Molecular dynamics analysis of potent inhibitors of M2 proton channel against
H1N1 swine influenza virus
Chia-Yu Laiab, Tung-Ti Changab, Mao-Feng Sunac, Hsin-Yi Chend, Fuu-Jen Tsaide, Jaung-Geng Linaand Calvin Yu-Chian Chenadf*
a
Laboratory of Computational and Systems Biology, School of Chinese Medicine, China Medical University, Taichung 40402, Taiwan, ROC;bDepartment of Chinese Pediatrics, China Medical University Hospital, Taichung 40402, Taiwan, ROC;cDepartment of Acupuncture, China Medical University Hospital, Taichung 40402, Taiwan, ROC;dDepartment of Bioinformatics, Asia University, Taichung 41354, Taiwan, ROC;eDepartment of Medical Genetics, China Medical University, Taichung 40402, Taiwan, ROC;
f
Computational and Systems Biology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA (Received 14 August 2010; final version received 13 November 2010)
The recent H1N1 (swine) influenza pandemic highlighted the urgent need of having effective anti-viral strategies. In addition to neuraminidase inhibitors, there is another class of anti-viral drug known as M2 inhibitors that were, in the past, effective in treating seasonal influenza. However, due to the emergence of M2 inhibitor-resistant influenza viruses, this class of drugs was not recommended for clinical usage in the latest influenza pandemic. In order to identify novel M2 inhibitors, we have performed molecular docking using a traditional Chinese medicine database (http://tcm.cmu.edu.tw/index.php). Docking and subsequent de novo designs gave 10 derivatives that have much better docking results than the control. Of these 10 derivatives, the top three, methyl isoferulate_1, genipin_1 and genipin_2, were selected for molecular dynamics simulation. During the simulation run, the top three derivatives all had stable interactions with M2 residues, Ser31 and Ala30. Methyl isoferulate_1 also has stable interaction to His37. Therefore, we recommend these three derivatives for further biomolecular experiments and clinical studies.
Keywords: H1N1; M2 proton channel; docking; molecular dynamics; traditional Chinese medicine (TCM)
1. Introduction
The influenza virus contains an M2 proton channel that is essential for the viral replication cycle. In addition to two viral surface glycoproteins, haemagglutinin and neurami-nidase, that are important in viral entry and viral release, M2 is the third class of membrane protein that activates at low pH and allows protons to enter the viral interior [1]. The influx of protons into the viral interior facilitates the dissociation of M1 matrix protein from viral RNA, which is crucial for the subsequent release of viral genome into the cytoplasm [2]. In terms of structure, M2 is a small, tetrameric protein that consists of a short N-terminal, a C-terminal tail and a transmembrane domain known to be responsible for proton transport [3,4].
In the past, adamantane-based M2 inhibitors were used to treat seasonal influenza. However, the prevalence of adamantane-resistant virus in recent influenza H1N1 pandemics has greatly curtailed the effectiveness of this class of anti-viral compounds [5,6]. Nevertheless, the recent emergence of neuraminidase inhibitor-resistant influenza viral strains has called attention to the need for a novel anti-viral strategy [6], and hence, research for novel
M2 inhibitors is as important as for new neuraminidase inhibitors [7].
In the hope of finding novel M2 inhibitors, we introduced a traditional Chinese medicine (TCM) database (http://tcm.cmu.edu.tw/index.php) into our research. Natural products, generally, have a large chemical diversity, thereby could be a great source for searching novel lead compounds. Furthermore, in the past, several novel anti-cancer or anti-inflammatory compounds have been discovered from medicinal plants [8 – 10]. Overall, we based our research on structure-based techniques and performed molecular docking and molecular dynamics (MD) simulation. Both molecular docking and MD simulation techniques were successfully implemented in drug design [11 – 20], and we have already used these techniques for drug design before [21 – 41].
2. Materials and methods 2.1 Molecular docking
The TCM ingredients obtained from the TCM database were docked into the M2 protein crystal. The starting M2
ISSN 0892-7022 print/ISSN 1029-0435 online
q2011 Taylor & Francis
DOI: 10.1080/08927022.2010.543972 http://www.informaworld.com
*Corresponding author. Email: ycc@mail.cmu.edu.tw; ycc929@mit.edu Vol. 37, No. 3, March 2011, 250–256
crystal structure was downloaded from the Protein Data Bank (PDB code: 3C9J) [42]. This high-resolution crystallographic structure solved by Stouffer et al. [42] has amantadine in the M2 protein in which the drug physically occludes the pore. Amantadine binding site is surrounded by residues (Val27, Ala30, Ser31 and Gly34) that are often mutated in the amantadine-resistant viral strains [43]. We, therefore, set the amantadine binding location in the crystal as our docking site.
The LigandFit program of Discovery Studio 2.5 0.9164 (Accelrys, Inc., San Diego, CA, USA) was used to dock the TCM ingredients. In preparation for docking, all water molecules were removed from the protein. Forcefield of Chemistry at Harvard Macromolecular mechanics (CHARMm) was applied on both the TCM constituents and amantadine before docking.
The scoring function outputs of amantadine were used as control. The docking results of other TCM constituents were then compared with the scores maintained. Compounds with docking scores higher than the control were taken for de novo evolution.
2.2 De novo evolution and Lipinski’s Rule of Five Compounds with higher docking scores than amantadine were chosen from the previous step. The Ludi algorithm is used in the de novo design. Fragments from the Ludi-formatted libraries are fitted into the interaction sites calculated by the
Ludi algorithm [44], and fragments with high Ludi score are fused or linked to the existing TCM scaffold.
Derivatives generated from de novo evolution were first screened with Lipinski’s Rule of Five before being docked back to M2 protein. Lipinski’s Rule of Five is a general guide for evaluating drug likeness [45].
2.3 MD simulation
Selected M2-derivative complexes were chosen for MD simulation using Standard Dynamics cascade of Discovery Studio 2.5. Each system was pre-applied with CHARMm force field and then energy was minimised with 500 and 500 steps of steepest descent and conjugate gradient minimis-ation. The system was heated to 310 K for 20 ps before entering 100 ps of equilibration phase. The production phase was conducted for 40 ns on a NVT ensemble at 310 K. A SHAKE algorithm was applied, and the step size was set to 2 fs throughout the entire simulation run. The non-bonded interaction cut-off was set at 10 A˚ , while spherical cut-off was used to calculate long range electrostatics. The trajectory snapshots were saved every 20 ps.
3. Results and discussion 3.1 Docking and de novo design
Compounds from the TCM database were docked into the amantadine binding site suggested by Stouffer et al. [42]. Table 1. Top six candidates and the control, amantadine.
Name Dock score
Binding
energy (Kcal/Mol) Ludi1 Ludi2 Ludi3 2 PMF04 Jain
Quinic acid 42.95 2 166.976 417 357 345 2 9.21 2.93 Genipin 42.18 2 107.598 417 362 320 2 12.8 2.27 Syringic acid 41.60 2 107.968 269 292 426 76 0.05 Cucurbitine 39.94 2 130.047 348 305 291 17.17 3.82 Fagarine 39.39 2 57.6988 324 297 590 68.95 2.37 Methyl isoferulate 38.68 2 126.665 383 340 317 2 3.86 1.33 Amantadine 33.87 2 49.0867 235 274 243 2 2.9 0.71
Table 2. Top 10 derivatives obtained from docking of de novo products.
Name Dock score
Binding
energy (Kcal/Mol) Ludi1 Ludi2 Ludi3 2 PMF04 JAIN
Methyl isoferulate_1 57.17 2 152.088 613 495 563 10.67 2.61 Genipin_1 55.61 2 161.667 593 491 447 2 9.82 3.47 Genipin_2 54.84 2 128.943 600 508 464 2 7.48 3.69 Methyl isoferulate_2 54.64 2 143.905 654 500 539 15.14 1.71 Genipin_6 53.60 2 136.371 519 445 407 2 4.91 3.73 Genipin_3 53.00 2 169.56 548 465 434 2 8.82 4.16 Limettin_9 52.49 2 85.3504 492 436 426 14.2 3.43 Limettin_7 52.24 2 87.649 399 349 445 22.51 1.76 Genipin_4 52.21 2 144.255 505 430 395 2 4.69 2.04 Genipin_8 51.72 2 103.383 471 419 373 2 9.72 3.91 Amantadine 33.87 2 49.0867 235 274 243 2 2.9 0.71
Amantadine that co-crystallised with the M2 protein was extracted from the crystal and was re-docked back to the protein to evaluate the binding pose and binding affinity. Dock Score implemented in DS 2.5, which evaluates ligand and receptor interaction energies, was used to rank the screening results. The screening results of amantadine and the top six TCM ingredients are shown in Table 1. All the top six TCM ingredients have dock score and the binding energy higher than the control, amantadine.
We have taken the docked TCM ingredients to generate derivatives. To screen out compounds that may have poor oral bioavailability, derivatives were first screened with Lipinski’s Rule of Five, before being docked back to M2 protein. Derivative docking results are summarised in Table 2. All the top 10 derivatives have elevated dock scores and low binding energy than amantadine and their parent compounds. The 2D structures and docking poses of the top three derivatives and the control are shown in Figures 1 and 2. The alignment of the top 10 derivative docking conformations is shown in Figure 3. All of the genipin derivatives have very similar binding confor-mation, as shown in Figure 3(b). This is a great contrast to limettin derivatives, in which each compound has very distinct binding conformation (Figure 3(c)).
By comparing the docking conformations of the top derivatives with the control, it is clear that there are no hydrogen bonds formed between the amantadine and the M2
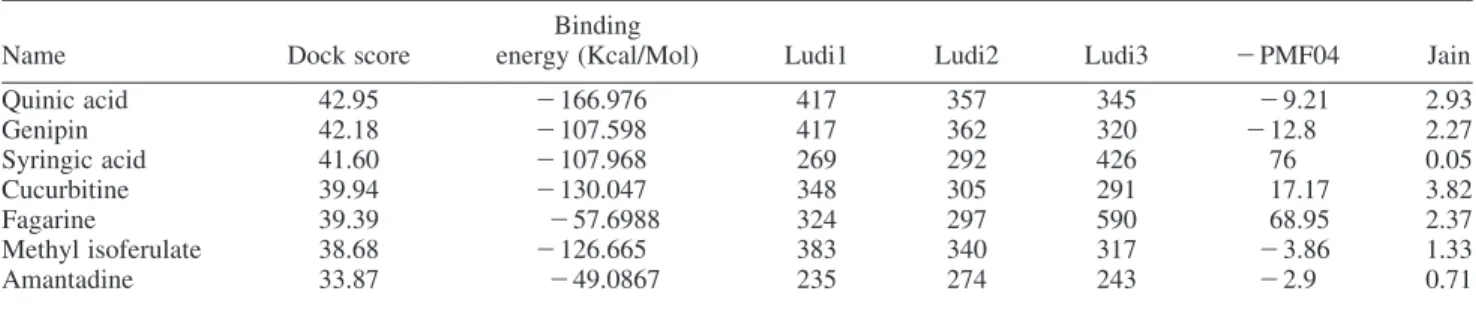
Figure 2. Docking conformations of (a) amantadine, (b) methyl isoferulate_1, (c) genipin_1 and (d) genipin_2. HO O O O N OH NH2 O OH O O H OH HO H O OH O O H H OH HO
(a) Amantadine (b) Methyl isoferulate_1
(c) Genipin_1 (d) Genipin_2
Figure 1. 2D structure of control amantadine and the top three derivatives.
protein, whereas several hydrogen bonds could be observed between the derivatives and M2 residues (Figure 2). As shown in Figure 2(b)– (d), methyl isoferluate_1, genipin_1 and genipin_2 all have hydrogen bond interactions with Ala30 and Ser31. Methyl isoferulate_1, in particular, forms interaction with His37, the residue responsible for the activation and selectivity of the channel [46]. This
interaction may be responsible for the elevated dock score of methyl isoferulate_1. Genipin_2 also has hydrogen bonding interaction with Val27, another key residue in amantadine binding site. Genipin_1 differs from genipin_2 in the addition of a methoxy group that can form hydrogen bonding interaction with Ala30 of chain A. On other hand, genipin_2 has an addition of hydroxyl group.
3.2 MD simulation
We selected the top three TCM derivatives for 40 ns MD simulation. The root mean square deviations (RMSDs) of the M2-amantadine and the M2-derivative complexes are shown in Figure 4. The RMSD values of all the structures were calculated by aligning all of the frames to the initial production structures. The RMSDs plot shows that each derivative complex reaches stability at a different time. The ligand RMSDs graph, however, shows that the TCM derivatives are relatively stable during the course of MD simulation (Figure 4). Thus, it is most likely that the large fluctuations in molecule RMSDs could be due to protein movement, but not changes in ligand conformations.
A summary of all the hydrogen bonds formed during the MD simulation is shown in Table 3. The main scaffold of amantadine is derived from adamantane, which is extremely hydrophobic. Therefore, it is not surprising for
Figure 3. Alignment of all the top 10 derivative products. The derivatives are divided by their parental compounds: (a) methyl isoferulate, (b) genipin and (c) limettin.
1.0 1.2 1.4 1.6 1.8 2.0 2.2 2.4 2.6 2.8 3.0 0 5 10 15 20 25 30 35 40 0.0 0.4 0.8 1.2 1.6 2.0 2.4 2.8 3.2 3.6 4.0 Amantadine Methyl isoferulate_1 Genipin_1 Genipin_2 Whole molecule RMSD Time (ns) Ligand RMSD
Figure 4. RMSDs of the M2 – ligand complexes (top) and the ligands (bottom). The unit is in angstrom (A˚ ).
amantadine to have a relatively few hydrogen bonds (Figure 5). All other derivatives have more ionisable sites that favour hydrogen bonding interactions with M2.
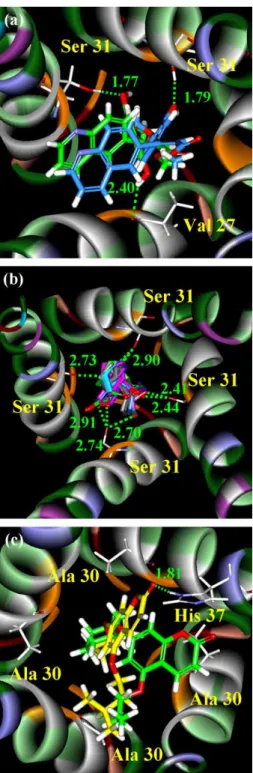
For methyl isoferulate_1, five hydrogen bonds were observed during the course of simulation. Hydrogen bonding to Ser31 of chain A and His37 of chain D appears to be most stable during the simulation. The hydrogen bonding interaction of methyl isoferulate_1 with Ser31 of chain C was originally thought to be negligible, until a detail examination of the hydrogen bonding pattern (Figure 6), which shows this hydrogen bond interaction was maintained during the production run at an average distance of 2.92 A˚ . For Genipin_1, four hydrogen bond interactions were observed with the interaction with Ser31 of chain D being the most interesting one (Figure 7). This interaction was found at the beginning of the simulation, Table 3. Summary of hydrogen bonds formed during the 40 ns MD simulation.
Max. distance (A˚ ) Min. distance (A˚ ) Ave. distance (A˚ ) % of occupancy
D:Ala30 O – Amantadine H12 3.98 1.93 2.77 32.30
D:Ala30 O – Amantadine H13 3.86 2.02 2.90 22.95
A:Ser31 HG – Methyl isoferulate_1 O24 2.37 1.70 2.33 72.75
A:Ser31 OG – Methyl isoferulate_1 H25 2.85 1.66 1.99 99.20
D:His37 HD1 – Methyl isoferulate_1 O7 3.22 1.62 1.95 99.95
C:Ala30 O – Methyl isoferulate_1 H43 3.25 1.82 2.42 67.10
C:Ser31 HG – Methyl isoferulate_1 O18 3.50 2.38 2.92 0.45
A:Ala30 O – Genipin_1 H12 3.49 1.86 2.50 54.25
D:SER31 OG – Genipin_1 H27 4.45 1.85 3.44 10.35
A:Ser31 HG – Genipin_1 O26 3.49 1.73 2.22 91.50
D:Ser31 HG – Genipin_1 O26 4.29 2.00 2.72 18.80
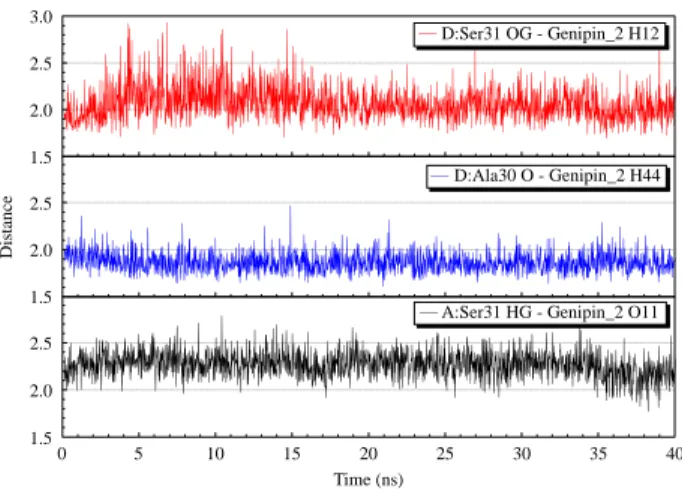
D:Ser31 OG – Genipin_2 H12 2.93 1.69 2.07 98.05
D:Ala30 O – Genipin_2 H44 2.47 1.61 1.87 100.00
A:Ser31 HG – Genipin_2 O11 2.79 1.77 2.27 95.45
0 5 10 15 20 25 30 35 40 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0 Time (ns) Distance
D:Ala30 O - Amantadine H12 D:Ala30 O - Amantadine H13
Figure 5. Hydrogen bonds formed between amantadine and M2 residues. The distance unit is in A˚ .
1.0 1.5 2.0 2.5 3.0 3.5
Distance C:Ala30 O - Methyl isoferulate_1 H43 1.0
1.5 2.0 2.5 3.0
3.5 D:His37 HD1 - Methyl isoferulate_1 O7 1.0 1.5 2.0 2.5 3.0 3.5 4.0
A:Ser31 HG - Methyl isoferulate_1 O24 A:Ser31 OG - Methyl isoferulate_1 H25
0 5 10 15 20 25 30 35 40 1.5 2.0 2.5 3.0 3.5
4.0 C:Ser31 HG - Methyl isoferulate_1:O18
Time (ns)
Figure 6. Hydrogen bond distance (unit in A˚ ) of interactions formed during the 40 ns MD simulation.
1.0 1.5 2.0 2.5 3.0 3.5 4.0 Distance A:Ala30 O - Genipin_1 H12 1.5 2.0 2.5 3.0 3.5 4.0 4.5
5.0 D:Ser31 OG- Genipin_1 H27
1.0 1.5 2.0 2.5 3.0
3.5 A: Ser31 HG - Genipin_1 O26
0 5 10 15 20 25 30 35 40 1.5 2.0 2.5 3.0 3.5 4.0 Time (ns)
D:Ser31 HG - Genipin_1 O26
Figure 7. Distance (unit in A˚ ) of hydrogen bonds formed between M2 residues and genipin_1 during the 40 ns MD simulation.
but was diminished after 8 ns. For Genipin_2, all interactions are found to be between Ser31 and Ala30 (Figure 8). All these results demonstrate that the derivatives are able to bind more tightly than amantadine to M2 protein and have great potential to become clinical M2 inhibitors.
4. Conclusions
Our docking and MD simulation gave 6 TCM compounds and 10 TCM derivatives that serve high potential for being M2 inhibitors. TCM derivatives, methyl isoferulate_1, genipin_1 and genipin_2 all have docking results better than amantadine and already have several hydrogen bonding interactions with key residues Ser31 and Ala30 prior to docking simulation. MD simulations of the top three derivatives show that interaction with Ser31 and Ala30 was maintained throughout the simulation time. In addition, methyl isoferulate_1 also has interaction with His37, which is critical for proton selectivity. All these results suggest that the top three derivatives mentioned before could be potential M2 inhibitors.
Acknowledgements
The research was supported by grants from the National Science Council of Taiwan (NSC 99-2221-E-039-013-), China Medical University (CMU98-TCM, CMU99-S-02) and Asia University (CMU98-ASIA-09). This study is also supported in part by Taiwan Department of Health Clinical Trial and Research Center of Excellence (DOH99-TD-B-111-004) and Taiwan Department of Health Cancer Research Center of Excellence (DOH99-TD-C-111-005). We are grateful to the National Center of High-performance Computing for computer time and facilities.
References
[1] A. Helenius, Unpacking the incoming influenza virus, Cell 69 (1992), pp. 577 – 578.
[2] L.H. Pinto and R.A. Lamb, The M2 proton channels of influenza A and B viruses, J. Biol. Chem. 281 (2006), pp. 8997 – 9000. [3] R.J. Sugrue and A.J. Hay, Structural characteristics of the M2
protein of influenza A viruses: Evidence that it forms a tetrameric channel, Virology 180 (1991), pp. 617 – 624.
[4] L.H. Pinto, G.R. Dieckmann, C.S. Gandhi, C.G. Papworth, J. Braman, M.A. Shaughnessy, J.D. Lear, R.A. Lamb, and W.F. DeGrado, A functionally defined model for the M2 proton channel of influenza A virus suggests a mechanism for its ion selectivity, Proc. Natl Acad. Sci. USA 94 (1997), pp. 11301 – 11306.
[5] N.A. Ilyushina, E.A. Govorkova, and R.G. Webster, Detection of amantadine-resistant variants among avian influenza viruses isolated in North America and Asia, Virology 341 (2005), pp. 102 – 106.
[6] P.K. Cheng, T.W. Leung, E.C. Ho, P.C. Leung, A.Y. Ng, M.Y. Lai, and W.W. Lim, Oseltamivir- and amantadine-resistant influenza viruses A (H1N1), Emerg. Infect. Dis. 15 (2009), pp. 966 – 968. [7] Q.S. Du, R.B. Huang, S.Q. Wang, and K.C. Chou, Designing
inhibitors of M2 proton channel against H1N1 swine influenza virus, Plos One 5 (2010), e9388.
[8] Y.J. Tang, J.S. Yang, C.F. Lin, W.C. Shyu, M. Tsuzuki, C.C. Lu, Y.F. Chen, and K.C. Lai, Houttuynia cordata Thunb extract induces apoptosis through mitochondrial-dependent pathway in HT-29 human colon adenocarcinoma cells, Oncol. Rep. 22 (2009), pp. 1051 – 1056.
[9] C.R. Su, Y.F. Chen, M.J. Liou, H.Y. Tsai, W.S. Chang, and T.S. Wu, Anti-inflammatory activities of furanoditerpenoids and other constituents from Fibraurea tinctoria, Bioorgan. Med. Chem. 16 (2008), pp. 9603 – 9609.
[10] Y.H. Hsieh, F.H. Chu, Y.S. Wang, S.C. Chien, S.T. Chang, J.F. Shanw, C.Y. Chen, W.W. Hsiao, Y.H. Kuo, and S.Y. Wang, Antrocamphin A, an anti-inflammatory principal from the fruiting body of Taiwanofungus camphoratus, and its mechanisms, J. Agric. Food Chem. 58 (2010), pp. 3153 – 3158.
[11] C.D. Yoo, S.C. Kim, and S.H. Lee, Molecular dynamics simulation study of probe diffusion in liquid n-alkanes, Mol. Simulat. 35 (2009), pp. 241 – 247.
[12] D.X. Li, G.L. Chen, B.L. Liu, and Y.S. Liu, Molecular simulation of
b-cyclodextrin inclusion complex with 2-phenylethyl alcohol, Mol. Simulat. 35 (2009), pp. 199 – 204.
[13] F. Luan, H.T. Liu, Y. Gao, and X.Y. Zhang, QSPR model to predict the thermal stabilities of second-order nonlinear optical (NLO) chromophore molecules, Mol. Simulat. 35 (2009), pp. 248 – 257. [14] J.H. Jing, G.Z. Liang, H. Mei, S.Y. Xiao, Z.N. Xia, and Z.L. Li,
Quantitative structure – mobility relationship studies of dipeptides in capillary zone electrophoresis using three-dimensional holographic vector of atomic interaction field, Mol. Simulat. 35 (2009), pp. 263 – 269.
[15] A.M. Al-Mekhnaqi, M.S. Mayeed, and G.M. Newaz, Prediction of protein conformation in water and on surfaces by Monte Carlo simulations using united-atom method, Mol. Simulat. 35 (2009), pp. 292 – 300.
[16] M.L. Mihajlovic and P.M. Mitrasinovic, Applications of the ArgusLab4/AScore protocol in the structure-based binding affinity prediction of various inhibitors of group-1 and group-2 influenza virus neuraminidases (NAs), Mol. Simulat. 35 (2009), pp. 311 – 324. [17] K. Roy and G. Ghosh, QSTR with extended topochemical atom (ETA) indices. 11. Comparative QSAR of acute NSAID cytotoxicity in rat hepatocytes using chemometric tools, Mol. Simulat. 35 (2009), pp. 648 – 659.
[18] P. Nimmanpipug, V.S. Lee, P. Wolschann, and S. Hannongbua, Litchi chinensis-derived terpenoid as anti-HIV-1 protease agent: Structural design from molecular dynamics simulations, Mol. Simulat. 35 (2009), pp. 673 – 680.
[19] P. Ghosh and M.C. Bagchi, Comparative QSAR studies of nitrofuranyl amide derivatives using theoretical structural proper-ties, Mol. Simulat. 35 (2009), pp. 1185 – 1200.
[20] Y.S. Zhao, Q.C. Zheng, H.X. Zhang, H.Y. Chu, and C.C. Sun, Homology modelling and molecular dynamics study of human fatty acid amide hydrolase, Mol. Simulat. 35 (2009), pp. 1201 – 1208. [21] C.Y. Chen, Insights into designing the dual-targeted HER2/HSP90
inhibitors, J. Mol. Graph Model. 29 (2010), pp. 21 – 31.
0 5 10 15 20 25 30 35 40 1.5 2.0 2.5 Distance Time (ns)
A:Ser31 HG - Genipin_2 O11 1.5 2.0 2.5 D:Ala30 O - Genipin_2 H44 1.5 2.0 2.5 3.0 D:Ser31 OG - Genipin_2 H12
Figure 8. Hydrogen bond interactions formed between M2 and genipin2 during the 40 ns simulation. The distance unit is in A˚ .
[22] H.J. Huang, K.J. Lee, H.W. Yu, H.Y. Chen, F.J. Tsai, and C.Y.C. Chen, A novel strategy for designing the selective PPAR agonist by the “Sum of Activity” model, J. Biomol. Struct. Dyn. 28 (2010), pp. 187 – 200.
[23] H.J. Huang, K.J. Lee, H.W. Yu, C.Y. Chen, C.H. Hsu, H.Y. Chen, F.J. Tsai, and C.Y.C. Chen, Structure-based and ligand-based drug design for HER2 receptor, J. Biomol. Struct. Dyn. 28 (2010), pp. 23 – 37.
[24] H.J. Huang, C.Y. Chen, H.Y. Chen, F.J. Tsai, and C.Y.C. Chen, Computational screening and QSAR analysis for design of AMP-activated protein kinase agonist, J. Taiwan Inst. Chem. E 41 (2010), pp. 352 – 359.
[25] C.Y.C. Chen, Virtual screening and drug design for PDE-5 receptor from traditional chinese medicine database, J. Biomol. Struct. Dyn. 27 (2010), pp. 627 – 640.
[26] C.Y.C. Chen, Bioinformatics, chemoinformatics, and pharmainfor-matics analysis of HER2/HSP90 dual-targeted inhibitors, J. Taiwan Inst. Chem. E 41 (2010), pp. 143 – 149.
[27] C.Y. Chen, H.J. Huang, F.J. Tsai, and C.Y.C. Chen, Drug design for influenza: A virus subtype H1N1, J. Taiwan Inst. Chem. E 41 (2010), pp. 8 – 15.
[28] C.Y.C. Chen, Weighted equation and rules – A novel concept for evaluating protein – ligand interaction, J. Biomol. Struct. Dyn. 27 (2009), pp. 271 – 282.
[29] C.Y.C. Chen, Computational screening and design of traditional Chinese medicine (TCM) to block phosphodiesterase-5, J. Mol. Graph Model. 28 (2009), pp. 261 – 269.
[30] C.Y. Chen, Y.H. Chang, D.T. Bau, H.J. Huang, F.J. Tsai, C.H. Tsai, and C.Y.C. Chen, Ligand-based dual target drug design for H1N1: Swine flu – A preliminary first study, J. Biomol. Struct. Dyn. 27 (2009), pp. 171 – 178.
[31] C.Y. Chen, Y.H. Chang, D.T. Bau, H.J. Huang, F.J. Tsai, C.H. Tsai, and C.Y.C. Chen, Discovery of potent inhibitors for phosphodi-esterase 5 by virtual screening and pharmacophore analysis, Acta Pharmacol. Sin. 30 (2009), pp. 1186 – 1194.
[32] C.Y.C. Chen, Pharmacoinformatics approach for mPGES-1 in anti-inflammation by 3D-QSAR pharmacophore mapping, J. Taiwan Inst. Chem. E 40 (2009), pp. 155 – 161.
[33] C.Y.C. Chen, De novo design of novel selective COX-2 inhibitors: From virtual screening to pharmacophore analysis, J. Taiwan Inst. Chem. E 40 (2009), pp. 55 – 69.
[34] C.Y.C. Chen, Chemoinformatics and pharmacoinformatics approach for exploring the GABA-A agonist from Chinese herb suanzaoren, J. Taiwan Inst. Chem. E 40 (2009), pp. 36 – 47.
[35] C.Y.C. Chen, Insights into the suanzaoren mechanism – From constructing the 3D structure of GABA-A receptor to its binding interaction analysis, J. Chin. Inst. Chem. Eng. 39 (2008), pp. 663 – 671.
[36] C.Y.C. Chen, Discovery of novel inhibitors for c-Met by virtual screening and pharmacophore analysis, J. Chin. Inst. Chem. Eng. 39 (2008), pp. 617 – 624.
[37] C.Y.C. Chen, Y.F. Chen, C.H. Wu, and H.Y. Tsai, What is the effective component in suanzaoren decoction for curing insomnia? Discovery by virtual screening and molecular dynamic simulation, J. Biomol. Struct. Dyn. 26 (2008), pp. 57 – 64.
[38] C.Y.C. Chen, A novel perspective on designing the inhibitor of HER2 receptor, J. Chin. Inst. Chem. Eng. 39 (2008), pp. 291 – 299. [39] C.Y.C. Chen, G.W. Chen, and W.Y.C. Chen, Molecular simulation of HER2/neu degradation by inhibiting HSP90, J. Chin. Chem. Soc.-Taiwan 55 (2008), pp. 297 – 302.
[40] Y.C. Chen and K.T. Chen, Novel selective inhibitors of hydroxyxanthone derivatives for human cyclooxygenase-2, Acta Pharmacol. Sin. 28 (2007), pp. 2027 – 2032.
[41] Y.C. Chen, The molecular dynamic simulation of zolpidem interaction with gamma aminobutyric acid type A receptor, J. Chin. Chem. Soc.-Taiwan 54 (2007), pp. 653 – 658.
[42] A.L. Stouffer, R. Acharya, D. Salom, A.S. Levine, L. Di Costanzo, C.S. Soto, V. Tereshko, V. Nanda, S. Stayrook, and W.F. DeGrado, Structural basis for the function and inhibition of an influenza virus proton channel, Nature 451 (2008), pp. 596 – 599.
[43] V.M. Deyde, X.Y. Xu, R.A. Bright, M. Shaw, C.B. Smith, Y. Zhang, Y.L. Shu, L.V. Gubareva, N.J. Cox, and A.I. Klimov, Surveillance of resistance to adamantanes among influenza A(H3N2) and A(H1N1) viruses isolated worldwide, J. Infect. Dis. 196 (2007), pp. 249 – 257.
[44] H.J. Bohm, The computer program LUDI: A new method for the de novo design of enzyme inhibitors, J. Comput. Aided Mol. Des. 6 (1992), pp. 61 – 78.
[45] C.A. Lipinski, F. Lombardo, B.W. Dominy, and P.J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Deliver Rev. 46 (2001), pp. 3 – 26.
[46] J. Hu, R. Fu, K. Nishimura, L. Zhang, H.X. Zhou, D.D. Busath, V. Vijayvergiya, and T.A. Cross, Histidines, heart of the hydrogen ion channel from influenza A virus: Toward an understanding of conductance and proton selectivity, Proc. Natl Acad. Sci. USA 103 (2006), pp. 6865 – 6870.