Y. C.; Cheng, M. C.; Wang, Y. Organometallics 1994, 13, 2150.
(2) Chang, C. W.; Ting, P. C.; Lin, Y. C.; Lee, G. H.; Wang, Y. J. Organomet. Chem. 1998, 553, 417.
(3) Chang, C. W.; Lin, Y. C.; Lee, G. H.; Huang, S. L.; Wang, Y. Organometallics 1998, 17, 2534.
(4) Chang, C. W.; Lin, Y. C.; Lee, G. H.; Huang, S. L.; Wang, Y. Organometallics 1999, 18, 982.
(5) Bruce, M. I. Chem. Rev. 1991, 91, 197.
(6) Bruce, M. I.; Swincer, A. G. Adv. Organomet. Chem. 1983, 22, 59.
(7) Davies, S. G.; McNally, J. P.; Smallridge, A. J. Adv. Organomet. Chem. 1990, 30, 1.
(8) Werner, H. Angew Chem., Int. Ed. Engl. 1990, 29, 1077. (9) Werner, H.; Rappert, T.; Wolf, J. Isr. J. Chem. 1990, 30, 377. (10) Werner, H.; Hohn, A.; Schulz, M. J. Chem. Soc., Dalton Trans. 1991, 777.
(11) Schafer, M.; Wolf, J.; Werner, H. J. Chem. Soc., Chem. Com-mun. 1991, 1341.
(12) Schneider, D.; Werner, H. Angew. Chem. 1991, 103, 710. (13) Werner, H.; Dirnberger, T.; Hohn, A. Chem. Ber. 1991, 124, 1957.
(14) Werner, H.; Weinhand, R.; Knaup, W. Organometallics 1991, 10, 3967.
(15) Rappert, T. O.; Mahr, N.; Wolf, J.; Werner, H. Organometallics 1992, 11, 4156.
(16) Nakanishi, S.; Goda, K.; Uchiyama, S.; Otsuji, Y. Bull Chem. Soc. Jpn. 1992, 65, 2560.
(17) Haquette, P.; Pirio, N.; Touchard, D.; Toupet, L.; Dixneuf, P. H. J. Chem. Soc., Chem. Commun. 1993, 163.
(18) Watatuski, Y.; Koga, N.; Yamazaki, H.; Morokuma, K. J. Am. Chem. Soc. 1994, 116, 8105.
(19) Kostic, N. M.; Fenske, R. F. Organometallics 1982, 1, 974. (20) Werner, H.; Wolf, J.; Muller, G.; Kru¨ ger, C. Angew. Chem., Int. Ed. Engl. 1984, 28, 431.
(21) Bruce, M. I.; Swincer, A. G. Aust. J. Chem. 1980, 33, 1471. (22) Ting, P. C.; Lin, Y. C.; Lee, G. H.; Cheng, M. C.; Wang, Y. J. Am. Chem. Soc. 1996, 118, 6433.
(23) Lo, Y. H.; Lin, Y. C.; Lee, G. H.; Wang, Y. Organometallics 1999, 18, 982.
10.1021/om000094l CCC: $19.00 © 2000 American Chemical Society Publication on Web 07/13/2000
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
have explored the effect of C5Me5(Cp*) on the chemistry
of the ruthenium cyclopropenyl system. In this paper, we report an unprecedented acetone insertion into the cyclopropenyl ring, giving a dihydrofuranyl ligand and
the electrophilic addition at Cβ of the five-membered
dihydrofuranyl ligand giving a carbene complex. Results and Discussion
Synthesis of Ruthenium Vinylidene Complexes. Treatment of [Ru]CtPh (1, [Ru] ) Cp*(dppp)Ru, Cp*
) η5-C
5Me5, dppp ) Ph2PCH2CH2CH2PPh2) with ICH2
-CN in CH2Cl2afforded the cationic vinylidene complex
[[Ru]dCdC(Ph)CH2CN]+(2a) in 85% yield (Scheme 1).
Unlike other vinylidene complexes,5which are generally
prepared at room temperature, 2a is prepared by
heating a CH2Cl2 solution of 1 and ICH2CN at reflux
for 1 day. The steric effect of the Cp* ligand may hinder the electrophilic addition, therefore requiring mild heat-ing and longer reaction times. Similarly, preparations
of vinylidene complexes [[Ru]dCdC(Ph)CH2R]+ (R )
CO2CH3, 2b; R ) C6F5, 2c; R ) C6H5, 2d; R ) p-C6H4
-CN, 2e; R ) p-C6H4CF3, 2f) have produced high yields.
All these reactions required mild heating and gave analytically pure complexes of various colors. The
characteristic deshielded CRresonances in the13C NMR
spectra of these vinylidene complexes appear as triplets
in the region of δ ) 343 ( 3. The31P NMR resonance
appears as a singlet at δ ) 35 ( 1 in CDCl3 at room
temperature due to the fluxional behavior of the
vin-ylidene ligand.24,25
Cyclopropenation of Vinylidene Complexes.
Deprotonation of 2a by n-Bu4NOH is followed by the
expected cyclization, yielding the cyclopropenyl complex [Ru]CdC(Ph)CHCN (3a) (Scheme 1). Use of acetone or acetonitrile as a solvent gives a good yield, and use of
other bases such as n-Bu4NF (1 M in THF), DBU
(1,8-diazabicyclo[5.4.0]undecene), or KOH (dissolved in a
minimum amount of H2O) also gives 3a in comparable
yield. The reaction gives yellow crystalline product in analytically pure form. Single crystals of 3a, suitable for X-ray diffraction analysis, were obtained when the reaction is carried out at lower concentration. Complex 3a is stable in air and soluble in CH2Cl2, CHCl3, THF,
and benzene, slightly soluble in acetone and diethyl
ether, but insoluble in n-hexane, CH3CN, and MeOH.
The 31P NMR spectrum of 3a displays two doublet
resonances at δ 48.13 and 45.15 with2J
P-P) 49.76 Hz
in CDCl3 arising from the presence of a stereogenic
center in the three-membered ring. In the 1H NMR
spectrum of 3a, the resonance of the methine proton
appears at δ 1.07, and in the13C{1H}NMR spectrum,
the triplet at δ 133.96 with2J
C-P) 5.74 Hz is assigned
to CR.
The deprotonation/cyclopropenation reaction also
oc-curs for 2b, giving [Ru]CdC(Ph)CHCO2CH3(3b) in 77%
yield. This reaction can only be carried out in acetone
and only by using n-Bu4NOH as a proton abstractor.
Previously we reported that deprotonation of the
analo-gous complex22[(η5-C
5H5)(PPh3)2RudCdC(Ph)CH2CO2
-CH3]+ gave the cyclopropenyl complex as a kinetic
product, which transformed to the furanyl complex in 1 h at room temperature. However, we did not observe such a transformation for 3b in benzene after 3 days at room temperature. This could be due to steric repulsion between Cp* and the phenyl substituent on the
cyclo-propenyl ligand. The 31P NMR spectrum of 3b also
displays two doublet resonances at δ 47.73 and 43.00
with2J
P-P) 50.18 Hz. In the1H NMR spectrum of 3b,
the resonance of the methine proton appears at δ 1.60. Facile deprotonation of 2a and 2b indicates the acidic nature of the methylene protons, which may be ascribed to the combined effect of the cationic character and electron-withdrawing substituents of the vinylidene complexes. However, a similar reaction is not observed
for complexes 2c-f with n-Bu4NF, DBU, or KOH.
Analogous Cp vinylidene complexes undergo a
depro-tonation reaction to yield cyclization products.22
There-(24) Allen, D. L.; Gibson, V. C.; Green, M. L.; Skinner, T. F.; Bashikin, J.; Grebenik, P. D. J. Chem. Soc., Chem. Commun. 1985, 895.
(25) Consiglio, G.; Morandini, F. Chem. Rev. 1987, 87, 761.
Scheme 1 Scheme 2
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
fore, we conclude that, in the vinylidene system with a Cp* ligand, a strong electron-withdrawing group on the vinylidene ligand is necessary to enhance acidity of the methylene protons for deprotonation.
Previous studies on metal cyclopropenyl
deriva-tives26-30include the synthesis of iron derivatives from
the reaction of metal carbonylate anions with cyclopro-penylium cations reported by Bartmann and Gomp-per,27,31as well as CO insertion and ring-opening studies
of such systems28,29 by Hughes and co-workers. We
reported the synthesis and chemical reactivity of several
neutral ruthenium cyclopropenyl complexes1,2,22-23 in
which the metal bonds to one C(sp2) atom of the
three-membered cyclopropenyl ring. A few structurally dif-ferent cyclopropenylidene complexes, mostly prepared
from dichlorocyclopropene,32,33 and a number of
π-cy-clopropene complexes34-37are also known. The acidity
of the aliphatic protons on the coordinated dppe ligand
in a cationic iron vinylidene complex38 has been
em-ployed for inducing the intramolecular cyclization be-tween the dppe and the vinylidene ligand.
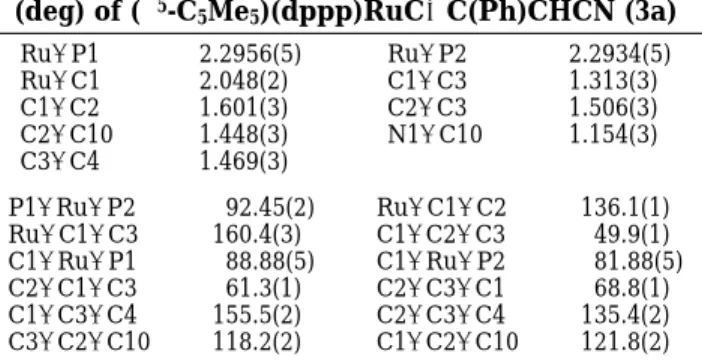
The molecular structure of 3a has been confirmed by a single-crystal X-ray diffraction study. An ORTEP diagram is shown in Figure 1, and selected bond distances and angles are listed in Table 1. This complex adopts a distorted three-leg piano stool geometry. The Ru-C1 distance of 2.048(2) Å is typical for a Ru-C
single bond, and the C1-C3 distance of 1.313(3) Å is a regular CdC double bond, indicating coordination of one
sp2carbon of the cyclopropenyl ligand. Both bond angles
Ru-C1-C3 and C1-C3-C4 (160.4(2)° and 155.5(2)°,
respectively) are greater than that of an idealized sp2
hybridization. The C1-C2 and C2-C3 bond lengths of 1.601(3) and 1.506(3) Å, respectively, are significantly different, conforming to the favorable cleavage of the C1-C2 bond.
Insertion of Acetone into the Cyclopropenyl Ring of 3a. If the bright yellow solution of 3a in acetone is stored at room temperature for 2 days, the color of
the solution turns yellow-brown. The31P NMR spectrum
of the mixture indicated formation of two new complexes in a ratio of 2:1. The major product,
[Ru]CdC(Ph)CH-(CN)C(CH3)2O (4), is formed by addition of an acetone
molecule to the three-membered ring of 3a, which is confirmed by single-crystal X-ray diffraction analysis.
Complex 4 is air stable and soluble in THF, CH2Cl2,
CHCl3, and benzene, moderately soluble in acetone,
acetonitrile, and methanol, and insoluble in hexane. Thermolysis of 4 at 60 °C afforded 1. The organic portion
from this reaction was not isolated. In the 31P NMR
spectrum of the mixture, two doublet resonances at δ
45.38 and 39.11 with JP-P) 52.12 Hz are assigned to
4. The1H NMR spectrum of 4 displays resonances at δ
1.09, 1.22, and 1.57 that are assignable to Cp*, 2CH3,
and CH, respectively.
Complex 4 was recrystallized from THF/hexane (1:3) to give single crystals, and its molecular structure was determined by a single-crystal X-ray diffraction study. An ORTEP diagram is shown in Figure 2, and selected bond distances and bond angles are given in Table 2. Complex 4 adopts a distorted three-leg piano stool geometry with the P1-Ru-P2, P1-Ru-C1, and P2-Ru-C1 angles being 90.78(8)°, 88.5(2)°, and 86.3(3)°, respectively. Insertion of an acetone molecule is clearly seen in the coordinated five-membered dihydrofuranyl ligand. The Ru-C1 distance of 2.101(7) Å is typical for a Ru-C single bond, and the C1-C3 distance of 1.371-(10) Å indicates a CdC double bond. The O1-C1 and O1-C4 single bond lengths of 1.431(8) and 1.455(9) Å, respectively, are comparable. The Ru-C1-C2 bond
angle of 136.7(5)° is slightly greater than an sp2
hybridization bond angle; thus O1-C1-C2 and Ru-C1-O1 bond angles of 108.6(6)° and 113.8(4)°, respec-tively, are smaller.
Acetone is the only reagent found to undergo an insertion reaction to give 4. Methyl isobutyl ketone, 2-butanone, and 3-pentanone have been used as sol-(26) Lowe, C.; Shklover, V.; Bosch, H. W.; Berke, H. Chem. Ber.
1993, 126, 1769.
(27) Gompper, R.; Bartmann, E. Angew. Chem., Int. Ed. Engl. 1978, 17, 456.
(28) DeSimone, D. M.; Derrosiers, P. J.; Hughes, R. P. J. Am. Chem. Soc. 1982, 104, 4842.
(29) Hughes, R. P.; Donaldson, W. A. J. Am. Chem. Soc. 1982, 104, 4846.
(30) Weiss, R.; Priesner, C. Angew. Chem., Int. Ed. Engl. 1978, 17, 457.
(31) Gompper, R.; Bartmann, E. Angew. Chem., Int. Ed. Engl. 1985, 24, 209.
(32) Miki, S.; Ohno, T.; Iwasaki, H.; Yoshida, Z. I. J. Phys. Org. Chem. 1988, 1, 333.
(33) Yoshida, Z. I. Pure Appl. Chem. 1982, 54, 1059. (34) Schrock, R. R. Acc. Chem. Res. 1986, 19, 342.
(35) Hughes, R. P.; Reisch, J. W.; Rheingold, A. L. Organometallics 1985, 4, 1754.
(36) Hughes, R. P.; Klaui, W.; Reisch, J. W.; Muller, A.; Rheingold, A. L. Organometallics 1985, 4, 1761.
(37) Mealli, C,; Midollini, S.; Moneti, S.; Sacconi, L.; Silvestre, J.; Albright, T. A. J. Am. Chem. Soc. 1982, 104, 59.
(38) Adams, R. D.; Davison, A.; Selegue, J. P. J. Am. Chem. Soc. 1979, 101, 7232.
Figure 1. ORTEP drawing of 3a with thermal ellipsoids
shown at the 30% probability level. For phenyl groups on dppp, only the ipso carbons are shown.
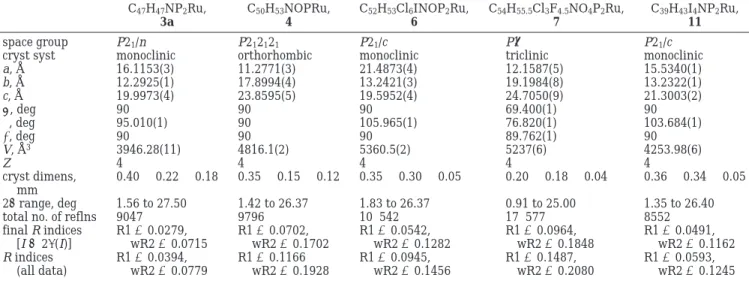
Table 1. Selected Bond Distances (Å) and Angles (deg) of (η5-C 5Me5)(dppp)RuCdC(Ph)CHCN (3a) Ru-P1 2.2956(5) Ru-P2 2.2934(5) Ru-C1 2.048(2) C1-C3 1.313(3) C1-C2 1.601(3) C2-C3 1.506(3) C2-C10 1.448(3) N1-C10 1.154(3) C3-C4 1.469(3) P1-Ru-P2 92.45(2) Ru-C1-C2 136.1(1) Ru-C1-C3 160.4(3) C1-C2-C3 49.9(1) C1-Ru-P1 88.88(5) C1-Ru-P2 81.88(5) C2-C1-C3 61.3(1) C2-C3-C1 68.8(1) C1-C3-C4 155.5(2) C2-C3-C4 135.4(2) C3-C2-C10 118.2(2) C1-C2-C10 121.8(2)
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
vents for the reaction of 2a with n-Bu4NOH. In these
solvent systems, 3a formed in good yield, but no insertion product was observed at room temperature.
Addition of PhNCO, PhNCS, (CF3)2CO, or formaldehyde
to a benzene solution of 3a resulted in ring opening, yielding 2a, and addition of propanal resulted in de-composition of 3a, leading to 1. The minor product of the reaction of 2a with base in the presence of acetone was not isolated due to decomposition to 1 during
chromatography. However, on the basis of the31P NMR
spectrum of the mixture displaying two doublet 31P
resonances at δ 28.67, 11.77 (JP-P) 52.21 Hz)
assign-able for this minor product and the mass spectrum of the mixture displaying only parent peaks of 4, the inverted insertion mode giving
[Ru]CdC(Ph)CH(CN)-OC(CH3)2(4′) is speculated.
Electrophilic Additions of Ruthenium Cyclo-pentenyl Complex 4. Treatment of 4 with organic
halides XCH2R afforded cationic cyclic carbene
com-plexes [[Ru]dCC(CH2R)(Ph)CH(CN)C(CH3)2O][X] (R )
CN, X ) I, 5a; R ) CO2CH3, X ) Br, 5b) in high yields.
Both31P NMR spectra display two doublet resonances:
δ 37.30, 31.45 with JP-P) 48.02 Hz and δ 37.38, 31.94
with JP-P ) 52.91 Hz, assignable to 5a and 5b,
respectively. The five-membered ring contains two ste-reogenic carbon centers, and two diastereomers were expected. However, for each compound, we see only one
set of two31P doublets, indicating high
diastereoselec-tivity. This is possibly due to the steric effect of the Cp* ligand. Spectroscopic data of 5 consist of a strongly
deshielded CRresonance as a triplet at around δ 290 in
the13C NMR spectrum.
Elimination of CH3CN from 5a in CDCl3 generated
the new cationic cyclic carbene complex [[Ru]dCC(Ph)d
C(CN)C(CH3)2O][I] (6) (Scheme 2). The reaction was
complete in 3 days at room temperature, and heating the solution accelerated the reaction. Complex 5b also transformed to 6 in solution after 1 week at room
temperature and in refluxing CHCl3after 1 day.
Com-plex 6 can also be obtained directly by thermolysis of 4
in the presence ICH2CN or BrCH2CO2CH3. The 31P
NMR spectrum of 6 displays a singlet at δ 37.1, and, in
the 13C NMR spectrum of 6, the deshielded triplet CR
resonance at δ 275.0 is characteristic of a carbene complex.
The molecular structure of 6 has been confirmed by a single-crystal X-ray diffraction study. An ORTEP diagram is shown in Figure 3, and selected bond distances and bond angles are given in Table 3. This complex adopts a three-leg piano stool geometry with the P1-Ru-P2, P1-Ru-C1, and P2-Ru-C1 angles being 91.97(5)°, 86.93(14)°, and 86.4(2)°, respectively. The Ru-C1 distance of 1.953(5) Å is typical for a Rud C double bond. In the five-membered ring, the C2-C3 bond distance of 1.378(7) Å indicates a double bond. The Figure 2. ORTEP drawing of 4 with thermal ellipsoids
shown at the 30% probability level. For phenyl groups on dppp, only the ipso carbons are shown.
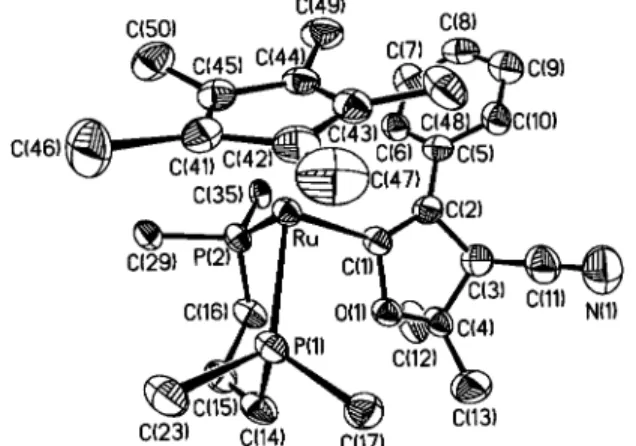
Table 2. Selected Bond Distances (Å) and Angles (deg) of (η5-C 5Me5)(dppp)RuCdC(Ph)CH(CN)C(CH3)2O (4) Ru-P1 2.321(2) Ru-P2 2.283(2) Ru-C1 2.101(7) C1-C2 1.37(1) C2-C3 1.55(1) C3-C4 1.55(1) C3-C11 1.50(2) C11-N1 1.13(1) C1-O1 1.431(8) C4-O1 1.455(9) C4-C12 1.46(1) C4-C13 1.45(1) Ru-C1-C2 136.7(5) Ru-C1-O1 113.8(4) C2-C1-O1 108.6(6) C1-C2-C3 108.0(7) C2-C3-C4 102.9(7) C1-C2-C5 134.6(8) C11-C3-C4 113.4(9) C11-C3-C2 110.8(8) C13-C4-O1 108.2(8) O1-C4-C12 110.9(7) O1-C3-C4 100.9(6) C1-Ru-P1 88.5(2) C1-Ru-P2 86.3(3) P1-Ru-P2 90.78(8) C3-C2-C5 117.2(7) C3-C4-C12 110.0(9) C3-C4-C13 118.0(8) C12-C4-C13 108.6(9)
Figure 3. ORTEP drawing of 6 with thermal ellipsoids
shown at the 30% probability level. For phenyl groups on dppp, only the ipso carbons are shown.
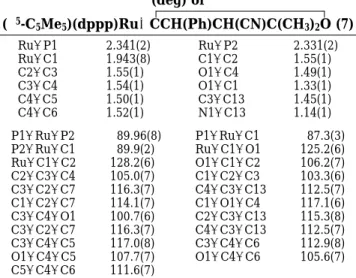
Table 3. Selected Bond Distances (Å) and Angles (deg) of (η5-C 5Me5)(dppp)RudCC(Ph)dC(CN)C(CH3)2O (6) Ru-P1 2.333(1) Ru-P2 2.333(1) Ru-C1 1.953(5) C1-C2 1.500(7) C2-C3 1.378(7) O1-C1 1.375(6) C3-C4 1.481(8) O1-C4 1.462(6) C3-C11 1.421(8) C11-N1 1.136(7) C4-C12 1.514(9) C4-C13 1.517(7) C1-Ru-P2 86.4(2) P1-Ru-C1 86.9(1) P1-Ru-P2 91.97(5) O1-C1-C2 103.1(4) Ru-C1-C2 137.1(4) O1-C1-Ru 119.7(3) C2-C3-C4 111.8(5) C1-O1-C4 116.3(4) C1-C2-C3 108.5(4) C1-C2-C5 131.2(5) C3-C2-C5 119.6(5) C2-C3-C11 126.6(5) C11-C3-C4 121.1(5) O1-C4-C3 99.9(4) C12-C4-C3 111.5(5) O1-C4-C12 108.3(5) C3-C4-C13 115.8(5) O1-C4-C13 108.8(4) C12-C4-C13 111.7(5)
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
shorter O1-C1 bond lengths of 1.375(6) Å, relative to that of O1-C4 of 1.462(6) Å, indicates contribution of the lone pair of oxygen, thus leading to a stronger
C(sp2)-O single bond. The C1-C2-C5 and
C2-C3-C11 bond angles of 131.2(5)° and 126.6(5)°, respectively,
are consistent with sp2 hybridization for C2 and C3.
Protonation of 4 with CF3COOH occurs also at Cβof
the five-membered ring, yielding the carbene complex
[[Ru]dCCH(Ph)CH(CN)C(CH3)2O][CF3COO] (7) (Scheme
2). This reaction is faster compared to the reaction of 4
with XCH2R (5 min vs 24 h at room temperature). The
protonation resulted in formation of two diastereomers, giving two sets of doublets of doublets at δ 38.63, 32.97
(JP-P) 48.03 Hz) and δ 37.09, 31.93 (JP-P) 48.93 Hz)
in a ratio of 20:1 in the31P NMR spectrum. Purification
by recrystallization gave only the major isomer. The
characteristic CR resonance of the major isomer of 7
appears as a triplet at δ 290.5 in the13C NMR spectrum,
and, in the1H NMR spectrum, two singlets appear at δ
5.13 and δ 1.59, assignable to the CHPh and CHCN groups of the five-membered ring, respectively. The FAB mass spectrum displays a parent peak at m/e 848.3
attributed to (M+ - CF3COO). In solution, 7 slowly
transformed to 6 by elimination of H2. After 14 days at
room temperature the transformation was not complete and the ratio of 7:6 was 4:1. Thermolysis of the solution caused decomposition of 7.
Addition of HgCl2to 4 resulted in immediate
forma-tion of the blue carbene complex
[[Ru]dCC(HgCl)(Ph)-CH(CN)C(CH3)2O][Cl] (8). The presence of two
stereo-genic carbon centers in the cyclic ligand resulted in two
sets of doublets of doublets at δ 37.8, 32.2 with JP-P)
50.85 Hz and δ 37.75, 31.83 with JP-P) 48.01 Hz in a
ratio of 10:1 in the 31P NMR spectrum. The minor
isomer escaped from purification by recrystallization
again, since only the major one was obtained. The13C
NMR spectrum of the major isomer of 8 consists of
strongly deshielded CRresonance as a triplet at δ 309.7
in CDCl3at room temperature. No transformation of 8
to 6 is observed in CDCl3 after 14 days. The lower
diastereoselectivity of the mercuric chloride addition to 4 relative to that in protonation is somewhat surprising, and we do not have rationalization for this observation. The molecular structure of 7 was confirmed by a single-crystal X-ray diffraction study. An ORTEP dia-gram is shown in Figure 4, and selected bond distances and bond angles are listed in Table 4. The Ru-C1 distance of 1.943(8) Å is typical for a Ru carbene system, and the C2-C3 bond length of 1.554(12) Å is a C-C single bond. The different O1-C1 and O1-C4 bond lengths of 1.33(1) and 1.49(1) Å, respectively, are
consistent with other similar carbene complexes.39
The Ru1-C1-C2 and O1-C1-Ru1 bond angles of 128.2(6)° and 125.2(6)°, respectively, are slightly greater
than an sp2hybridization bond angle. The O1-C1-C2,
C1-C2-C3, C2-C3-C4, and C3-C4-O1 bond angles of 106.2(7)°, 103.3(6)°, 105.0(7)°, and 100.7(6)°, respec-tively, are slightly smaller than a typical pentagonal
angle (108°). Formation of carbene complexes 5, 7, and 8 occurs by selective addition of electrophile (XCH2R,
H+, and HgCl2) to the nucleophilic Cβ of the
five-membered ring in 4. Possibly because of steric crowding
around Cβand Cγ, it is inclined to form a more stable
carbene complex 6 by elimination of a small molecule. Reactions of Ruthenium Cyclopropenyl Com-plex 3a. Reactions of CF3COOH with 3a and 3b readily
gave 2a and 2b, respectively, indicating the basic character of the methine carbon of the three-membered ring. This protonation is different from the acid-induced demethoxylation of the iron cyclopropenyl complex
Cp(CO)2Fe[C3(OMe)(Ph)2], containing a methoxy group
on the three-membered ring.31
Treatment of 3a with HgCl2produced the vinylidene
complex [[Ru]dCdC(Ph)CH(CN)(HgCl)][Cl] (9) in 86%
yield. As expected, the31P NMR spectrum of 9 displays
two doublets at δ 37.88 and 32.82 with JP-P) 50.39
Hz. Formation of 9 occurs by selective cleavage of the cyclopropenyl single bond near the metal center.
At-tempts to carry out cyclopropenation of 9 by using n-Bu4
-NOH, n-Bu4NF, and DBU resulted in cleavage of the
C-Hg bond, yielding 3a (Scheme 3).
The addition of 5 equiv of Me3SiN3(TMSN3) to 3a in
THF at room temperature initiated a color change from the light yellow solution of 3 to red initially, then (39) Bruce, M. I.; Swincer, A. G.; Thomson, B. J.; Wallis, R. C. Aust.
J. Chem. 1980, 33, 2605. (b) Garner, J.-A. M.; Irving, A.; Moss, J. R. Organometallics 1990, 9, 2836. (c) Adams, H.; Bailey, N. A.; Grayson, M.; Ridgway, C.; Smith, A. J.; Taylor, P.; Winter, M. J.; Housecroft, C. E. Organometallics 1990, 9, 2621.
Figure 4. ORTEP drawing of 7 with thermal ellipsoids
shown at the 30% probability level. For phenyl groups on dppp, only the ipso carbons are shown.
Table 4. Selected Bond Distances (Å) and Angles (deg) of (η5-C 5Me5)(dppp)RudCCH(Ph)CH(CN)C(CH3)2O (7) Ru-P1 2.341(2) Ru-P2 2.331(2) Ru-C1 1.943(8) C1-C2 1.55(1) C2-C3 1.55(1) O1-C4 1.49(1) C3-C4 1.54(1) O1-C1 1.33(1) C4-C5 1.50(1) C3-C13 1.45(1) C4-C6 1.52(1) N1-C13 1.14(1) P1-Ru-P2 89.96(8) P1-Ru-C1 87.3(3) P2-Ru-C1 89.9(2) Ru-C1-O1 125.2(6) Ru-C1-C2 128.2(6) O1-C1-C2 106.2(7) C2-C3-C4 105.0(7) C1-C2-C3 103.3(6) C3-C2-C7 116.3(7) C4-C3-C13 112.5(7) C1-C2-C7 114.1(7) C1-O1-C4 117.1(6) C3-C4-O1 100.7(6) C2-C3-C13 115.3(8) C3-C2-C7 116.3(7) C4-C3-C13 112.5(7) C3-C4-C5 117.0(8) C3-C4-C6 112.9(8) O1-C4-C5 107.7(7) O1-C4-C6 105.6(7) C5-C4-C6 111.6(7)
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
becoming a light orange after ca. 30 min, and, after 1 h, turning yellow again. The final product was recovered
as [Ru]N3(10) (Scheme 3) in 78% yield. Recently40we
reported reactions of Cp ruthenium cyclopropenyl
com-plexes with TMSN3. The reaction of 3a with TMSN3
may proceed similarly, i.e., via electrophilic addition of
a TMS group to the sp3carbon of the cyclopropenyl ring
followed by hydrolysis. Nucleophilic addition of an N3
anion at CRand electrophilic addition of a second TMS
unit at Cβ followed by loss of N2 then gives an
N-coordinated nitrile complex. Substitution of the nitrile
ligand by N3-occurs finally after 1 h to give 10. The
proton source of the reaction is believed to come from
the small amount of water in solvent and in TMSN3(no
attempt was made to dry TMSN3 due to potential
hazards) and protons incorporated into the product through hydrolysis of the TMS substituents. No attempt was made to isolate the organic product.
Interestingly, treatment of yellow complex 10 with
excess ICH2CN at room temperature afforded the brown
N-coordinated iodoacetonitrile complex{[Ru]NC-CH2I}
-[I3] (11) (Scheme 3). Surprisingly, no iodide addition was
observed; instead the coordinated N3-is readily replaced
by ICH2CN without cleavage of the C-I bond. In the
1H NMR spectrum of 11, two singlet resonances at δ
4.50 and 1.54 are assigned to CH2and Cp*, respectively.
The31P NMR spectrum displays a singlet resonance at
δ 35.01. In the 13C NMR spectrum, resonances at δ
119.4, 50.9, and 9.7 are assigned to CN, CH2, and Cp*,
respectively. Complex 11 is stable in air and soluble in
most polar solvents such as CH2Cl2, acetone,
aceto-nitrile, THF, and MeOH and moderately soluble in n-pentane and n-hexane.
The molecular structure of 11 was confirmed by a single-crystal X-ray diffraction study. An ORTEP dia-gram is shown in Figure 5, and selected bond distances and bond angles are given in Table 5. The metal center
is coordinated to the nitrile nitrogen atom of ICH2CN,
and the counteranion is I3-. The Ru-N1 bond length of
2.027(5) Å is typical for a Ru-N dative bond. The N1-C2 bond length of 1.155(8) Å is typical for a C-N triple bond. The C2-C3 and C3-I1 bond lengths of 1.453(10) and 2.188(9) Å indicate a C-C single bond and C-I
single bond, respectively. The N1-C2-C3 angle of 176.1(9)° is close to linear, indicating a C(sp) hybridiza-tion. The C2-C3-I1 angle of 109.8(5)° is typical for that
of an idealized C(sp3) hybridization.
Conclusions
Facile preparation of two neutral C5Me5-dppp-Ru
cyclopropenyl complexes was achieved by deprotonation of cationic vinylidene complexes with
electron-with-drawing substituents such as -CN and -CO2CH3 in
acetone. Treatment of cationic vinylidene complexes containing relatively weaker electron-withdrawing
sub-stituents such as C6F5, Ph, p-C6H4CN, and p-C6H4CF3
with base failed to give similar result. Protonation of the ruthenium cyclopropenyl complexes regenerated the vinylidene complexes, showing the nucleophilic nature
of the antecedent Cγcarbon of the cyclopropenyl ligand.
Thus other electrophiles could also be added to this Cγ
site by reactions with cyclopropenyl complexes. No conversion of 3b to the ruthenium furanyl complex was observed regardless of much higher strain energy of the cyclopropenyl ring. An acetone insertion reaction was observed in the cyclopropenyl complex 3a, yielding a neutral five-membered dihydrofuranyl complex 4 in moderate yield.
Electrophilic addition to 4 affords the cationic carbene complexes 5a and 5b. Complexes 5a and 5b are trans-formed to 6 by elimination of a small organic molecule. The significantly different reactivity of ruthenium
com-plexes with a Cp* ligand from those with a C5H5ligand
may due to the electronic and steric effects. Treatment
of 3 with (CH3)3SiN3 afforded 10, and subsequent
reaction of 10 with ICH2CN produced a novel
N-coordinated nitrile complex 11.
Experimental Section
General Procedures. All manipulations were performed under nitrogen using vacuum-line, drybox, and standard (40) Chang, K. H.; Lin, Y. C. Chem. Commun. 1998, 1441.
Scheme 3
Figure 5. ORTEP drawing of 11 with thermal ellipsoids
shown at the 30% probability level. For phenyl groups on dppp, only the ipso carbons are shown.
Table 5. Selected Bond Distances (Å) and Angles (deg) of [(η5-C 5Me5)(dppp)RuNCCH2I][I3] (11) Ru-P1 2.334(2) Ru-P2 2.334(1) Ru-N1 2.027(5) N1-C2 1.155(8) C2-C3 1.45(1) C3-I1 2.188(9) P1-Ru-P2 86.09(5) P1-Ru-N1 89.0(1) P2-Ru-N1 87.2(1) Ru-N1-C2 173.1(5) N1-C2-C3 176.1(9) C2-C3-I1 109.8(5)
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
Schlenk techniques. CH2Cl2 was distilled from CaH2, and
diethyl ether and THF were distilled from sodium diphen-ylketyl. All other solvents and reagents were of reagent grade and were used without further purification. NMR spectra were recorded on Bruker AC-200 and AM-300WB FT-NMR spec-trometers at room temperature (unless stated otherwise) and are referenced to residual protons in the solvents (CDCl3, δ
7.24: acetone-d6, δ 2.04). FAB mass spectra were recorded on
a JEOL SX-102A spectrometer. Complex [Ru]CtCPh, 1, was prepared following the methods reported in the literature.41
Elemental analyses and X-ray diffraction studies were carried out at the Regional Center of Analytical Instruments located at the National Taiwan University.
Synthesis of [[Ru]dCdC(Ph)CH2CN][I] (2a). A Schlenk flask was charged with ICH2CN (240 µL, 3.3 mmol) and 1 (250
mg, 0.33 mmol) in 10 mL of CH2Cl2. The mixture was heated
to reflux for 24 h. After the solution was allowed to cool, the solvent was reduced to about 3 mL under vacuum and was added to 20 mL of a vigorously stirred diethyl ether solution. The pink powders thus formed were filtered and washed with 20 mL of n-hexane and 20 mL of diethyl ether and dried under vacuum to give the product 2a (260 mg, 85% yield). Spectro-scopic data for 2a are as follows: 1H NMR (CDCl3) δ
7.47-6.82 (m, 25H, Ph), 3.22 (s, 2H, CH2), 2.65 (m, 2PCH2), 1.93 (m, PCH2CH2), 1.58 (s, 15H, 5CH3);31P NMR (CDCl3) δ 34.35; 13C NMR (CDCl 3) δ 341.8 (t, CR, JC-P) 17.9 Hz), 133.3-127.8 (Ph), 119.4 (Cβ), 118.6 (CN), 104.7 (Cp), 29.4 (t, PCH2, JC-P) 17.9 Hz), 21.3 (PCH2CH2), 17.4 (CH2CN), 10.3 (5CH3); MS (m/z, Ru102) 790.2 (M+- I), 750.2 (M+- I - CH 2CN), 649.1
(M+- I - CH2CN - CCPh). Anal. Calcd for C47H48NP2RuI:
C, 61.57; H, 5.28; N, 1.53, Found: C, 62.37; H, 5.45; N, 1.44. Complexes [[Ru]dCdC(Ph)CH2R][Br] (R ) CO2CH3, 2b
(85% yield); R ) C6F5, 2c (82% yield); R ) Ph, 2d (79% yield);
R ) p-C6H4CN, 2e (83% yield); R ) p-C6H4CF3, 2f (87% yield))
were prepared from the reaction of 1 with BrCH2CO2CH3,
BrCH2C6F5, BrCH2C6H5, BrCH2(p-C6H4CN), and
BrCH2(p-C6H4CF3), respectively, using a procedure similar to that of
2a. Spectroscopic data for 2b are as follows: 1H NMR (CDCl 3) δ 7.62-6.84 (m, 25H, Ph), 3.56 (s, 3H, OCH3), 3.27 (s, 2H, CH2), 2.71 (m, 2PCH2), 2.02 (m, PCH2CH2), 1.56 (s, 15H, 5CH3);31P NMR (CDCl3) δ 35.10;13C NMR (CDCl3) δ 345.1 (t, CR, JC-P) 14.0 Hz), 171.5 (CO2), 133.3-121.5 (Ph and Cβ), 104.1 (Cp), 52.0 (OCH3), 33.3 (CH2), 29.1 (t, PCH2, JC-P) 18.4 Hz), 21.7 (PCH2CH2), 10.4 (5CH3); MS (m/z, Ru102) 823.3 (M+ - Br), 750.2 (M+- Br - CH2CO2CH3), 649.1 (M+- Br - CH2CO2
-CH3- CCPh). Anal. Calcd for C48H51O2P2BrRu: C, 63.85; H,
5.69. Found: C, 64.22; H, 5.83. Spectroscopic data for 2c are as follows: 1H NMR (CDCl 3) δ 7.44-6.76 (m, 25H, Ph), 3.97 (s, 2H, CH2), 2.80 (m, 2PCH2), 2.65 (m, PCH2CH2), 1.59 (s, 15H, 5CH3);31P NMR (CDCl3) δ 35.86;13C NMR (CDCl3) δ 341.8 (t, CR, JC-P) 14.5 Hz), 134.1-123.7 (Ph), 119.4 (Cβ), 104.2 (Cp), 29.1 (t, PCH2, JC-P) 18.8 Hz), 21.8 (PCH2CH2), 20.1 (CH2C6F5), 10.5 (5CH3); MS (m/z, Ru102) 931 (M+- Br), 750.2 (M+ Br -CH2C6F5), 649.1 (M+- Br - CH2C6F5- CCPh). Anal. Calcd
for C52H48F5P2BrRu: C, 61.78; H, 4.79. Found: C, 62.35; H,
4.93. Spectroscopic data for 2d are as follows: 1H NMR (CDCl 3) δ 7.90-6.86 (m, 25H, Ph), 3.82 (s, 2H, CH2), 2.70 (m, 2PCH2), 2.17 (m, PCH2CH2), 1.63 (s, 15H, 5CH3);31P NMR (CDCl3) δ 35.77;13C NMR (CDCl 3) δ 345.4 (t, CR,JC-P) 10.1 Hz), 137.7-125.7 (Ph and Cβ), 103.7 (Cp), 32.7 (CH2C6H5), 29.2 (t, PCH2, JC-P) 19.2 Hz), 21.9 (PCH2CH2), 10.7 (5CH3); MS (m/z, Ru102) 841.4 (M+- Br), 750.2 (M+- Br - CH 2C6H5), 649.1 (M+- Br
- CH2C6H5- CCPh). Anal. Calcd for C52H53P2RuBr: C, 67.82;
H, 5.80. Found: C, 68.25; H, 5.93. Spectroscopic data for 2e are as follows: 1H NMR (CDCl 3) δ 7.62-6.79 (m, 25H, Ph), 3.94 (s, 2H, CH2), 2.77 (m, 2PCH2), 2.13 (m, PCH2CH2), 1.59 (s, 15H, 5CH3);31P NMR (CDCl3) δ 35.46;13C NMR (CDCl3) δ 344.6 (t, CR, JC-P) 14.1 Hz), 134.1-124.8 (Ph), 119.2 (Cβ), 118.7 (CN), 103.8 (Cp), 29.2 (t, PCH2, JC-P) 18.9 Hz), 21.8 (PCH2CH2), 32.6 (CH2C6H4CN), 10.6 (5CH3); MS (m/z, Ru102) 946.2 (M+- Br), 750.2 (M+- Br - CH 2C6H4CN), 649.1 (M+
- Br - CH2C6H4CN - CCPh). Anal. Calcd for C53H52NP2
-RuBr: C, 67.30; H, 5.54; N, 1.48. Found: C, 68.35; H, 5.67; N, 1.35. Spectroscopic data for 2f are as follows: 1H NMR (CDCl
3) δ 7.59-6.83 (m, 25H, Ph), 3.89 (s, 2H, CH2), 2.77 (m, 2PCH2), 2.05 (m, PCH2CH2), 1.61 (s, 15H, 5CH3);31P NMR (CDCl3) δ 35.61;13C NMR (CDCl 3) δ 345.0 (t, CR, JC-P) 16.3 Hz), 142.5-125.1 (Ph and Cβ), 103.8 (Cp), 32.4 (CH2C6H4CN), 29.2 (t, PCH2, JC-P) 19.5 Hz), 21.9 (PCH2CH2), 10.6 (5CH3); MS (m/z, Ru102) 989.2 (M+- Br), 750.2 (M+- Br - CH 2C6H4CF3),
649.1 (M+ - Br - CH2C6H4CF3- CCPh). Anal. Calcd for
C53H52F3P2RuBr: C, 64.37; H, 5.30. Found: C, 64.58; H, 5.49.
Synthesis of [Ru]CdC(Ph)CHCN (3a). To a 15 mL acetonitrile solution of complex 2a (1.70 g, 1.854 mmol) was added an aliquot of nBu4NOH (1 M in MeOH, 2 mL) . The
mixture was stirred at room temperature for 30 min to give bright yellow precipitates. The precipitate was filtered and washed with 2× 10 mL of acetonitrile and dried under vacuum to give the product 3a (1.33 g, 91% yield). Spectroscopic data for 3a are as follows: 1H NMR (CDCl
3) δ 7.72-7.03 (m, 25H, Ph), 2.65 (m, 2PCH2), 1.93 (m, PCH2CH2), 1.35 (s, 15H, 5CH3), 1.07 (s, 1H, CH);31P NMR (CDCl 3) δ 48.13, 45.52 (AB, JP-P) 49.76 Hz);13C NMR (CDCl 3) δ 135.2-122.7 (Ph, and Cβ), 134.0 (t, CR, JC-P) 5.74 Hz), 114.0 (CN), 94.0 (Cp), 29.9 (t, PCH2, JC-P) 28.1 Hz), 24.1 (PCH2CH2), 13.7 (CH), 10.0 (5CH3); MS (m/z, Ru102) 790.1 (M++ 1), 751.2 (M+-CHCN), 649.1 (M+
-CHCN-CCPh). Anal. Calcd for C47H47NP2Ru: C, 71.55; H,
6.01; N, 1.78. Found: C, 72.31; H, 6.37; N, 1.55.
Synthesis of [Ru]CdC(Ph)CH(CO2CH3) (3b). To a 10 mL acetone solution of complex 2b (500 mg, 0.554 mmol) was added an aliquot of (nBu)4NOH (1 M in MeOH, 1.0 mL). The
mixture was stirred at room temperature for 30 min to give bright yellow precipitates, which were filtered and washed with 2× 5 mL of acetone, then dried under vacuum to give the product 3b (340 mg, 77% yield). Spectroscopic data for 3b are as follows: 1H NMR (CDCl 3) δ 7.63-6.89 (m, 25H, Ph), 3.43 (s, 3H, OCH3), 2.43 (m, 4H, PCH2), 2.02 (m, PCH2CH2), 1.60 (s, 1H, CH), 1.34 (s, 15H, 5CH3);31P NMR (CDCl3) δ 47.73, 43.00 (AB, JP-P) 50.18 Hz);13C NMR (CDCl3) δ 183.6 (CO2), 134.0-119.4 (Ph and CR), 104.2 (Cβ), 89.1 (Cp), 52.1 (OCH3), 26.8 (t, JC-P) 13.7 Hz, PCH2), 21.5 (PCH2CH2), 10.0 (CH), 10.5 (5CH3), 9.7 (CH); MS (m/z, Ru102) 823.1 (M++ 1), 750.2
(M+- CHCO2CH3), 649.1 (M+- CHCO2CH3- CCPh). Anal.
Calcd for C48H50O2P2Ru: C, 70.14; H, 6.13. Found: C, 71.23;
H, 6.37.
Synthesis of [Ru]CdC(Ph)CH(CN)C(CH3)2O (4). To a 15 mL acetone solution of 2a (500 mg, 0.634 mmol) was added an aliquot of nBu4NOH (1 M in MeOH, 1.0 mL). The mixture
was stirred at room temperature for 5 min to give a bright yellow solution of 3a. The solution was stirred at room temperature for 5 days, and then the solvent was reduced to 2 mL under vacuum. To the residue, 30 mL of MeOH was added, and yellow precipitates thus formed were filtered and washed with 2 × 5 mL of MeOH. The31P NMR spectrum
indicates the presence of two products in a ratio of 2:1. Thus the product was passed through a silica gel-packed column eluted with 1:1 ethyl acetate/hexane, and only a yellow band was collected. After the solvent was removed under vacuum the yellow band gave a single product, 4 (280 mg, 52% yield). The minor product decomposed in the column. Spectroscopic data for 4 are as follows: 1H NMR (CDCl
3) δ 7.58-6.17 (m, 25H, Ph), 2.86 (t, PCH2, JH-P) 9.85 Hz), 2.54 (m, PCH2CH2), 2.86 (t, PCH2, JH-P) 13.57 Hz), 1.57 (s, 1H, CH), 1.22 (s, 6H, 2CH3), 1.08 (s, 15H, 5CH3);31P NMR (CDCl3) δ 45.43, 39.11 (AB, JP-P) 52.21 Hz);13C NMR (CDCl3) δ 145.2 (OC(CH3)2), 135.1-123.6 (Ph, CRand Cβ), 113.1 (CN), 93.3 (Cp), 31.9 (t, PCH2, JC-P) 18.7 Hz), 22.1 (PCH2), 9.6 (CH), 10.4 (5CH3); MS (m/z, Ru102) 848.3 (M++ 1), 790.2 (M+- CH 3COCH3), 751.2
(41) Oshima, N.; Suzuki, H.; Moro-oka, Y. Chem. Lett. 1984, 1161.
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
(M+ - CH
3COCH3 - CHCN), 649.1 (M+ - CH3COCH3
-CHCN - CCPh). Anal. Calcd for C50H53NOP2Ru: C, 70.90; H,
6.31; N, 1.65. Found: C, 71.33; H, 6.45; N, 1.65.31P NMR data
of the minor product 4′: 31P NMR (CDCl
3) δ 28.67, 11.77 (JP-P
) 50.23 Hz).
Synthesis of [[Ru]dCC(Ph)(CH2CN)CH(CN)C(CH3)2 O]-[I] (5a). A Schlenk flask was charged with ICH2CN (520 µL, 0.590 mmol) and 4 (100 mg, 0.118 mmol) in 10 mL of CH2Cl2.
The mixture was stirred at room temperature for 8 h. The solvent was reduced to about 2 mL under vacuum, and the residue was added to 20 mL of vigorously stirred diethyl ether. The brown powder thus formed was filtered and washed with 2× 5 mL of diethyl ether and dried under vacuum to give the product 5a (98.5 mg, 82% yield). Spectroscopic data for 5a are as follows: 1H NMR (CDCl 3) δ 7.74-6.18 (m, 25H, Ph), 5.28 (s, 2H, CH2), 2.84 (m, 2H, CH2), 2.59, 2.13 (m, 2H, PCH2), 1.46 (s, 1H, CHCN), 1.14 (s, 6H, 2CH3), 1.17 (s, 15H, 5CH3);31P NMR (CDCl3) δ 37.30, 31.45 (AB, JP-P) 48.02 Hz);13C NMR (CDCl3) δ 290.6 (t, CR, JC-P ) 11.6 Hz), 139.0-127.7 (Ph), 116.1, 115.2 (CN), 102.0 (Cp), 97.5 (OC(CH3)2), 70.0 (CH2), 28.8 (dd, JC-P) 29.9, 6.4 Hz, PCH2), 27.6 (dd, JC-P) 30.7, 5.3 Hz, PCH2), 22.6, 21.1 (2CH3), 20.4 (CH2), 14.0 (s, CHCN), 10.3 (5CH3); MS (m/z, Ru102) 887.3 (M+- I), 649.1 (M+- I - CH3
-COCH3 - CHCN - CCPh - CH2CN). Anal. Calcd for
C52H55N2OP2RuI: C, 61.60; H, 5.47; N, 2.76. Found: C,
62.03; H, 5.64; N, 2.42.
Complex [[Ru]dC-C(Ph)(CH2CO2CH3)CH(CN)C(CH3)2
O]-[Br] (5b) (100 mg, 88% yield) was prepared from 4 (100 mg, 0.118 mmol) with BrCH2CO2CH3(56 µL, 0.590 mmol) using a
procedure similar to that of 5a. Spectroscopic data of 5b are as follows: 1H NMR (CD 3COCD3) δ 7.77-6.18 (m, 25H, Ph), 3.81 (s, 2H, CH2), 3.76 (s, 3H, OCH3), 3.34 (t, 2H, JH-H) 13.42 Hz, PCH2), 2.95 (m, 2H, PCH2CH2), 2.18 (t, 2H, JH-H) 13.42 Hz, PCH2), 1.79 (s, 6H, 2CH3), 1.59 (s, 1H, CH), 1.17 (s, 15H, 5CH3);31P NMR (CDCl3) δ 37.38, 31.94 (JP-P) 52.91 Hz);13C NMR (CDCl3) δ 291.1 (t, CR, JC-P) 12.2 Hz), 167.7 (CO2), 139.1-127.7 (Ph), 119.4 (Cβ), 115.3 (CN), 102.0 (Cp), 97.4 (OC(CH3)2), 70.3 (CH2), 28.9 (dd, JC-P) 30.8, 5.5 Hz, PCH2), 27.3 (dd, JC-P) 29.1, 6.0 Hz, PCH2), 21.1 (PCH2CH2), 20.5 (2CH3), 13.7 (CHCN), 10.2 (5CH3); MS (m/z, Ru102) 920.3 (M+ - Br), 649.1 (M+- Br - CH 3COCH3- CHCN - CCPh - CH2
-CO2CH3). Anal. Calcd for C53H58NO3P2RuBr: C, 63.66; H, 5.85;
N, 1.40. Found: C, 64.18; H, 6.01; N, 1.23.
Synthesis of [[Ru]dCC(Ph)dC(CN)C(CH3)2O][I] (6). A Schlenk flask was charged with ICH2CN (510 µL, 0.590 mmol)
and 4 (100 mg, 0.118 mmol) and 10 mL of CH2Cl2. The mixture
was heated to reflux for 24 h. Then the solvent was reduced to about 2 mL under vacuum, and the residue was added to 20 mL of vigorously stirred n-hexane. The brown powders thus formed were filtered and washed with 2× 5 mL of diethyl ether and dried under vacuum to give 6 (75 mg, 65% yield). Spectroscopic data for 6 are as follows: 1H NMR (CDCl
3) δ 7.71-6.18 (m, 25H, Ph), 2.84 (t, 2H, JH-H ) 14.15 Hz, PCH2CH2), 2.62, 2.08 (2m, 4H, PCH2), 1.14 (s, 6H, 2CH3), 1.06 (s, 15H, 5CH3);31P NMR (CDCl3) δ 36.78.13C NMR (CDCl3) δ 275.0 (t, CR, JC-P) 12.2 Hz), 163.0 (Cβ), 139.6-128.9 (Ph), 120.6 (Cγ), 114.1 (CN), 103.5 (Cp), 102.1 (OC(CH3)2), 29.1 (t, JC-P) 18.3 Hz, PCH2), 22.3 (PCH2CH2), 12.1 (5CH3), 11.5 (2CH3); MS (m/z, Ru102) 846.2 (M+- I), 649.1 (M+- I - CH3
-COCH3- CCN - CCPh). Anal. Calcd For C50H52NOP2IRu: C,
61.73; H, 5.39; N, 1.44. Found: C, 62.17; H, 5.52; N, 1.17. Synthesis of [[Ru]dCCH(Ph)CH(CN)C(CH3)2O][CF3 -COO] (7). A Schlenk flask was charged with CF3COOH (95 µL, 1.18 mmol) and 4 (100 mg, 0.118 mmol) in 10 mL of CH2
-Cl2. The mixture was stirred at room temperature for 1 h. The
solvent was reduced to about 2 mL under vacuum, and 20 mL of diethyl ether was added to the residue. The brown powder thus formed was filtered and washed with 2× 5 mL of diethyl ether and dried under vacuum to give 7 (85 mg, 75% yield).
Spectroscopic data for 7 are as follows: 1H NMR (CD
3COCD3) δ 7.63-6.16 (m, 25H, Ph), 5.13 (s, 1H, CHPh), 3.20 (t, 1H, JH-P ) 10.19 Hz, CH2), 2.93 (t, 1H, JH-P) 13.14 Hz, CH2), 2.82-2.45 (m, 2H, PCH2), 2.19-2.08 (m, 2H, PCH2), 1.59 (s, 1H, CHCN), 1.24, 1.09 (s, 3H, 2CH3), 1.15 (s, 15H, 5CH3);31P NMR (CDCl3) δ 38.63, 32.97 (2d, JP-P) 48.03 Hz);13C NMR (CDCl3) δ 290.5 (t, JC-P) 12.7 Hz, CR), 138.2-128.3 (Ph and Cβ), 116.3 (CN), 102.2 (Cp), 97.3 (OC(CH3)2), 70.2 (CHPh), 30.7, 30.2 (2CH3), 28.7 (dd, JC-P) 28.6, 6.0 Hz, PCH2), 28.3 (dd, JC-P) 28.6, 5.8 Hz, PCH2), 27.8 (CHCN), 20.3 (PCH2CH2), 10.1 (5CH3); MS (m/z, Ru102) 849.3 (M+- CF3COO), 649.1 (M+
-CH3COCH3- CHCN - CCPh - H). Anal. Calcd for C52H54
-NO3F3P2Ru: C, 64.99; H, 5.39; N, 1.44. Found: C, 65.38; H,
5.61; N, 1.25.
Synthesis of [[Ru]dCC(Ph)(HgCl)CH(CN)C(CH3)2 O]-[Cl] (8). A Schlenk flask was charged with HgCl2(160 mg,
0.59 mmol) and 4 (100 mg, 0.118 mmol) in 10 mL of CH2Cl2.
The mixture was stirred at room temperature for 30 min. Excess HgCl2was filtered, and the solvent was reduced to
about 2 mL under vacuum. The residue was added to a 20 mL solution of vigorously stirred diethyl ether. The black-blue powder thus formed was filtered and washed with 2× 5 mL of diethyl ether and dried under vacuum to give 8 (115 mg, 90% yield). Spectroscopic data for 8 are as follows: 1H NMR
(CD3COCD3) δ 7.67-6.92 (m, 25H, Ph), 3.20 (s, 1H, CHCN), 2.43 (m, 2H, PCH2CH2), 2.00 (t, 2H, JH-P) 15.80 Hz, PCH2), 1.80 (t, 2H, JH-P) 13.80 Hz, PCH2), 1.61 (s, 6H, 2CH3), 1.21 (s, 15H, 5CH3);31P NMR (CDCl3) δ 37.83, 32.23 (JP-P) 50.85 Hz);13C NMR (CDCl 3) δ 309.7 (t, JC-P) 14.6 Hz, CR), 137.3-125.1 (Ph and Cβ), 118.5 (CN), 92.5 (Cp), 89.2 (OC(CH3)2), 30.7 (t, JC-P) 23.1 Hz, 2PCH2), 27.8, 27.4 (2CH3), 21.4 (PCH2CH2), 10.3 (CHCN), 9.3 (5CH3); MS (m/z, Ru,102Hg202) 1049.3 (M+ -Cl), 649.1 (M+- Cl - CH3COCH3- CHCN - HgCl - CCPh).
Anal. Calcd for C50H53Cl2P2RuHg: C, 55.17; H, 4.91. Found:
C, 56.33; H, 5.02.
Synthesis of [[Ru]dCdC(Ph)CH(CN)(HgCl)][Cl] (9). A Schlenk flask was charged with HgCl2(170 mg, 0.634 mmol)
and 3a (100 mg, 0.127 mmol) in 10 mL of CH2Cl2. The mixture
was stirred at room temperature for 30 min. Excess HgCl2was
filtered, and the solvent was reduced to about 2 mL under vacuum. The residue was added to a 20 mL solution of vigorously stirred diethyl ether. The pink powder thus formed was filtered and washed with 2× 5 mL of diethyl ether and dried under vacuum to give 9 (116 mg, 86% yield). Spectro-scopic data for 9 are as follows: 1H NMR (CDCl
3) δ 7.84-6.54 (m, 25H, Ph), 3.66 (s, 1H, CH), 2.95 (m, 2H, PCH2), 2.81 (m, 2H, PCH2), 2.70 (m, 2H, PCH2CH2), 1.59 (s, 15H, 5CH3);31P NMR (CDCl3) δ 37.88, 32.82 (2d, JP-P) 50.39 Hz);13C NMR (CDCl3) δ 340.7 (t, JC-P) 14.6 Hz, CR), 134.1-128.0 (Ph and Cβ), 122.1 (CN), 104.6 (Cp), 31.2 (d, JC-P) 35.3 Hz, PCH2), 29.9 (CH), 28.7 (d, JC-P) 32.9 Hz, PCH2), 21.8 (PCH2CH2), 10.6 (5CH3); MS (m/z, Ru,102Hg202) 991.3 (M+- Cl), 649.1 (M+
- Cl - CHCN - HgCl - CCPh). Anal. Calcd for C47H47NP2
-Cl2RuHg: C, 52.23; H, 4.47; N, 1.32. Found: C, 52.55; H, 4.75;
N, 1.20.
Synthesis of [Ru]N3(10). A Schlenk flask was charged with (CH3)3SiN3(78 µL, 0.590 mmol) and 3a (100 mg, 0.127
mmol) and 10 mL of THF. The mixture was stirred at room temperature for 3 h. The solvent was reduced to about 2 mL under vacuum and added to 20 mL of vigorously stirred
n-hexane. The yellow powder thus formed was filtered and
washed with 2× 5 mL of n-hexane and dried under vacuum to give the product 10 (68 mg, 78% yield). Spectroscopic data for 10 are as follows: 1H NMR (CDCl
3) δ 7.65-6.94 (m, 25H, Ph), 2.56 (t, 4H, JH-P ) 13.33 Hz, PCH2), 2.31 (m, 2H, PCH2CH2), 1.37 (s, 15H, 5CH3);31P NMR (CDCl3) δ 38.26.13C NMR (CDCl3) δ 138.6-127.5 (Ph, CRand Cβ), 89.6 (Cp), 27.9 (t, JC-P) 13.8 Hz, PCH2), 21.2 (PCH2CH2), 9.6 (5CH3); MS (m/z, Ru102) 692.3 (M++ 1), 675.3 (M+- N - 1), 649.3 (M+
-Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
N3). Anal. Calcd for C37H41N3P2Ru: C, 64.33; H, 5.98; N, 6.08.
Found: C, 64.46; H, 6.10; N, 5.93.
Synthesis of [[Ru]NCCH2I][I3] (11). A Schlenk flask was charged with ICH2CN (100 µL, 1.45 mmol) and 10 (100 mg,
0.145 mmol) in 10 mL of CH2Cl2. The mixture was stirred at
room temperature for 3 h. The solvent was reduced to about 2 mL under vacuum and added to 20 mL of vigorously stirred
n-pentane. The orange-yellow powder thus formed was filtered
and washed with 5 mL of n-pentane and dried under vacuum to give 11 (66 mg, 56% yield). Spectroscopic data for 11 are as follows: 1H NMR (CDCl 3) δ 8.07-6.83 (m, 25H, Ph), 4.50 (s, 2H, CH2), 2.59 (m, 2H, PCH2), 2.43 (m, 2H, PCH2CH2), 2.00 (m, 2H, PCH2), 1.54 (s, 15H, 5CH3);31P NMR (CDCl3) δ 35.01; 13C NMR (CDCl 3) δ 133.8-124.7 (Ph, CRand Cβ), 119.4 (CN), 93.2 (Cp), 50.9 (CH2), 29.4 (t, JC-P) 8.22 Hz, PCH2), 20.3 (PCH2CH2), 9.7 (5CH3); MS (m/z, Ru102) 816.2 (M++ 1 - I3), 776.1 (M+- I 3- CH2CN), 649.3 (M+ - I4- CH2CN). Anal.
Calcd for C39H42NP2RuI4: C, 39.18; H, 3.54; N, 1.17. Found:
C, 40.25; H, 3.73; N, 1.06.
X-ray Analysis of 3a. Single crystals of 3a suitable for X-ray diffraction study were grown as mentioned above. A single crystal of dimensions 0.32× 0.08 × 0.05 mm3was glued
to a glass fiber and mounted on a SMART CCD diffractometer. The data were collected using 3 kW sealed-tube molybdenum KR radiation (T ) 295 K). Exposure time was 5 s per frame.42
SADABS43 (Siemens area detector absorption) absorption
correction was applied, and decay was negligible. Data were
processed and the structures were solved and refined by the SHELXTL44program. The structure was solved using direct
methods and confirmed by Patterson methods refining on intensities of all data (20 610 reflections) to give wR1 ) 0.0579 and wR2 ) 0.122145 for 20 610 unique observed reflections
(I > 2σ(I)). Hydrogen atoms were placed geometrically using the riding model, with thermal parameters set to 1.2 times that for the atoms to which the hydrogen is attached and 1.5 times that for the methyl hydrogens. The procedures for the structure determination of 4, 6, 7, and 11 were similar to that of 3a. Crystal data of these complexes are listed in Table 6.
Acknowledgment. Financial support by the Na-tional Science Council of Taiwan, the Republic of China, is gratefully acknowledged. We thank Nathan T. Allen for helpful discussions.
Supporting Information Available: Details of the struc-tural determination for complexes 3a, 4, 6, 7, and 11 including crystal and intensity collection data, positional and anisotropic thermal parameters, and all of the bond distances and angles. This material is available free of charge via the Internet at http://pubs.acs.org.
OM000094L
(42) SAINT (Siemens Area Detector Integration) program; Siemens Analytical X-ray: Madison, WI, 1995.
(43) The SADABS program is based on the method of Blessing; see: Blessing, R. H. Acta Crystallogr., Sect. A 1995, 51, 33.
(44) SHELXTL: Structure Analysis Program, version 5.04; Siemens Industrial Automation Inc.: Madison, WI, 1995.
(45) GOF ) [∑[w(F2o- F2c)2]/(n - p)]1/2, where n and p denote the number of data and parameters. R1 ) (∑||Fo| - |Fc||)/∑|Fo|, wR2 ) [∑ -[w(F2
o- F2c)2]/∑[w(F2o)2]]1/2where w ) 1/[σ2(F2o) + (aP)2+ bP] and P ) [(max; 0, F2o) + 2F2c]/3.
Table 6. Crystal and Intensity Collection Data for (C5Me5)(dppp)RuCdC(Ph)CHCN (3a),
(C5Me5)(dppp)RuCdC(Ph)CH(CN)C(CH3)2O (4), [(C5Me5)(dppp)RudC-C(Ph)dC(CN)C(CH3)2O][I]‚2CHCl3(6), [(C5Me5)(dppp)RudCCH(Ph)CH(CN)C(CH3)2O][CF3COO]‚1/2CF3COOH (7), and [(C5Me5)(dppp)RuNCCH2I][I3]
(11) C47H47NP2Ru, 3a C50H53NOPRu, 4 C52H53Cl6INOP2Ru, 6 C54H55.5Cl3F4.5NO4P2Ru, 7 C39H43I4NP2Ru, 11 space group P21/n P212121 P21/c P1h P21/c
cryst syst monoclinic orthorhombic monoclinic triclinic monoclinic
a, Å 16.1153(3) 11.2771(3) 21.4873(4) 12.1587(5) 15.5340(1) b, Å 12.2925(1) 17.8994(4) 13.2421(3) 19.1984(8) 13.2322(1) c, Å 19.9973(4) 23.8595(5) 19.5952(4) 24.7050(9) 21.3003(2) R, deg 90 90 90 69.400(1) 90 β, deg 95.010(1) 90 105.965(1) 76.820(1) 103.684(1) γ, deg 90 90 90 89.762(1) 90 V, Å3 3946.28(11) 4816.1(2) 5360.5(2) 5237(6) 4253.98(6) Z 4 4 4 4 4 cryst dimens, mm 0.40× 0.22 × 0.18 0.35× 0.15 × 0.12 0.35× 0.30 × 0.05 0.20× 0.18 × 0.04 0.36× 0.34 × 0.05 2θ range, deg 1.56 to 27.50 1.42 to 26.37 1.83 to 26.37 0.91 to 25.00 1.35 to 26.40
total no. of reflns 9047 9796 10 542 17 577 8552
final R indices [I > 2σ(I)] R1 ) 0.0279, wR2 ) 0.0715 R1 ) 0.0702, wR2 ) 0.1702 R1 ) 0.0542, wR2 ) 0.1282 R1 ) 0.0964, wR2 ) 0.1848 R1 ) 0.0491, wR2 ) 0.1162 R indices (all data) R1 ) 0.0394, wR2 ) 0.0779 R1 ) 0.1166 wR2 ) 0.1928 R1 ) 0.0945, wR2 ) 0.1456 R1 ) 0.1487, wR2 ) 0.2080 R1 ) 0.0593, wR2 ) 0.1245
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
![Table 5. Selected Bond Distances (Å) and Angles (deg) of [(η 5 -C 5 Me 5 )(dppp)RuNCCH 2 I][I 3 ] (11)](https://thumb-ap.123doks.com/thumbv2/9libinfo/8673115.196287/6.918.131.398.72.375/table-selected-bond-distances-å-angles-dppp-runcch.webp)