行政院國家科學委員會專題研究計畫 成果報告
利用重要功能位置預測直系同源關係
計畫類別: 個別型計畫 計畫編號: NSC93-2311-B-009-004- 執行期間: 93 年 08 月 01 日至 94 年 07 月 31 日 執行單位: 國立交通大學資訊科學學系(所) 計畫主持人: 胡毓志 計畫參與人員: 林勁伍,黃子緯 報告類型: 精簡報告 報告附件: 出席國際會議研究心得報告及發表論文 處理方式: 本計畫可公開查詢中 華 民 國 94 年 8 月 29 日
行政院國家科學委員會補助專題研究計畫
■成果報告
□期中 進度報告
利用重要功能位置預測直系同源關係
計畫類別:■ 個別型計畫 □ 整合型計畫
計畫編號:NSC 93-2311-B-009-004-
執行期間: 93 年 8 月 1 日至 94 年 7 月 31 日
計畫主持人:胡毓志
共同主持人:
計畫參與人員: 林勁伍,黃子緯
成果報告類型(依經費核定清單規定繳交):■精簡報告 □完整報告
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
■出席國際學術會議心得報告及發表之論文各一份
□國際合作研究計畫國外研究報告書一份
處理方式:除產學合作研究計畫、提升產業技術及人才培育研究計畫、
列管計畫及下列情形者外,得立即公開查詢
□涉及專利或其他智慧財產權,□一年□二年後可公開查詢
執行單位:交通大學資訊科學系
中 華 民 國 94 年 8 月 29 日
行政院國家科學委員會專題研究計畫成果報告
國科會專題研究計畫成果報告撰寫格式說明
Preparation of NSC Project Reports
計畫編號:NSC
92-2213-E-009
-098-執行期限:93 年 8 月 1 日至 94 年 7 月 31 日
主持人:胡毓志 交通大學資訊科學系
計畫參與人員:
林勁伍,黃子緯
交通大學資訊科學系
一、中文摘要 準確預測基因功能在蛋白質體的時代扮演 著重要的角色。在替未知基因作功能註解 時,最可靠的方式是透過其他物種的直系 同源基因來萃取功能資訊。我們提出了一 個以重要位置為基礎的方法來辨識直系同 源關係。本研究在同源基因的多序列排比 上找出可以區分不同旁系同源基因的重要 位置,並且利用這些位置作為特徵,對於 未知基因進行直系同源關係的預測。本研 究應用於數個家族,例如一個高度探討的 桿 菌 轉 錄 子 家 族 LacI family ,α-proteasome family , glycoprotein hormone family 以 及 growth hormone family 等來驗證本研究對於直系同源基因 的預測準確度,並且與其他的親緣分析方 法,諸如 ClustalW,BLAST,INPARANOID 等,做有系統之比較。 關鍵詞: 功能重要位置,直系同源,旁系同 源 Abstract
Making accurate functional predictions for genes plays an important role in the era of proteomics. The most reliable functional information is extracted from orthologs in other species when annotating an unknown gene. Here a site-based approach is proposed to predict orthologous relations. We explore functionally important sites in the multiple sequence alignment of orthologous and paralogous proteins and use these sites to build a model that is able to classify orthologous relations of unknown proteins. Our method provides substantial information for guiding experiments such as site-directed mutagenesis to elucidate the orthologous relations. We tested our prediction system on the bacterial transcription factor PurR/LacI family, the α-proteasome family, the glycoprotein hormone family and the growth hormone family to demonstrate its ability to predict orthologs. In addition, we also compared it with other current similar
methods such as ClustalW, BLAST and INPARANOID.
Keywords: functionally important sites, ortholog, paralog
Introduction
Rapid sequencing has generated lots of data to be annotated. This is typically done by searching sequence databases for the best-fit homolog and then assigning its functional annotation to novel proteins/genes. Although the homologous relations have been identified for most of the sequences, as the advance of functional genomics, an accurate and efficient functional prediction method is required to distinguish between orthologs and paralogs [6]. Since incorrect prediction of orthologous relations may result in misjudgment of cellular function and erroneous metabolic pathway reconstruction [5, 9], careful discrimination between orthologs and paralogs has drawn much attention recently.
Several approaches have been developed to detect orthologous sequences. Cotter et al. used closely related sequences as outgroup sequences to refine the BLAST search [4]. However, selecting proper outgroup sequences requires domain knowledge that is not always available. Others applied statistical resampling techniques to multiple sequence alignments to verify the reliability of phylogenetic tree [18]. Storm and Sonnhammer introduced the support value for evaluating sequence orthology [16]. One drawback of the methods above is that they highly depend on the correctness of calculated phylogenetic trees. Unlike previous works, we develop a novel orthology prediction method based on the functionally important sites of orthologs. The motivation behind our method is that active protein residues are under evolutionary pressure to maintain their functional integrity. They undergo fewer mutations than less functionally important amino acids. Consequently, functionally important sites may be used to better characterize orthologous relations. The
orthologous relation of an unknown protein sequence is then inferred from the important sites found. We assume that some important residues are conserved in orthologous proteins to maintain their identical function while divergent in paralogous proteins to reflect their specificity. We explore functionally important sites in the multiple sequence alignment of orthologous and paralogous proteins and use these sites to build a model that is able to classify orthologous relations of unknown proteins.
System
We refer the functionally important sites of an orthologous family to those residues: (1) well conserved within orthologs and (2) divergent among paralogs. Residues with both properties in a multiple sequence alignment of homologs (orthologs and paralogs) are considered important and will be used to construct the classification model of orthologous subfamilies. Given an alignment of homologous proteins that have been properly partitioned into orthologous subfamilies, we evaluate the degree of inter-paralog divergence and intraortholog conservation of each site by calculating the adjust Rand Index [10] and the entropy. Given an unknown protein x and a set of homologs already divided into I ortholog subfamilies that are paralogous to each other, our goal is to classify x to the most appropriate subfamily based on the important sites found. Our procedure of classification is as follows:
(1) Calculate the similarity of x to each sequence j in subfamily i, respectively.
(2) Calculate the similarity of x to entire subfamily i.
(3) Assign x to the subfamily with the
highest similarity.
Experimental Results
We tested our method on the PurR/LacI family and the protein kinase AGC family to verify its ability to identify functionally important sites. We also applied our method to the AGC family, the glycoprotein hormone family, the α-proteasome family and the somatotropin hormone family to demonstrate its performance in the prediction of orthologous relations. Sensitivity and positive predictive value(PPV) are commonly used to measure prediction performance. They are defined as follows:
Higher sensitivity of a prediction algorithm reflects its ability to cover more true positives, and higher positive predictive value indicates the ability to better avoid false positives. However, for most prediction algorithms, it is difficult to obtain a high score of both sensitivity and positive predictive value because these two measures generally contradict each other. To consider both measures at the same time, we further combine them into an F-score [12] to evaluate prediction performance. The definition of F-score on prediction is as follows:
Identification of Functionally Important Sites
We compared our method with Mirny and Gelfand’s [8] in the identification of functionally important sites in two families.
There are twelve important sites in the PurR/LacI family, nine of which are binding sites (DNA or ligand) and the others interact with other residues or form special conformation. Our method successfully identified the twelve important sites and four putative sites that are next to or in the proximity of the binding sites.
There are 39 important sites in the AGC family, including the substrate-inhibitor binding sites, the Mg2ATP binding sites, and some residues that are close to or interact with these binding sites [13, 15, 3]. Our method identified 22 sites, ten (Trp84, Glu127, Phe129, Glu170, Thr183, Phe187, Thr197, Leu198, Pro202 and Leu205) of which are substrate-inhibitor binding sites or ATP binding sites, two (Lys189 and Cys199) of which are related to the protein structure, and five (Arg56, Met120, Leu132, Pro169 and Ala188) of which are next to particular binding sites. Seventeen sites identified by our method have been biologically verified and published in literature.
The results of sensitivity and positive predictive value are summarized in Table 1. The sensitivity and positive predictive value of our method are 1.000 and 0.750 in PurR/LacI family; 0.436 and 0.773 in AGC family. In both cases, our method obtains
better F-scores than Mirny and Gelfand’s method [14, 13]. Furthermore, our method requires much less CPU time than Mirny and Gelfand’s, which is hindered by the complex resampling procedure. Simulated on an AMD Athlon 1.0GHZ machine with 512 MB RAM, our computational time was in the order of minutes compared with hours of Mirny and Gelfand’s.
Prediction of Orthologous Relations
We tested our method on the AGC family, the glycoprotein hormone family, the
α-proteasome family and the somatotropin
hormone family to demonstrate its performance in the prediction of orthologous relations. For comparison, we applied CLUSTALW [17], profile HMMs [13], PSIBLAST [1] and Meta-MEME [2, 7] to the same data. A three-fold cross validation was used to evaluate the predictive accuracy. In each run, we used one third of the data for testing, and the remaining data for training. The results were summarized in Table 2. It shows that our method is comparable with others. Profile HMMs had an almost perfect prediction for the AGC family, the glycoprotein hormone family, and the
α-proteasome family, but they were short of comprehensible interpretations of the orthologous relations found. Unlike others, our method makes a prediction based on the functionally important sites carrying biological meanings. The orthologous relations with the functional sites predicted by our method can be further analyzed by site-directed mutageneses. Associations between functionally important residues and evolutionary relations can be established.
Discussion
We have proposed a method capable of not only identifying functionally important sites in a set of homologous proteins, but also predicting orthologous relations for new protein sequences. It first identifies the putative functionally important residues related to specificity among paralogous proteins and then it uses these residues to construct a model to classify unknown protein
sequences.
For the PurR/LacI family, our method not only successfully identified all the binding sites, but also highlighted the residues that are responsible for protein conformation. As for the AGC family, we found 17 residues that are located in the binding domains or interact with other important sites to form particular conformation related to the kinase function.
Our method identified several active sites in the cleft between the two lobes with the adenine ring of ATP deeply buried at the base of the cleft. Many of the important sites we identified interact with other residues to form the interaction network.
In addition to demonstrating the ability of our method to detect functionally important sites, we also systematically evaluated its performance in the prediction of orthologous relations on four families. Compared with other approaches, our method is more accurate and efficient in general.
Unlike most previous works, besides the prediction of orthologous relations, our method also suggests useful associations between functionally important sites and orthologous families. This type of information may provide biologists with new research topics and eventually become useful domain knowledge.
Our current method can be further improved in two directions. Firstly, as multiple sequence alignment is essential to the identification of important sites, we can improve the quality of sequence alignment by incorporating more background knowledge to ensure the correctness of the alignment. Secondly, associations between important sites and their physicochemical properties can be further exploited to refine the predictive accuracy
References
[1] S. Altschul, T. Madden, A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. Lipman. Gapped blast and psi-blast: a new generation of protein database search programs. Nucleic
Acids Research, 25:3389–3402, 1997.
[2] T. Bailey and C. Elkan. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proceedings
of the Second International Conference on Intelligent Systems for Molecular Biology,
2:28–36, 1994.
[3] R. Brinkworth, R. Breinl, and B. Kobe. Structural basis and prediction of substrate specificity in protein serine/threonine kinases.
Proceedings of the National Academy of Sciences of the United States of America,
100(1):74–79, 2002.
[4] R. Cotter, D. Caffrey, and D. Sgields. Improved database searches for orthologous sequences by conditioning on outgroup sequences. Bioinformatics, 18(1):87–91, 2002. [5] R. Doolittle, D. Feng, S. Tsang, G. Cho, and E. Little. Determining divergence times of the major kingdoms of living organisms with a protein clock. Science, 271:470–477, 1996. [6] W. Fitch. Distinguishing homologous from analogous proteins. Systematic Zoology, 19:99–113, 1970.
[7] W. Grundy, T. Bailey, C. Elkan, and M. Baker. Metameme: Motif-based hidden markov models of protein families. Computer
Applications in the Biosciences, 13(4):397–
406, 1997.
[8] S. Hannenhalli and R. Russell. Analysis and prediction of functional sub-types from protein sequence alignments. Journal of
Molecular Biology, 303:61–76, 2000.
[9] S. Henikoff, E. Greene, S. Pietrokovski, P. Bork, T. Attwood, and L. Hood. Gene families: The taxonomy of protein paralogs and chimeras. Science, 278:609–614, 1997.
[10] L. Hubert and P. Arabie. Comparing partitions. Journal of Classification,
2:193–218, 1985.
[11] L. Kuo, J. Lee, P. Cheng, and J. Pai. Bayes inference for technological substitution data with data-based transformation. Journal
of Forecasting, 16:65–82, 1997.
[12] D. Lewis and W. A. Gale. A sequential algorithm for training text classifier.
Proceedings of the 17th Annual International ACM-SIGIR Conference on Research and Development in Information Retrieval, pages
3–12, 1994.
[13] L. Li, E. Shakhnovich, and L. Mirny. Amino acids determining enzyme-substrate specificity in prokaryotic and eukaryotic protein kinases. Proceedings of the National
Academy of Sciences of the United States of America, 100(8):4463–4468, 2003.
[14] L. Mirny and M. Gelfand. Using orthologous and paralogous proteins to identify specificity-determining residues in bacterial transcription factors. Journal of
Molecular Biology, 321:7–20, 2002.
[15] C. Smith, E. Radzio-Andzelm, Madhusudan, P. Akamine, and S. Taylor. The catalytic subunit of camp-dependent protein
kinase: prototype for an extended network of communication. Progress in Biophysics and
Molecular Biology, 71:313–341, 1999.
[16] C. E. Storm and E. Sonnhammer. Automated ortholog inference from phylogenetic trees and calculation of orthology reliability. Bioinformatics, 18(1):92–99, 2002.
[17] J. Thompson, D. Higgins, and T. Gibson. Clustal w: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice.
Nucleic Acids Research, 22(22):4673–4680,
1994.
[18] Y. Yuan, O. Eulenstein, M. Vingron, and P. Bork. Towards detection of orthologues in sequence database. Bioinformatics, 14(3):285–289, 1998.
行政院國家科學委員會補助國內專家學者出席國際學術會議報告
94 年 8 月 29 日報告人姓
名
胡 毓 志
服務
機構
及職稱
交通大學資訊科學系
副教授
時間
會議
地點
06/20/2005-06/23/2005
Las Vegas, USA
本會核定
補助文號
NSC 93-2311-B-009-004-
會議
名稱
(中文) 數學暨工程醫學生物應用國際研討會
(英文)2004 International Conference on Mathematics and
Engineering Techniques in Medicine and Biological Sciences
發表
論文
題目
1.(中文)
核糖核酸分群與二級結構預測(英文)
RNA Clustering and Secondary Structure Prediction附
件
報告內容應包括下列各項: 一、參加會議經過
於 06/20 辦理註冊報到,隨即參加 Opening Address,於 06/20-06/23 期間,參加與會學 者之論文發表,同時,06/22 發表論文。並與多位國外學者討論相關研究議題。由於 Human Genome Project 即完成,會議中有數篇有關後基因體研究之論文,其中不乏有關 基因發掘,蛋白質結構分析,調控訊號檢視等等,對於我國內生物資訊的發展,將提供 非常多的助益與新的發展方向。 二、與會心得 本次參加人數及國家眾多,其研究領域更包括計算機科學、醫學、生物學等之應用,藉 由討論及論文發表,獲得寶貴經驗,對於未來研究提供了新的方向。其中更結識他國友 人,經由研討,可明白其他國家的發展經驗。 國內已有許多研究單位的多位學者及專家致力於生物晶片的開發,這將使我國未來有自 主能力採集重要基因組的基因表現資料,這不但可減少購買設備之成本,也使我國生物 科技產業邁前一大步。雖然微矩陣相關技術及硬體設備的蓬勃發展僅是近年的事,然 而,其應用的潛力己深獲肯定,可預見的是,在不久的將來,大量的基因表現資料將如 同 DNA 及蛋白質序列般,不斷地被產生及發表,如何能從這些不同類型的生化資料中 發掘有用的訊息將是重要課題。硬體必須要有軟體的配合,硬體所產生的實驗數據資料 有賴軟體的分析,藉由這次與會學習的經驗,我們可以得知國外研究之重點,作為我國 在生物科技的發展依據。 三、考察參觀活動(無是項活動者省略) 四、建議 數學與工程的應用極廣,由於生物科技是目前國內新興研究發展之重要產業,懇請國科 會及相關單位,能多支持與獎勵國內學者多參與此類國際研討會,除了增加我國在國際 相關領域的能見度,同時,提供相互學習之機會。此外,建議由國科會主導,召集國內 各大學與民間企業支援,以召開國際性生物資訊與相關科技研討會,邀請國內外學者共 同參與,這是直接提昇我國在生技發展地位的最有效做法。 五、攜回資料名稱及內容 The Proceedings of METMBS.
RNA Clustering and Secondary Structure Prediction
Yuh-Jyh Hu
Computer and Information Science Department
National Chiao Tung University
1001 Ta Hsueh Rd., Hsinchu, Taiwan
[email protected]
TEL: +886-3-573-1795
FAX: +886-3-572-1490
Abstract
RNA plays a crucial role in post-transcriptional regulation. Similar to transcriptional regulation, post-transcriptional regulation is often accomplished by the binding of proteins to specific motifs in mRNA molecules. Unlike DNA binding proteins, which recognize motifs composed of conserved sequences, RNA protein binding sites are more conserved in structures than in sequences. A lot of works have been done for RNA structure prediction; however, most of them focus on single RNA structure prediction instead of finding characteristic structure motifs within a RNA family. Though some current approaches can now identify common structure motifs from a set of RNAs, they typically assume the given set forms a single family, which is not necessarily correct. We propose a new adaptive method that conducts structure prediction and clustering simultaneously. Its performance is demonstrated on several real RNA families.
Introduction
RNA molecules are the key players in the biochemistry of the cell, playing many important roles in regulation, catalysis and structural support. Like proteins, their functions generally depend on their structures. Although structural genomics, the systematic study of all macro-molecular structures in a genome, is currently focused more on proteins, thousands of genes produce transcripts exerting their functions without ever producing protein products [1]. It can be easily argued that the comprehensive understanding of the biology of a cell requires the knowledge of identity of all functional RNAs (both non-coding and protein-coding) and their
molecular structures. Since it is often difficult to acquire the 3D spectrum data of RNA molecules for structure determination, versatile and reliable computational methods that can predict RNA structures are highly desirable.
Many functional RNAs have evolutionarily conserved secondary structures in order to fulfill their roles in a cell. For protein-coding RNAs, some of the functions can be presented by functional motifs. For example, several best-understood structurally conserved RNA motifs are found in viral RNAs, such as the TAR and RRE structures in HIV and the IRES regions in Picornaviridae [2]. Apparently, structural information is very useful in characterizing a class of functional RNAs. Based on characteristic structures, we can likely identify novel functional RNAs or partition given RNAs into biologically meaningful families. Several systems have been developed to find consensus structural elements within a family of functionally related RNAs [3-5]; however, there is little work on clustering of unaligned RNAs based on characteristic secondary structures. Given a set of unaligned RNA sequences without prior knowledge of the number or identity of families in the set, our goal is to automate both clustering and secondary structure prediction simultaneously. In this paper, we propose an adaptive approximation approach combined with a genetic programming-based structure prediction method to identify from unaligned RNAs reasonable clusters associated with characteristic secondary structure elements. To demonstrate its performance, we tested it on several real datasets.
RNA Clustering and Structure Prediction
Unlike previous studies of RNA secondary structure prediction whose input is either a single RNA sequence or a known class of functionally related sequences, our new method is instead applied to a set of unaligned RNA sequences which consist of an unknown number of classes. In order to find a reasonable partition for a given set of unaligned RNAs without knowing beforehand how many clusters actually existing in this set, we assume that each cluster is likely a functional family that contains characteristic structure motifs. Based on this assumption, our new method is focused on finding significant consensus structure motifs that can be used to characterize the families of RNAs. Since the number of clusters and its size are unknown in advance, we take a generate-and-test strategy that iteratively adjusts the hypothesized cluster size until some significant consensus structure elements can be found associated with this cluster. After a cluster is obtained, all its members are then removed from the given RNAs. We repeat the same separate-and-conquer strategy to identify other clusters from the remaining RNAs.
Generate-and-Test
The generate-and-test strategy we use is an adaptive approximation approach that systematically revises the hypothesized cluster size. During the generate-and-test process, the cluster size is defined by a range between an upper bound U and a lower bound L. Without any prior information of clusters, the cluster size is initialized within a range between an upper bound
U=n and a lower bound L=0, that is, we first assume that all the given RNA sequences consist in
an entire family. To the entire family, a genetic programming-based structure prediction method is applied to look for the fittest consensus structure motifs. If the specificity of the structure motifs associated with a cluster exceeds or equals some pre-specified threshold, the hypothesis of the cluster is accepted, and the cluster along with the associated structure elements will be reported. On the other hand, low specificity suggests that the current hypothesized cluster size is too big to be real and needs to be decreased. In this case, we reduce the current hypothesized cluster, and search the fittest consensus structure motifs and evaluate their specificity again. If the specificity is still lower than the threshold, we further decrease the cluster size. The same process for cluster size reduction can be repeated till we find a cluster with structure motifs of high-specificity. On the contrary, if the specificity is over or equal to the threshold, one of the two possibilities holds: (1) the current cluster is real, and any more sequences added will be harmful to the specificity of consensus structures, or (2) the current cluster found is only a subset of a bigger real cluster. To verify which event actually happens, we increase the cluster size and a new search for the fittest consensus structure motifs is conducted. As each update generates a tighter range for cluster size, we expect the cluster size will eventually converge to the appropriate one.
Secondary Structure Element Prediction by Genetic Programming
The objective here is to learn the structure elements that can be used to distinguish the given functionally related sequences from the random sequences. We modify the fitness function of our previous work [6] on RNA consensus secondary structure prediction to find significant structure
elements from a dataset that may contain multiple variable-sized clusters of unaligned sequences. The fitness function is used to measure the quality of individuals (i.e. candidate structure elements) in a population. The higher the fitness of an individual, the better its chances of survival to the next generation. In the previous work, the input dataset was assumed to be a single class of functionally related RNA sequences. We were interested in those structure elements that can reflect the characteristics conserved in a family, e.g. the RNA protein binding sites. Derived from the F-score, the fitness function was aimed to balance the importance of two measures, recall (i.e. sensitivity) and precision (i.e. positive predictive value) [4]. It assigns higher values to those structural motifs commonly shared by the given family of RNAs, and rarely contained in random sequences. For a given set of RNA sequences that form a single family only, the fitness function used in [4,6] can effectively guide the evolutionary process in genetic programming. Nevertheless, when the input dataset contains multiple functional classes, the recall measure may dominate the calculation of F-score if the fitness function treats the entire dataset as a single class. This will mislead the system to find over-general elements shared by most sequences. To alleviate the bias, we define a new measure of recall, and present the fitness function as below, where p is the number of positive examples containing motifi, Q is the total number of positive examples, R is the total number of examples containing motifi, and U is the upper bound of the hypothesized range for cluster size.
) ( Pr ) ( Re ) ( Pr ) ( Re 2 ) ( i i i i i motif ecision motif call motif ecision motif call motif Fitness + ∗ ∗ = ≥ < = if , 1 if , ) ( Re U p U p motif call i Q p R p motif ecision( i)= Pr
By taking cluster size into account, we can better constrain the search space and allow conserved clusters to emerge more likely instead of being buried in bigger but much less coherent clusters.
Consensus Structure Specificity and Separate-and-Conquer Strategy
The GP (Genetic Programming)-based structure prediction method can find the fittest secondary structure elements according to a given range of the cluster size, while the significance of the cluster found along with its characteristic structure elements highly depends on the range we choose. With proper adjustment of cluster size through the generate-and-test procedure combined with the GP-based prediction method, we can identify a meaningful cluster and the associated characteristic structure elements.
The adaptive adjustment of cluster size in the generate-and-test procedure is controlled by the consensus structure specificity. It is defined as the Laplace prior precision. The Laplace prior approach has also been applied to inductive leaning to evaluate the significance of inductive rules [7]. The Laplace prior precision of cluster Ci is given by the formula:
2 1 ) ( Pr Pr + + = i i i C in examples of number total C in examples positive of number C ecision ior Laplace
conserved clusters whose size is too small. For example, the Laplace prior precision of a cluster of 50 positive examples and five negative examples is better than that of a cluster of only five positive examples. Note that the Laplace prior precision is only used to determine the significance of a cluster found, unlike the F-score, which is used to direct the optimization process to find the best structure elements under the constraints of the cluster size. Based on the comparison of the Laplace prior precision with a pre-specified threshold, we adjust the range of cluster size accordingly, and then re-run the GP-based method to predict new structure elements and a new cluster they characterize.
Once a significant cluster is found, we separate all its members out of the given dataset of RNA sequences. We then apply the same procedure to those that still remain in the dataset until the entire set is emptied. This separate-and-conquer strategy is effective when no prior knowledge of the identities of the clusters is given. It can automatically partition the given dataset into meaningful clusters, and also identify their characteristic structure elements.
Experimental Results
Two types of quality were considered to evaluate the performance of our method. One is to measure the agreement between the predicted clusters and the actual cluster identities; the other, to quantify the agreement between the predicted structure elements and the actual structure assignment. Since no other current approaches known to perform clustering and structure prediction in parallel, no comparative study can be done. Instead we applied the widely-used precision and recall to measure the first quality; the Matthews correlation coefficient [8], to measure the second quality.
For each sequence in the data set, two secondary structure assignments were compared by counting the number of true positives Pt (base pairs exist in actual assignment and are predicted), true negatives Nt (base pairs do not exist in actual assignment and are not predicted), false positives Pf (base pairs do not exist in
actual assignment but are predicted) and false negatives Nf (base pairs exist in
actual assignment but are not predicted), respectively. The Matthews correlation coefficient can then be computed as:
C= PtNt−PfNf
(Nt+Nf)(Nt+Pf)(Pt+Nf)(Pt+Pf)
Given that the sequence length is sufficiently large, the Matthews correlation coefficient can be approximated in the following way [5].
C ≈ Pt
Pt+Nf ⋅
Pt
Pt+Pf
With the published/curated alignments, we can calculate the Matthews correlation coefficient. Higher correlation coefficients mean more accurate structure predictions.
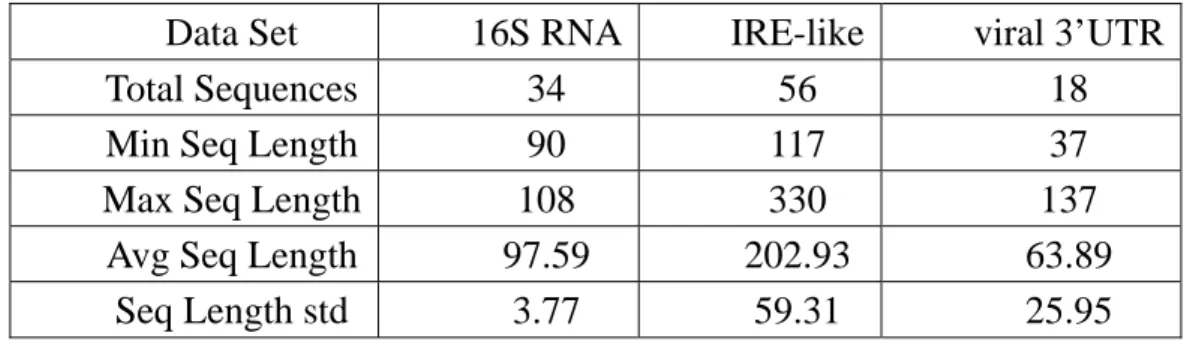
Our algorithm is designed to automatically partition a given set of unaligned RNA sequences into meaningful clusters, each with characteristic conserved secondary structure elements. The number of real clusters and the distribution of cluster size may affect the prediction of partitions and characteristic structure elements. To measure their effect on the performance, we tested our method on different datasets with various RNA families. We used three families, including 16S RNA, IRE (Iron Response Element) and viral 3’UTR as summarized in Table 1, to prepare the test datasets. They have been used in previous experiments and published in literature [4,5]. The sequence data and the correct structure elements can be accessed at public databases [9,10]. The 16S RNA dataset contains 34 archaea 16S ribosomal sequences originally derived from a set of 311 sequences extracted from the SSU rRNA database. The archaea set of 311
sequences was further reduced to 34, filtering out the sequences that miss base assignments or are greater than 90% identical. The IRE dataset was constructed by Gorodkin et al. [5] from 14 sequences from the UTR database. They modified the IREs and their UTRs to make the search more difficult. By iteratively shuffling the sequences and randomly adding one nucleotide to the IRE conserved region, they built a set of 56 IRE-like sequences from the 14 IRE UTRs. The third data set includes 18 viral 3'UTRs each of which contains a pseudoknot. Seven of the RNA sequences are the soil-borne rye mosaic viruses; the others are the soil-borne wheat mosaic viruses.
On the basis of the three real families of RNA sequences, we tested our method on each possible pair of the families, i.e. 16S RNA/IRE, 16S RNA/viral 3’UTR, and IRE/viral 3’UTR. In each run of the experiment, no information regarding the number of families or the family size was given to the algorithm beforehand. One purpose of this experiment is to analyze the effect incurred by the distribution of cluster size in a dataset. Furthermore, as the real conserved structure elements differ in various families, we can also observe how the interleaving of distinct structure motifs within a single dataset may affect the prediction process. The results are presented in Table 2, and some partial predicted secondary structures are shown in Figure 1.
Conclusion
In this paper, we propose a new approach that can perform structure prediction and clustering simultaneously for RNA analysis. The predicted results provide biologists with reasonable hypotheses and suggest further biological verifications. The performance of the new strategy has been demonstrated on several real RNA functional families. The system can be extended in the following directions. First, in case domain knowledge is available, we expect the results can be better improved by incorporating the background knowledge into the optimization process to effectively constrain the search space. Second, the discovery of important clusters in data usually goes through a repeated process cycle of finding clusters, interpreting results and augmenting data. No current unsupervised clustering system can produce maximally useful results if operated alone [11]. We plan to design a human-machine interface, so that biologists can easily monitor the system status and adapt the system parameter settings. Third, the algorithm itself is highly modular and most of the modules are independent of each other. This property may lead to a parallel-processing version of the system to significantly reduce its computational time.
Data Set 16S RNA IRE-like viral 3’UTR
Total Sequences 34 56 18
Min Seq Length 90 117 37
Max Seq Length 108 330 137 Avg Seq Length 97.59 202.93 63.89
Seq Length std 3.77 59.31 25.95
Table 1. Summary of the RNA families used in experiments. The first row shows the total number of sequences in each data set. Row 2 to 4 present the minimum, the maximum and the average sequence length respectively. The fifth row gives the standard deviation of sequence length.
(a)
IRE+viral 3’UTR Recall Precision Matthews
IRE 0.97 0.99 0.97
viral 3’UTR 0.71 0.95 0.79 (b)
16S RNA+viral 3’UTR Recall Precision Matthews
16S RNA 0.97 0.95 0.83
viral 3’UTR 0.77 0.98 0.77 (c)
IRE+16S RNA Recall Precision Matthews
IRE 0.73 0.99 0.85
16S RNA 0.81 0.73 0.67
Table 2. Summary of the experimental results. Table (a), (b) and (c) present the result for the dataset containing IRE and viral 3’UTR, 16S RNA and viral 3’UTR, IRE and 16S RNA, respectively. ***** IRE ***** > seq_D15071.1 41 45 47 51 58 62 63 67 t g c g g u c c u g g c c a g u g a g c u g g g c c g c predicted: . ( ( ( ( ( . ( ( ( ( ( . . . ) ) ) ) ) ) ) ) ) ) published: . ( ( ( ( ( . ( ( ( ( ( . . . ) ) ) ) ) ) ) ) ) ) ***** 16S RNA ***** > U51469 13 20 23 31 37 46 52 61 g u u u c a u u g a a g u u u g c u u u u a g u g a g g u g a c g u c u a a u u g g c g u u a u c g 62 67 75 78 85 a a c u u g u g g u a a g c g a c a a g g g a a a a predicted: . ( ( ( ( ( ( ( ( . . ( ( ( ( ( ( ( ( ( . . . ( ( ( ( ( ( ( ( ( ( . . . ) ) ) ) ) ) ) ) ) ) . . . ) ) ) ) ) ) ) ) ) . . ) ) ) ) ) ) ) ) . . published: . ( ( ( ( ( ( ( ( . . ( ( ( ( ( ( ( ( ( ( ( ( ( . . ( ( ( ( ( ( ( ( ( . . . ) ) ) ) ) ) ) ) ) . . ) ) ) ) ) ) ) ) ) ) ) ) ) . . ) ) ) ) ) ) ) ) . . ***** viral 3’UTR ***** > PKB183 14 16 18 24 25 27 32 38 a c g u c g u g c a g u a c g g u a a a c u g c a c a u predicted: . ( ( ( . [ [ [ [ [ [ [ ) ) ) . . . . ] ] ] ] ] ] ] . . published: . ( ( ( . [ [ [ [ [ [ [ ) ) ) . . . . ] ] ] ] ] ] ] . .
Figure 1. A partial result of the predicted RNA motifs. The numbers above the sequences are the indices of the nucleotides. The predicted and the published motifs are both shown for reference.
References
1. The Genome Sequencing Consortium (2001) “Gene content of the human genome”, Nature, 409, p860-921.
2. Hofacker, I., Priwitzer, B. and Stadler, P. (2004) “Prediction of locally stable RNA secondary structures for enome-wide surveys”, Bioinformatics, 20, p186-190.
3. Eddy, S. and Durbin, R. (1994) “RNA sequence analysis using covariance models”, Nucleic Acids Res., 22, p2079-2088.
sequences", Nucleic Acids Res., 30, p3886-3893.
5. Gorodkin, J., Stricklin, S. L. and Stormo, G. D. (2001) “Discovering common stem-loop motifs in unaligned RNA sequences”, Nucleic Acids Res., 29, 2135-2144. 6. Hu, Y. (2003) "GPRM: a genetic programming approach to finding common RNA secondary
structure elements", Nucleic Acids Res., 31, p3446-3449.
7. Clark, P and Boswell, R. (1991) “Rule Induction with CN2: some recent improvements”, in Proceedings of the Fifth European Conference on Machine Learning, p151-163.
8. Matthews, B.W. (1975) “Comparison of the predicted and observed secondary structure of T4 phage lysozyme”, Biochem. Biophys. Acta, 405, 442-451.
9. Batenburg, F.H.D. van, Gultyaev, A.P. and Pleij, C.W.A. (2001) “PseudoBase: structural information on RNA pseudoknots”, Nucleic Acids Res., 28, 1, 201-204. 10. Hu, Y. (2002) "The NCTU BioInfo Archive of biological data sets for bioinformatics research
and experimentation", Bioinformatics Vol 18, No 8, p1145-1146.
11. Cheeseman, P. and Stutz. J. (1996) “Bayesian Classification (AUTOCLASS): Theory and Results”, in Advances in Knowledge Discovery and Data Mining, p153-180, AAAI.