國 立 臺 灣 大 學 農 業 化 學 研 究 所
博 士 論 文
衰 化 種 子 活 勢 指 標 分 析 :
衰 化 綠 豆 存 活 幼 苗 R N A 解 螺 旋
的 選 殖 及 特 性
Vigour analysis of accelerated aging seeds:
Cloning and characterization of a RNA helicase from the
viable seedlings of aged mung bean.
指導老師:陳慶三 教授
研究生:李信昌 撰
學號:D85623601
I. 序論
1.1 植物之老化及衰化
1.2 種子壽命
1.3 種子衰化
1.4 種子活勢定義及觀念
1.5 種子活勢測定
1.6 衰化種子生理與生化的變化
1.7 衰化種子發芽初期的變化
1.8 RNA 解旋
的特性
1.9 RNA 解旋
在生物體內扮演的角色
1.10 RNA 解旋
結構學方面的研究
1.11 RNA 解旋
可能作用機制的探討
1.12 RNA 解旋
在植物學方面的研究
1.1 植物之老化及衰化 (Senescence and aging in plants)
老化 (senescence) 可以定義為導致組織器官或整個植株死亡之生理變化過 程。許多植物(例如水稻)於開花結實後整個植物體死亡的過程可稱之為老化, 又如有些落葉植物在秋、冬季節葉片會變黃脫落,葉片從綠色到趨近脫落的死亡 過程亦稱之為老化,Senescence 通常存有內源性可控制的退化過程,伴隨著計劃 性細胞死亡(programmed cell death)(1, 2)。從文獻中可以看到另一個相近似的名
稱 ”aging”經常容易與老化混淆,嚴格的說 aging 與 senescence 意義不同,Aging 係指植物隨著時間變化,所產生之生理變化過程,其結果並不一定是死亡,中文 譯名可稱之為衰化,Aging 過程屬於被動性、非調節性及退化性,其造成的主要 原因是由於外源性因子,Aging 本身不會直接造成植物死亡,但是會降低植物於
惡劣環境下的適應力,提高死亡的機率(2)。例如植物隨著時間增加,每年植株高
度生長降低;煙草癒傷組織之生長與分化能力隨著年齡增加而降低;種子儲藏過 程中其發芽能力之降低都可稱之為 aging。然而 senescence 與 aging 彼此間由於
生化上之性質並非瞭解很多,通常無法將二者很明確的劃分(2)。
引導細胞趨於死亡的模式可區分為三 (Davies and Sigee, 1984)(3),包含有:
(1) 計劃性細胞死亡(programmed cell death, PCD);(2) 壞死(necrosis);(3)慢 性退化(chronic degeneration)。Senescence 屬於 PCD,細胞循一定的步驟走向 死亡。而 Necrosis 則是細胞於短時間內大量累積傷害所造成的死亡,例如急速凍 傷、機械傷害或病源菌感染等。Chronic degeneration 則是細胞隨著長時間地慢慢 累積傷害而後死亡,也就是 aging。Aging 和 Necrosis 的差別在於外源性傷害強 度的大小,細胞因累積損傷的增加而提高死亡機率;然而在另一方面,細胞亦藉 由本身修補機制來恢復活力,種種因素取決於環境壓力(environmental stress) 之 大小。曾有報告指出細胞於較低的環境壓力下(例如低溫冷凍),此時細胞走向 死亡之模式為 senescence(2)。
1.2 種子壽命 (The life of seeds)
種子壽命是指自種子採收至失去發芽能力所經過的時間。一粒種子除非讓它 發芽,否則無法判別其生或死。在適當的條件下若可以發芽,因為種子已長成幼 苗,所以可能低估該種子的壽命;若不克發芽,又非休眠,則該種子已死,而且 死於何時無從查知,因此也無法測量其壽命。種子壽命因種子種類不同而異,大 多數種子其壽命都很短,約 5-30 年。探究種子的壽命,有兩個截然不同的方式, 一是測驗某批古老種子是否具有生命,而這種子的年代可以知曉;這個方法基本 上還是如前所論,無法正確地測量種子的壽命。另一個方法則是由經控制的實 驗,將一批種子分次於不同的日期測量發芽率,以記錄該批種子壽命衰退的歷 程,更進一步,由所得的數據推算該種子在某環境下的可能壽命(4, 7 )。 第一個有系統研究種子壽命的是 Beal (1905)(5)。他在 1879 年發表 21 種種子中只有 Oenothera biennis、Rumex crispus、與 Verbascum blatteria 三種可以存活 70 年以上。Beal 於 1879 年在美國密西根農學院校園埋下 23 種植物的種子,種 子拌砂裝於玻璃瓶,瓶口朝下埋於土中,令種子處於土中濕潤狀態。每隔五年 (前 期)、十年 (後期) 取樣一次進行發芽試驗。這個試驗進行到 1980 年時恰好是第 一百年,到 2030 年該試驗才全部完成。供試的 20 餘種種子當中,約五種種子 在首次五年挖出時即已完全不具生命,壽命在 30∼50 年之間者約 11 種,壽命為 80∼100 年者約五種。由這類試驗的結果很明顯地可以看出來,埋於土中可以生 存較久的,皆是具有休眠性的種子;一般作物種子在一年內即在土內發芽死去。 種子壽命的長短係受遺傳與環境因子的影響,而種子水分含量與種子的壽命 有密切的關係。種子水分含量受到種子的成分組成、本身的水分含量與貯藏時之 相對濕度之影響。種子水分含量愈高,則其種子壽命愈短,水分含量愈低,種子 壽命愈長。Robert (1960) 發現種子水分含量與 50%活力 (half- viability, P50) 喪失 所需時間之對數成一直線的關係;而貯藏時之溫度對種子壽命亦有明顯的影響, 溫度高時,種子貯藏壽命短,而溫度低時壽命長。Roberts (1972)(6)由許多試驗
的結果建立水分含量與溫度對種子壽命影響的數學模式:log P50=Kv-C1m-C2t 式中 P50表示 50%種子喪失活力所需的時間,m 為種子水分含量,t 為貯存時的 溫度,而 kv、C1 與 C2 分別為常數。利用該數學模式可準確的預測種子的壽命。 但該模式只考慮到水分含量與貯藏溫度,其他可能的影響因子,如種子貯藏前的 狀況與貯藏時氧氣含量並未考慮。Harrington (1973) 也提出簡單預測種子壽命的 方法,他認為:(一)種子水分含量 (4-14%) 每降低 1%,種子壽命加倍;(二)貯 藏之溫度 (0-50℃) 每降低 5℃,種子壽命加倍。 正儲型種子 (orthodox seeds) 為可乾燥、可冷凍儲藏的種子,例如綠豆、紅 豆等。賈思勰齊民要術,卷一,收種篇:「凡五榖種子浥鬱則不生,生者亦尋死」 浥者濕也,鬱乃熱氣,說明種子在高溫濕潤的環境下容易敗壞。到目前為止,我 們所瞭解影響種子壽命的最大因素,還是浥鬱這兩個字。所謂「生者亦尋死」, 應當指那些倖存的不正常幼苗,雖然發芽,但因其活勢 (vigour) 太弱,即使可 發芽,也是不正常的幼苗,發芽後不久就死去之意。正儲型種子成熟採收之初, 接近 100% 發芽率。固定不變的儲存條件下經過一段時間後,其活勢仍然維持很 高的狀態,發芽率的降低相當緩慢。然後由某階段開始,發芽率的下降突然趨快, 直到整批種子的發芽率相當低時,才又轉緩直到全部喪失生命為止 (下圖)(7)。 正儲型種子壽命的分佈
一般而言,儲藏條件越惡劣 (即溫度越高或種子含水率越高),高活度的期間越 短,發芽率下降的速度也越快。種子存活曲線 (前圖中 a、b) 很接近常態分布的 反向累積頻率曲線,亦即表示種子族群在儲藏時間內的死亡頻率 (前圖中 a`、b`, 或者說壽命頻率) 的分布亦是接近常態,也就是說一批種子中只有少數的種子壽 命很短,也只有少數的種子壽命很長,種子壽命接近於平均值的最多。圖中的 ñ 為平均壽命,在常態分布的狀況下,也代表發芽率由 100% 降到 50% 的儲藏 時間。ó 為死亡頻率分布的標準偏差,亦即發芽率由 50% 降到 15.9% (或 84.1 % 降到 50% ) 所需的儲藏時間,因為這段時間所涵蓋的死亡頻率恰為 34.1% 常態分布的特性。 異儲型種子(recalcitrant seeds):乾燥後會喪失活勢的種子。許多熱帶果樹如 芒果、蓮霧、荔枝、榴璉、可可椰子、波羅蜜;工藝作物如橡膠樹,以及栗、胡 桃、榛等種子,經乾燥後發芽率皆會下降,皆是異儲型。齊民要術,卷四種栗篇: 有關種子的注釋:「栗初熟時出殼,即於屋埋著濕土中,埋必深勿令凍徹。若路 遠者以韋囊盛之;見風日則不復生矣」。這段文字對栗子不耐低溫、乾燥的特性, 描述地十分清楚。 中間型種子(intermediate seeds)種子在-20℃下的壽命不超過一年,又若太 乾燥,在 0℃時的儲藏壽命反而不如在 15℃的久。這些特性偏向於異儲型者;若 種子含水率較高,而溫度也高於 0℃,則溫度越高儲藏壽命越短,這種特性又符 合正儲型的特性,因此稱做中間型,木瓜、油棕、咖啡的種子屬之。
1.3 種子衰化 (Seed aging)
種子 aging 過程中,種子品質(quality)逐漸下降,其主要原因是受到貯藏溫 度、貯藏環境相對濕度、種子本身組成及貯藏時間所影響。其造成的結果包括 DNA 的損傷造成發芽率的降低(8),RNA 代謝過程發生變化(9),改變種子蛋白質 的生成代謝(10, 11),降低發芽初期種子 polyA (+) RNA 的含量(12, 13, 14),細胞膜完 整性降低導致種子滲漏( leakage)程度增加(15),發芽初期乙烯的合成下降(16),脂質的過氧化作用造成種子惡化(17)
種子 aging 被認為是降低種子 viability 和 vigour 的主要原因,包含退化性的 累積造成種子惡化及發芽率降低(18, 19)。影響種子壽命的主要的兩個環境因子包 括溫度及種子本身的水份(20)。以發芽率來測定種子 viability 為國際間公認的主 要方法,然而發芽率試驗的主要限制是無法偵測高發芽率種子彼此間的品質差 異。因此高發芽率種子,並不代表是高品質的種子,為何有這樣的差異呢?主要 原因是在標準的發芽條件下,品質稍差的種子在良好的發芽環境下,仍可以維持 高發芽率。一旦這些種子在較差的環境下發芽或是經由逆境(stress)處理後, 高品質種子與品質較差的種子相較下,仍可維持較高的發芽率。 研究報告指出,相同品種之不同批次、年份種子,在理想的環境下有很高且 相近的發芽率,一但置於田間試驗,或是長時間的儲藏,或是運送,種子發芽就 有顯著的不同(Table 1)(21)。
Table 1: Field, storage and transport performance of seed lots which germination data indicate are of similar quality
Field-Pisum sativum L.1 Seed lots 1 2 3 4 Germination (%) 93 92 95 97 Field emergence (%) 84 71 68 82 Storage-Trifolium pratense L.2 Seed lots 1 2 3 4
Germination before storage (%) 90 90 90 90 Germination after 12 month’s
storage (%)
Transport-Bromus willdenowii Kunth3.
Seed lots
1 2 3 4
Germination before transport (%) 94 96 93 90 Germination after transport
overseas (%)
87 19 74 53
1
adapted from data of Castillo et al. (1993) 2
adapted from data of Wang and Hampton (1991) 3
Hampton, unpublished.
如何在高發芽率種子間判定何者為高活力的種子,種子學家驗證的方法為 seed vigour testing。發芽率為測定種子之最大的發芽能力,其結果可以用來評估 種子播灑於田間的種植效益(22, 23, 97),發芽率的測定在二方面很成功,一方面是 重復性很高,而另一方面則是提供種子於理想環境下其發芽的能力之訊息。種子 aging 會導致發芽率的下降、胚軸(axis)不易突破種皮、對於不良的環境適應力 下降、以及影響初生幼苗的生長(19)。因此有學者研究指出,當種子發芽率低於 90%以下時,種子本身已經有惡化(deterioration)的情形發生(24)。曾有報告指 出實驗室發芽率低的黃豆種子,其在田間的發芽率也顯著的下降,二者間似乎有 良好的相關性(25)。然而在其他的學者研究卻發現,發芽率有明顯差異( 44~98%) 之豌豆(garden pea),其在田間之發芽率比較卻無明顯不同,因此在豌豆的實驗 上,實驗室和田間發芽率試驗,二者無明顯相關(24)。會造成以上差異的主要原 因,是由於發芽率的試驗有其限制性,也就是當一群種子本身就有很高的發芽率 時,我們無法以發芽率來評估個別種子品質上的不同。種子於自然與正常情形下 有其一定的存活曲線,發芽曲線些微下降之種子,此時可能表示種子惡化情形已 經很嚴重(26),在此情形下,學者利用種子內部活力( seed vigour testing)指標的
1.4 種子活勢定義及觀念 (Definition and concept of seed vigour)
Seed vigour 並非只有使用單一指標來測定種子活力,而是藉由多源性指標來 評估種子於儲藏、運送及田間的表現情況(24, 27, 28)。依據 ISTA(International seed
testing association)對 seed vigour 之定義為”決定種子活力程度之所有表現特性, 這些特性決定了種子活性程度、發芽率和發芽速率,其特性表現良好者稱為高活 力種子”(15)。種子 vigour 檢測標準包括以下三方面: 1. 種子發芽率、發芽速率、及幼苗生長有無一致性。 2. 種子於田間生長情形有無一致性。 3. 種子於儲藏、運送後的表現,特別是保留種子之發芽能力有無一致。 種子惡化通常在成熟後便開始進行,並持續延續至採收、加工處理及儲藏過程, 過程中會受到遺傳及環境因子的影響,種子惡化過程有階段性及連續性,其先後 序列很難加以區分。然而導致種子惡化的主要原因被認為是細胞膜物理性及生理 性的破壞(19, 21);種子酵素;呼吸和荷爾蒙的改變;RNA 及蛋白質合成受損;損 害遺傳物質;累積有毒代謝物質(30)。此結果使得種子逐漸降低表現能力,包括 降低發芽速率及發芽一致性、降低種子於逆境下的適應能力、影響幼苗的生長及 表現(21),逆境下除了影響植物的生長及表現外,並會影響作物的生長及產量(19, 21)。從圖一(19)我們可以發現,種子 vigour 的流失早於發芽率,因此高發芽率的 種子,生理性年齡(種子惡化程度)亦有不同,種子因 vigour 不同,因此有不 同表現情形。High vigour 種子與 low vigour 種子在理想環境下的發芽率也許差異 不大,然而在逆境下,high vigour 種子有較好的表現能力。
高發芽率種子其 vigour 的狀態取決於儲藏環境,種子於儲藏過程中所遭遇的 逆境(例如儲藏環境溫度上升或是濕度無法調控),會下降種子 vigour。而種子 經由運送到不同地方,運送過程所遭遇到的環境變異(溫度、濕度嚴重變化), 亦會下降種子 vigour。從 Table 1(21)我們可得知 high vigour 種子不管於田間種植、
1.5 種子活勢測定 (Measuring seed vigour)
種子 vigour 測定法的困難度在於是否可以用量化的方式區別高發芽率種子群 彼此間惡化的程度,其測定方法需符合簡便、有再現性及能夠描述種子於田間的 實際播種結果或是種子本身的儲藏潛能(storage potential)(16)。 過去四十年內,有許多測定 vigour 的方法被加以提出,但只有少數方法為國 際間所通用,其主要原因是 vigour test 的再現性、靈敏度、種子適用性等變因會 影響試驗本身的準確性(21)。然而種子 vigour 為種子品質之重要因子的觀念,已 經被大多數人所接受。Vigour 試驗可區分為三類(24): 1. 以種子發芽現象為基礎。 2. 測量種子生理或生化指標。 3. 多樣性試驗程序。 第一類評估種子 vigour 的方法較為大眾所熟知,例如測量種子發芽速率、幼 苗的生長和發育、低溫發芽試驗、加速 aging 試驗(accelerated aging, AA)及種 子控制惡化試驗(controlled deterioration, CD)等(21)。這些 vigour 試驗法都有操作方便、數據再現性高等優點,廣為一般”種子測試”試驗室所採用,然而依據國 際種子檢查協會(ISTA)標準,這些 vigour 試驗和種子實際在田間的種植,彼 此間仍有差異性。依據 Hamptonh 和 Coolbear 綜合以上的 vigour 試驗發現以加速 aging 試驗和控制惡化試驗,對於種子 vigour 的評估有較佳的效果,其應用性可 幫助瞭解播種潛力、儲藏及運送後的品質以及可以廣泛應用於農業、園藝及林業 種子(31)。
第二類方法為測量種子生理或生化指標,最常使用的方法為電導度試驗 (conductivity test)(83, 98)和 tetrazolium 試驗(TZ test)(21, 96)。其他的試驗策略包
括種子呼吸活力的測定、ATP 含量測定及麩胺酸去羧 活性(glutamic acid
decarboxylase activity;GADA)的測定(32)。除了電導度試驗具有方便、快速及
滿足種子 vigour 試驗的需求外,其他方法仍有改良的空間。
法來進行試驗,例如將豌豆( Pisum sativum L.)於田間的預期播種效益( expected field emergence, E. F. E.)列入計算,或是將小麥(Triticum aestivum L.)與玉米 (Zea mays L.)置於不同之複雜逆境下分析種子 vigour,此外還必需考慮不同品 種間的差異及不同環境下所收穫的種子(24)。
以上的三類方法為依據 ISTA 針對種子 vigour 試驗所歸訥的結果,這些 vigour 試驗本身並無強迫性,也就是種子間並無特定的方法來測定 vigour,然而其測定 結果不僅可提供種子於播種及儲藏的活力指標,更能夠幫助我們區別不同種子群 於內在及外在因子影響下,種子 vigour 表現效益。
1.6 衰化種子生理與生化的變化
種子收穫後,隨著儲藏期間增加造成種子活力逐漸降低,此種活力降低的過 程稱之為種子衰化 (seed aging)。Bewley 和 Black 列舉種子因衰化所產生的惡化 現象有以下幾個特徵,包括有種皮顏色發生變化、胚根延緩突出影響幼苗之生 長、發芽率降低、對於不良環境的耐受力降低、不正常幼苗數目增多、對於輻射 線敏感、衰化種子容易發霉並且於衰化過程中有熱的產生(6, 33)。種子收穫後如果 貯藏於不良環境下,往往製造出許多衰化或老化的種子,這些惡化結果造成種子 活力下降,發芽率下降,導致幼苗存活的機率降低。除此之外,衰化或老化過程 中,尚有下列現象:種子細胞膜受到氧族自由基或脂質過氧化作用而改變(34)、 DNA 引起變異或喪失完整性、蛋白質合成產物降低、代謝作用異常、細胞的微 細構造發生改變(35)。種子活力喪失則其應用價值降低,如何使種子在儲藏期間 活力不喪失,是研究者想要瞭解之問題。
1.7 衰化種子發芽初期的變化
剛收穫之種子發芽快速,呼吸作用、ATP 之形成、各類 RNA 與蛋白質之合 成於種子發芽後不久即開始進行。衰化種子之發芽較新鮮種子為慢,蛋白質與 RNA 之合成速率也低於新鮮種子(36),衰化種子其 DNA 開始複製的時間也延遲(37)。衰化之大麥種子其糊粉層α-amylase 之活性降低,其他酵素,如 protease、 β-amylase、phosphatase、catalase、peroxidase 與 dehydrogenase 等的活性亦會隨 著衰化程度之增加而降低(35)。 衰化種子發芽初期蛋白質合成能力降低,顯示種子衰化過程中會發生許多生 化變化,這些變化可以由膜的間隔化消失;粒線體呼吸作用酵素、ATP 形成系 統 功 能 喪 失 ; 核 糖 體 rRNA 完 整 性 消 失 ; 轉 譯 作 用 酵 素 aminoacyl-tRNA synthetases、transfer enzyme I 與 II 功能喪失;細胞核 DNA 模版完整性喪失; 轉錄作用能力喪失;無法合成 mRNA 以及長壽命 mRNA 功能降低。從微細構造 層次來考慮,具有活力與衰化之種子間有明顯且一致的差異性。例如,死亡之黑 麥種子其胚細胞內之粒線體外膜與破裂之內膜分離,細胞膜收縮與細胞壁分離, 顯示細胞膜之半透性 (semi-permeability) 之特性受到破壞,核膜亦表現不正常與 破裂。這些現象說明了種子衰化過程胚細胞之膜系統發生了明顯的變化。 衰化種子置於水中,細胞質內成分會滲漏到水中,這也顯示細胞膜之完整性 喪失,其滲漏的程度會隨著衰化程度之增加而增加。雖然未衰化之種子亦會表現 滲漏現象,然而新鮮種子之胚發芽時,此滲漏出之物質又可以經過主動運輸的方 式被吸收到胚,由電子顯微鏡觀察之結果,顯示未衰化之種子於發芽初期能夠恢 復膜之透過性,修補貯藏時膜完整性損壞之部分(40, 41)。自由基與不飽和脂肪酸 發生過氧化作用 (peroxidation) 造成膜完整性之喪失(17, 82)。而自由基之形成亦 可以由不飽和脂肪酸含量隨著衰化進行而降低得到證明(17),而衰化種子發芽能 力降低之原因,即可能是由於其無法修補膜完整性損害之部份所造成。 衰化種子浸潤後呼吸作用下降與衰化種子粒線體膜完整性喪失有密切之因果 關係(38)。衰化種子發芽時,粒線體完整性之喪失不僅使呼吸速率下降,同時也 使 ATP 之形成降低。而蛋白質之合成過程是需要利用 ATP,此可解釋為什麼衰 化 種 子 發 芽 初 期 蛋 白 質 合 成 下 降 。 通 常 衰 化 種 子 粒 線 體 之 脫 氫 酵 素 (dehydrogenase) 活性下降(21)。因此,脫氫酵素功能之喪失常用來做為檢定種子 活力之依據。將種子浸於 1%之 2, 3, 5-triphenyltetrazolium chloride (TTC) 中,脫
氫酵素可使無色之 TTC 轉變為紅色之 formazan (42, 43),而衰化種子因脫氫酵素 活性降低,因此將 TTC 轉為 formazan 的能力降低。由染色之深淺部位,即可 以得知種子衰化程度之大小。
黑麥種子衰化過程中已證實 rRNA 之完整性會喪失,由不同衰化程度之種子 中抽取 rRNA,發現 18S rRNA 與 25S rRNA 被降解的程度,隨著衰化程度之增 加而增加(1, 6)。98% 活力之種子其 rRNA 都沒被降解,而活力低的種子其核糖
體之 rRNA 已經被降解,活力愈低,rRNA 被降解的程度愈大(37)。 rRNA 之
降解主要是由於 RNase 持續作用的結果,由於不同衰化程度之種子其 RNase 活 性無差異,因此,rRNA 之降解程度係決定於 rRNA 被 RNase 降解速率之大 小。RNase 不同於脫氫酵素,它在貯藏過程中之乾燥種子內非常穩定。由貯藏 103 年之小麥種子中抽取 RNase, 該酵素仍有活性(39)。
理論上,種子發芽初期在新的 mRNA 尚未合成時,蛋白質是利用原有之 mRNA,該 mRNA 在種子發育過程中合成,種子脫水後該 mRNA 繼續存在種 子內,當種子吸水發芽時該 mRNA 恢復活性合成蛋白質。許多學者因而推測衰 化種子中此長壽命 mRNA 可能受到損害而無法利用來合成蛋白質,但是,比較 0% 活力胚與 90% 活力胚之 poly A-rich mRNA 發現兩者都可以用來合成蛋白 質(39)。0% 活力胚之 poly A-rich mRNA 合成蛋白質的能力約為 90% 活力胚的
69%(39)。此外,0% 活力胚之 poly A-rich mRNA 的分子量較低。這些結果顯示
由於 poly A-rich mRNA 雖具有合成蛋白質的能力,但其 poly A分子變短,因 此合成蛋白質的能力降低。然而 0% 活力之種子根本不能合成蛋白質,因此種 子內原先存在的長壽命 mRNA 之功能喪失並不是唯一影響種子衰化之原因,必 定還有其他因子參與。
衰化過程中,種子合成各類 RNA (包括 mRNA) 之能力下降(36),在黑麥種子 的衰化過程中 31S rRNA (rRNA 之前身物)修飾為 25S 與 18S rRNA 之速率下 降,0% 活力種子發芽初期所合成之 RNA 其分子量均比 4-5S 為小。此種小分 子量 RNA 之合成原因很多,其原因之一可能是由已斷裂的 DNA 做為模版而
合成。因此種子衰化過程中 DNA 之變化亦是有趣且重要的問題。黑麥種子胚細 胞內每一個核所含之 DNA 總量在衰化過程中不會發生改變,然而衰化過程中 DNA 分子的完整性卻發生明顯的變化,高分子量 DNA 隨著衰化程度之增加而減 少,且具有活力之種子其 DNA 平均分子量大於不具活力之種子(37)。DNA 之斷 裂可能使得作為模版之能力降低,同時進行錯誤的轉錄作用,會使得細胞無法合 成蛋白質。由於衰化過程中 DNA 總量不變,因此 DNA 之斷裂很可能是由於 endodeoxyribonuclease 所引起。不具活力之黑麥胚 DNase 活性高於 95%活力之 種子(44)。由於乾燥種子於貯藏過程或衰化期間不可能合成此種酵素,因此該酵 素活性的增高,很可能是由原已存在之酵素經活性化產生。一些試驗結果也顯 示,95%活力之種子胚內含有 DNase 之抑制劑 (該抑制劑對熱敏感),當種子活 力消失時該抑制劑的活性消失(45)。種子衰化過程中,斷裂的 DNA 不斷累積,從 上述之討論可以推想,具有活力之種子含有穩定的 DNase 與該酵素之抑制劑。 因此,DNase 之活性在發芽前無法表現。當種子衰化時,DNase 抑制劑喪失活 性,因此,在吸水時 DNase 活性明顯的表現,再加上種子吸水後缺少 DNA 修 補系統或修補酵素,因此 DNA 之完整性喪失。 種子成熟後,其活勢大小主要由儲藏環境的溫度、濕度及時間所控制,種子 活勢流失愈少,發芽成幼苗的機率就愈大。在作物生產循環 (production cycle) 中,由種子發展成幼苗的過程,是一個重要的時期。欲獲得高發芽率和整齊的種 苗,所用種子必需具成熟、充實及高活勢。不當的溫度、缺水等生長逆境,以及 病原菌及蟲害,皆會影響發芽率及幼苗植相的建立。為了要得到最大的田間效 益,種子在播種前必需經過一些處理,例如精選、萌爆、浸種及添加殺蟲劑、殺 菌劑、植物生長調節劑、養份等等。正確的種子處理技術,確保其生產成本,尤 其對於高經濟的園藝、作物種子,因而需要投注特別的種子處理技術來確保其投 資及保證有最好的植株發育。一但種子受到水份浸潤啟動發芽時,我們相信,決 定種子”生(viable)”或”死(death)”的基因、酵素或相關因子便展開了激烈的 拔河比賽,能夠幸存下來的幼苗,可能驅動了某些與活力有關的關鍵基因或酵素
之表現。
1.8 RNA 解旋
的特性
解旋 廣泛的存在於生物體內,從細菌到人類及許多的病毒都曾被發現, 解旋 包含有 RNA 解旋 及 DNA 解旋 ,根據胺基酸序列相似性的比對分類, 可將解旋 分成三個大家族 (superfamilies, SF1~3) 及二個小家族 (families, F4, F5) (46)。其中 DEAD box protein 屬於 SF2,SF1 與 SF2 皆具有 7 個重要的 motif,
與 RNA、ATP 的結合,及 ATP 的水解,RNA 的解旋活性有關。SF3 家族來自於 小 DNA 及 RNA 病毒,只有 3 個保留區域 (conserved motifs)。F4 為 DnaB-like 蛋白質,包含有 5 個保留區域。F5 則以 DNA-RNA 解旋 轉錄終止因子 Rho (transcription termination factor) 為代表。從序列上來看,SF1 與 SF2 具有 7 個相 似的保留區域,和其餘 3 個家族的差異較大。若由解旋的方向來分類,則有 3’ →5’及 5’→ 3’兩種方向,大部份 SF1 和 SF2 屬於 3’→5’,而 F4 和 F5 則多為 5’ → 3’的解旋方向。
在 DNA 複製 (replication)、修復 (repair)、RNA 合成、剪裁 (splicing)、轉 錄 (transcription) 及轉譯 (translation) 等過程中,雙股螺旋的 DNA 或 RNA 必須 被解開成單股,方能進行以上的反應,解旋 負責將雙股螺旋的 DNA 或 RNA 解開成單股的酵素。解旋 最早在 1976 年被 Abdel-Monem 等人鑑定出來(47)。此 類酵素解開雙股螺旋的時候需要以 NTP 的結合及水解作為能量來源,同時他們 也能連續地在核酸長鍊上快速移動 (500~1000 bp/sec),所以也被歸類為 motor protein (48)。具有功能的解旋 的形式通常為寡聚合物 (oligomer) 或六聚合物 (49)。
1.9 RNA 解旋
在生物體內扮演的角色
解旋 除了有上述功能外,針對酵母菌 (Saccharomyces cerevisiae),RNA 解旋 更扮演了多元化的角色,包括參與 rRNA的合成,pre- mRNA的修飾,tRNA 的成熟,細胞核到細胞質的運送,RNA 代謝,如 Figure 1(50)所示:
RNA 解 旋 能 夠 解 開 雙 股 RNA 是 來 自 於 水 解 NTP (nucleoside triphosphate),特別是水解 ATP 以獲得能量來源。在大部份份情形下,RNA 解旋
其 ATPase 的活性是受到短鏈 RNA 所誘導產生。相對於 DNA 解旋 而言,DNA 解旋 一般是解旋較長、也較規則的雙股 DNA,而 RNA 解旋 是經由單一步驟 解旋較短之雙股 RNA,然而其詳細的作用機制及 RNA 解旋 天然受質尚未被發 現。
不同的 RNA 解旋 在細胞內有其不同的功能及作用部位,RNA 解旋 參 與的功能包含有 (a) 核糖體合成,(b) 轉錄作用,(c) Pre- mRNA 修飾,(d) RNA 的成熟,(e) RNA 的運送,(f) 轉譯作用,(g) RNA 的分解(52)。其中參與 translation
的 eIF4A 屬於 DEAD box protein family,具有 RNA 解旋 核心區重要的 7 個 motif,這些 motif 與 ATPase 活性、解旋 的活性以及與 RNA 結合有關。不同的 RNA 解 旋 其 詳 細 的 作 用 位 置 如 Figure 2 ( 51 ) 黃 色 標 識 處 。
RNA 解旋 參與的部位以黃色表示,整個輪廓圖如前頁 (Figure 1) 所示,(a) 核糖體合成,(b) 轉錄作用,(c) Pre- mRNA修飾,(d) RNA的成熟,(e) RNA的運 送,(f) 轉譯作用,(g) RNA的分解。
1.10 RNA 解旋
結構學方面的研究
解旋 至今被解出的結構包括 Bacillus stearothermophilus 的 DNA 解旋 ,PcrA(53, 54),Escherichia coli 的 DNA 解旋 ,Rep(51)及 C 型肝炎病毒的 RNA
解旋 ,NS3(35, 56, 57, 58),前二者屬於 SF1,而 NS3 則屬於 SF2,這 3 個結構的
結晶都是單元體。HCV NS3 解旋 是 SF2 目前唯一結構被測定出來的,其結構 包含有 3 個 domains (1~3),其中 domain 1、2 的胺基酸序列雖然沒有相似性,但 確有類似的結構相似性 (structural homology),二者的核心部份都是由平行交疊
的β-sheet 所組成,周圍包圍著數條α-helices,這樣的結構和 1992 年發表的 E.coli
的 DNA 重組蛋白 RecA 很相似(59),所以稱為 RecA- like domain。Domain 3 則都
是由α-helice 所構成。
除了前述已解出的 PcrA (DNA 解旋 ) 及 HCV NS3 解旋 (RNA 解旋 ) 外,對於細胞內 RNA 解旋 的結構仍不清楚,然而 DNA 解旋 與 RNA 解旋 因有結構的相似性,因此二者解開雙股核酸的機制應該也很類似。HCV NS3 解旋 有三個 domains,組成 Y 字型,Domain 1 含有 NTPase I 與 NTPase II, Domain 2 包含 motif VI其與 RNA的作用有關,Domain 3 則無任何保留區。Domain 1 與 Domain II 由 motif III 如同絞鍊般將二個 motif 連在一起,motif III 容易彎 曲,使得 Domains I 與 II 能夠轉動(50)。
從研究轉譯啟動子 eIF4A (DEAD- box-protein family)發現,當 ATP、RNA 與 eIF4A 結合後,Domain I 與 II 會使得 cleft 關閉,而 ATP 經水解產生 ADP 後, 又使得 cleft 打開,並且伴隨解開雙股核酸。因此藉由 eIF4a 結構上的改變,影 響了 RNA 與蛋白質或蛋白質與蛋白質之間的排列,使得雙股 RNA 分子能夠解 旋成單股。NS3 解旋 核心,其七個重要 motif 在蛋白質結構上的位置如下圖所 示(50):
(J. de la Cruz., 1999, TIBS)
1.11 RNA 解旋
可能作用機制的探討
從文獻回顧可得知 RNA 解旋 存在於所有的細胞生物和病毒基因組,對於 各 種 RNA 的 生 合 成 代 謝 扮 演 了 重 要 的 角 色 。 解 旋 最 早 在 1976 年 被 Abdel-Monem 發現後,研究者分別從細菌,酵母菌及病毒選殖到許多種類的解
旋 。 但 是 由 於 其 種 類 繁 多 , 又 有 許 多 不 同 的 作 用 機 制 , 其 中 又 以 DEAD/DExH-box 這些蛋白質對於解開 RNA 螺旋構造,變成了熱門的研究題材。 從 1996 年嗜熱脂肪芽孢桿菌 (Bacillus stearothermophilus) 解讀出 DNA解旋 - PcrA 之結構,1997 年解讀出大腸桿菌 (Escherichia coli) 的 DNA 解旋 -Rep, 1998 年解讀出屬於 RNA HCV NS3 解旋 ,我們對於解旋 在生物體內所扮演 的角色,才有一些可能的推測與探討。
權威期刊「自然」的姐妹刊物「自然結構生物學」最新的研究報告指出解旋 可能的作用機制有”主動解開雙股 ”(Active duplex unwinding)”及被動解開雙 股 ” (Passive duplex unwinding)” 兩種可能的模式,Active duplex unwinding 又可 分為”Rolling model mechanism” (60)及”Snow-plough or inchworm mechanism”(58)。
在 active rolling model 需要解旋 以多元體 (至少為雙體) 的行式方能進 行。每一個次單元體 (subunit) 都具有和單、雙股螺旋結合的能力,但無法同時 結合,並利用 NTP 結合及水解去調控每個次單元體對不同 DNA 的親和力。從下 頁圖上可知寡聚合體解旋 交互結合在單股及雙股的核酸分子上,而 ATP 和蛋 白質的結合體 (aATP ) 和雙股核酸的結合力很強,一旦水解變成 (aADP)則與雙股 核酸的結合力減弱,此時 (bADP ) 再藉由 ATP 的水解獲得磷酸根成為 (bATP) 結 合上核酸分子,其過程如同滾輪般滾動,因而解開雙股 DNA。而在另一個 snow-plough model 不嚴格要求解旋 為寡聚合體的形式,單體亦可,而且每個 單體都能和單雙股螺旋同時結合,如同鏟雪機般在雙股螺旋上滑動,滑動的同時 並解開雙股螺旋。從附圖圖中可知 RNA 解旋 延著核酸開岔處,藉由 ATP 水解 所獲得的能量把雙股核酸分子間的氫鍵打開,如同鏟雪般向內滑動。相對地,在 passive unwinding model 中,解旋 會結合在單股核酸分子,藉由 ATP 水解供應 能量向內移動打開雙股 RNA。
1.12 RNA 解旋
在植物學方面的研究
RNA 解旋 廣泛分佈於各種生物體,在植物方面的研究包括煙草(tobacco)
(60, 61, 62, 63)、阿拉伯芥(Arabidopsis thaliana)(64, 65, 66, 67, 68)和豌豆(69, 70)。經由電
腦軟體預測可得到為數眾多的”putative computer predicted helicases”,然而只有少 數經實驗證明具有 ATP-dependent helicase activity, 例如 human p68(71)、rabbit
reticulocyte eIF-4A(72)、PPVCI(73)、a DEAH box RNA helicase from HeLa cell(74)、
vaccinia virus RNA helicase(75)、 Drosophila vasa protein(76)、 E.coli CsdA(77) and
DbpA(78)、 Xenopus xp54(79)和 An3(80, 81)。
在植物方面具有活性的 RNA 解旋 只有阿拉伯芥(Arabidopsis thaliana)的
AtDRH1(66)和豌豆(69, 70),其在植物體內扮演的生物功能(Biological function)
II. 研究目地
2.1 綠豆品系選擇與種子加速衰化條件的建立
2.2 種子衰化指標的分析
2.3 mRNA 差異展現法
2.4 VrRH1 基因全長選殖、基因表現與酵素活性分析
2.5 VrRH1 生物功能之推測
2.1 綠豆品系選擇與種子加速衰化條件的建立
本實驗初期所使用的種子年份包括 92 年台南三號綠豆、94 年及 98 年台南五 號綠豆。其中台南三號光皮綠豆,為育成台南五號粉質綠豆之父本,本實驗室因 多年來專注於台南五號綠豆醣 的研究,發現發芽初期的幼苗,至少有 13 種 以上的醣 會被誘發,大量表現於發芽後第 4~5 天。其後並完成 â-半乳醣 異構 的純化與生化功能的探討(84)及基因的選殖。 三種不同年份種子其間隔最大年份雖達 6 年,但於實驗室理想狀況的發芽條 件下,其發芽率幾近於 96~98%,但是種子間的發芽情況並非很整齊,因此引發 我們研究的興趣,種子或是初期發芽幼苗間彼此的差異,是否存有差異的蛋白質 及基因的表現?而這些差異因子的表現是否就是種子的活勢 (vigour) 因子?最 後影響種子發芽率的總體表現。許多學者從 80 年代開始,利用蛋白質化學許多 實驗利器 (例如: 2-dimension gel electrophoresis)(9, 10, 14) 企圖找尋發芽初期的相關蛋白質,並無很好的結果。本實驗室近年來由於由研究「抗豆象基因的選殖、
基因表現及生物活性探討」(95),累積許多經驗,因此我們想分頭並進,分離出使
得衰化幼苗可以繼續生長的蛋白質或是基因,以期進一步研究其生化、生理或是 生物學上的功能。
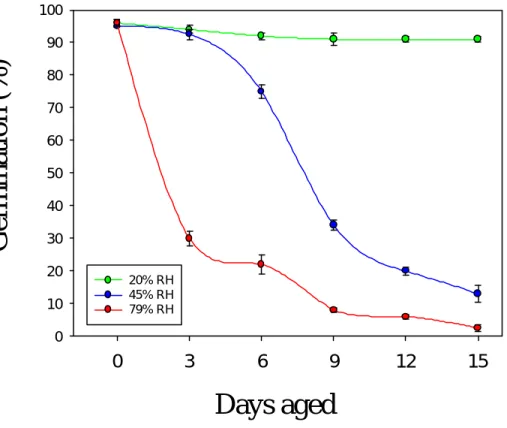
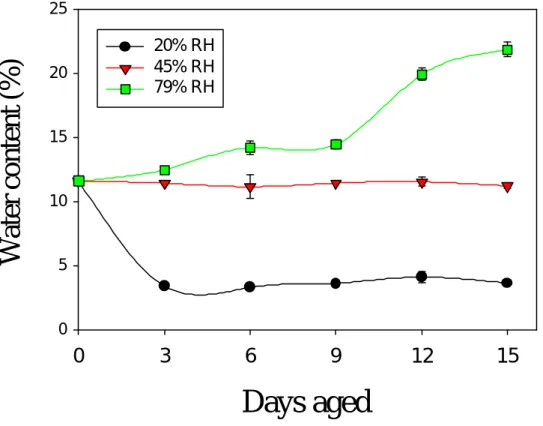
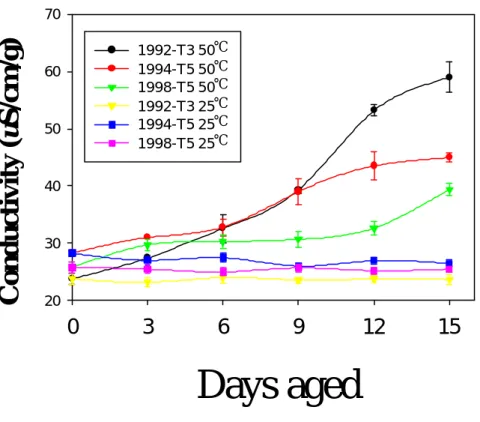
為了拉大高活勢群種子 (high vigour seeds) 與低活力群種子 (low vigour seeds) 彼此間的差距,實驗室常用的方法為高溫 (40~50℃)、高濕法 (80~100% RH, relative humidity)(21)。我們選定 1994 年台南五號綠豆為材料,分別以室溫 (25 ℃)及高溫 (50℃)搭配不同的相對濕度處理種子,以瞭解種子於逆境處理後,發 芽初期基因與蛋白質的表現。
2.2 種子衰化指標的分析
鑑 定 種 子 品 質 最 常 使 用 的 方法 為 測 定 發 芽 率 , 其 為 活 力 指 標 ( viability index);以及發芽速率,其為活勢指標(vigour index)。種子衰化程度愈高,發 芽率降低,種子發芽速率亦隨之降低,結果造成種子不發芽或不正長苗的增多。我們參考文獻(),選則正儲藏形種子常用的活勢指標分析,來分析綠豆種子於不
同程度衰化情形下,種子 vigour 的大小。Vigour index 常用法包括有 tetrazolium chloride test (TTC 法) ,其原理是當種子活力愈強,其去氫 的活性愈高,因此 可藉由已知的 TTC 染色圖譜,利用圖譜判讀不同批次種子活力比較,快速地篩 選出活力強的種子群。另一類的活體染料是伊凡斯藍 (Evan’s Blue),因其只能進 入死亡或受傷的細胞並染色,而細胞膜完整的細胞則阻止染料進入而保持未染色 的狀態,常用來作為處理植物組織的活體染料。除了染色法外,另一個常用的指 標為電導度法,種子衰化常導致細胞膜的不完整,細胞質滲漏到胞外的情形隨之 提高,造成導電度的上升,可用電導度計加以測量。衰化種子評估指標,除了上 述幾種作法,亦有學者利用不同衰化種子發芽初期,乙烯釋出量會隨著種子衰化 程度的不同而有所不同來加以測定。本實驗則是觀察不同衰化程度下,種子於密 閉試管內,乙烯釋放情形之分析。
2.3 mRNA 差異展現法
一般高等生物的細胞內約含有十萬個不同的基因,在全部生命過程的不同階 段,只有 10~15%的基因在個別的細胞中表現,若我們想要瞭解這些過程中有那 一些基因被表現或關閉,依上述理論約有一萬到一萬五千種 mRNA,如何找到 這些差異的基因呢?針對 RNA 這一層次一般人熟知的方法有 cDNA 扣除雜交法 (cDNA subtractive hybridization method) 或 是 mRNA 差 異 展 現 法 (mRNA differentia l display method, DDRT-PCR)(85)。我們採用 Sokolov 於 1994 年(86)修飾Liang & Pardee 的 DDRT-PCR(85),其方法為首先以 6 個寡核 酸進行 RT-PCR, 所合成之 cDNA 再以不同任意引子的搭配組合進行 PCR,最後以洋菜膠體電泳 進行差異片斷的溶離(86)。此方法無須使用放射性物質,能夠經由染色於洋菜膠 體電泳找到明顯差異之 cDNA 片段,從文獻指出所得到的片斷幾乎都是 Northern 正相關,由於其 cDNA 量多、回收方便,可方便後續基因的選殖定序及分析。 本實驗為瞭解衰化種子與正常種子不發芽或是二者發芽初期,mRNA 的表現
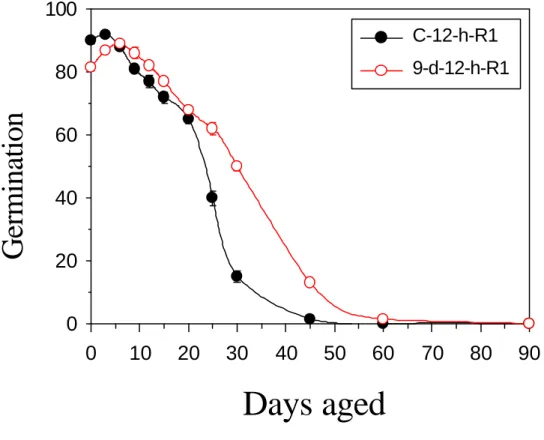
是否有所不同?因此採用「mRNA 差異展現技術」篩選衰化 9 天種子(9-d-aged seeds),或是衰化 9 天種子可發芽、正常生長的幼苗(9-d-aged-12 h seedling)相
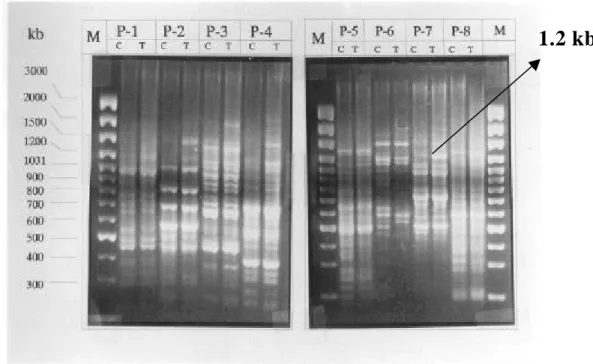
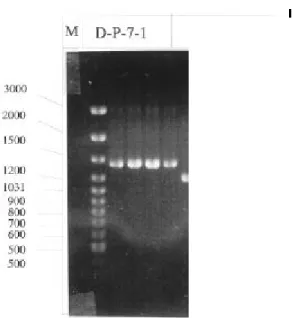
較於正常種子(Control seeds)、正常幼苗所誘導出之基因(Control seedling), 經「mRNA 差異展現技術」後,於 DNA 洋菜膠體電泳後之可能示意圖如下: □ 我們的目標是篩選處理衰化種子差異基因或衰化種子幼苗發芽初期差異的基 因,如上圖ü號所示,而被篩選出之 VrRH1 基因如符號þ所示。 未浸潤前之正常與衰化種子,從衰化種子選殖到許多 rRNA 片斷;然而經 浸潤 12 小時後,從可存活的衰化幼苗,除了篩得很多的 rRNA 片斷外,我們幸 運篩得一個 1.2 kb 之 RNA helicase 片斷外,因此實驗焦點集中在正相關基因-
VrRH1(Vigna radiata RNA helicase 1)全長基因序列的選殖及表現。
2.4 VrRH1 基因全長選殖、基因表現與酵素活性分析
為了瞭解篩選出之差異片段基因之功能為何?首先必需選殖全長基因,所採 用的方法為 5’/3’ RACE 法,其次對於基因全長的表現,有許多蛋白質表現系統 12 hr imbibition Control seeds 9-d-aged seeds Control seedling 9-d-aged-12 h seedlingü
ü
þ
ü
提供使用者快速的純化方法,以本文所篩選到的基因 VrRH1 為例,1998 年於阿 拉伯芥中已有日本 Okanami 等人成功地將所篩到的 RNA helicase-AtDRH1 構築 入 pET-28(+)系統,並於大腸菌 (BL21/ DE3) 成功地加以表現及純化(66)。我們
修 飾 其 蛋 白 質 表 現 系 統 及 純 化 流 程 , 得 到 VrRH1 基 因 表 現 的 融 合 蛋 白 質”His-VrRH1”。
VrRH1 經 GCG 序列分析比對及活性分析後,證明屬於 DEAD box protein,
屬於 RNA helicase SF2 (SF2, superfamily 2 之簡稱),然而為數眾多的 RNA helicase 的推測序列,並不一定有酵素活性,為了研究從 E. coli 表現之 VrRH1 是否有 helicase 活性,我們以人工合成的雙股 RNA 基質 (ds RNA) 及 ã[P32]ATP 分析 VrRH1 helicase 活性及 ATPase 活性的分析。VrRH1 表現蛋白並且作為抗原免疫 兔子,進行抗血清的製備。
2.5 VrRH1 生物功能之推測
除了證明 VrRH1 基因表現所得到的融合蛋白 His-VrH1 是否有活性外,我們 亦追蹤在活力高低種子、種子發芽初期以及不同部位,VrRH1 基因的表現情形。 一般而言,種子為了抵抗惡劣的環境,外表通常有特殊的構造來保護種子,使其 活力不至於喪失,如果當種子長期處於不適的環境中,特別是儲存於高溫、高濕 的環境,便會造成種子 vigour 快速地喪失,一旦有合適的萌芽環境,雖然有水 份浸潤啟動發芽,衰化嚴重、活力喪失到無法恢復的種子只有走向死亡一途;而 活力喪失較少,能夠加以彌補的種子,雖遭受阻礙但終能長成幼苗。衰化種子為 何仍能發芽?發芽後,植株生化、生理及外觀產生了何種變化?會不會影響到下 一代的種子?一連串的疑問目前在種子方面的研究瞭解不是很多,如能深入加以 研究,應該可以幫助對於衰化種子其活力的再回復,提供一條思考的途徑。III. 材料與方法
3.1 種子加速衰化條件建立
3.2 種子含水率之測定
3.3 種子品質分析
3.4 mRNA 差異展現法
3.5 T-vector 選殖及定序
3.6 VrRH1 cDNA 全長的選殖
3.7 VrRH1 基因的表現與純化
3.8 His-VrRH1 融合蛋白質活性分析
3.9 蛋白質分析方法
3.10 His-VrRH1 抗體製備
3.11 蛋白質電泳轉印
3.12 VrRH1 免疫組織定位
3.13 VrRH1 北方點墨法分析
3.14 原態 VrRH 純化
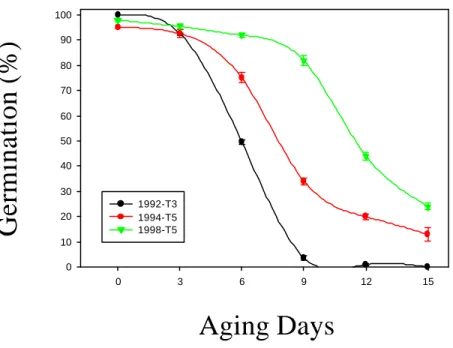
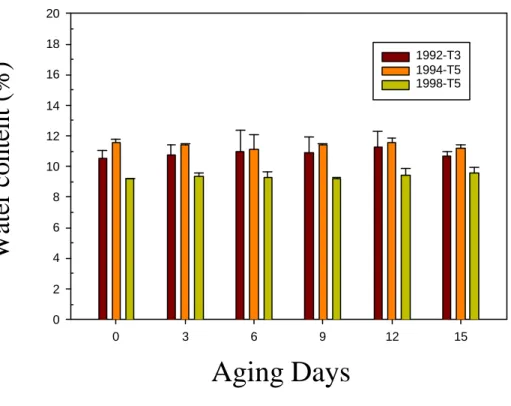
3.1 種子加速衰化條件建立
取置於 -20℃貯藏之 1992、1994 和 1998 之台南五號綠豆種子(Vigna radiata VC 3890 Sel #5)進行加速衰化處理。處理條件為將種子置於塑膠材質培養皿上, 以 50 粒綠豆為一組,進行四重複。不同年份樣品分別以室溫(25℃)或是高溫 (50℃)處理,其環境相對濕度分別為 20% RH(relative humidity)、45% RH、 79% RH 和 100% RH 直到 21 天。為了控制不同環境相對濕度,我們利用不同飽 合度之硫酸銨置放於容器內來調整相對濕度的變化(87),並以濕度計來加以監 測。衰化種子每三天進行取樣,進行種子發芽試驗及含水率測定。3.2 種子含水率之測定
種子含水量的測定為參考國際種子檢查規則,採用低恆溫烘乾法(92),將綠豆 樣品均勻分佈於容器表面,放入種子前後,容器和蓋子均應稱重,容器置蓋子上 迅速放入溫度為 103 ± 2℃的烘箱內烘乾 17 ± 1 小時。烘乾時間由烘箱溫度回升 至所需溫度時算起,測定時室內周圍空氣的相對濕度須低於 70%。結果計算方法 如下: 水分含量之重量百分比以下列公式計算至小數點一位: M1-容器和蓋子的重量,以公克表示。 M2-乾燥前容器、蓋子和內含物的重量,以公克表示。 M3-乾燥後容器、蓋子和內含物的重量,以公克表示。 一個樣品的測定,取二重覆的算數平均值為其結果,若二者間的差異超過 0.2%, 則另取二重覆重新測定 (M2-M3) × 100 (M2-M1)3.3 種子品質分析
3.3.1 發芽率測定 取 50 粒種子為一組,使用捲紙法進行四重覆之發芽實驗,以測定種子活力。 方法為將種子平均散佈於拭手紙上,並將拭手紙捲成圓筒狀,置於夾鍊帶內,加 入去離子水維持樣品濕潤,移入 28℃恆溫生長箱培養二天,由於幼苗胚軸伸長 達 0.5 cm 以上者可繼續生長。因此我們以胚軸長度大於 0.5 cm 以上,作為判斷 幼苗繼續生長的界線。 3.3.2 種子活勢測定 3.3.2.1 發芽速率之測定 取 50 粒種子為一組,使用捲紙法進行四重覆之發芽速率實驗,以測定種子活 勢。其方法為不同衰化程度種子,浸潤 12 小時後,測量胚軸突出於種皮之長度。 3.3.2.2 Tetrazolium testTetrazolium(TZ)試驗方法為配置 2,3,5-triphenyl tetrazolium 之氯化物或溴化 物基質液,其方法為將定量的 tetrazolium 溶於 pH 7.0 之磷酸緩衝液內,配置成 1% (w/v) 使用(21, 92, 96)。取 50 粒種子為一組進行四重覆實驗,種子樣品先以去
離子水浸潤 6~8 小時,脫去種子外皮後浸潤於基質液中於 37℃避光反應 30 分鐘 後,再以去離子水沖洗二次,每次 10 分鐘。
3.3.2.3 Evan’s Blue test
Evan’s Blue test 方法是利用生理食鹽水配置 0.25% (w/v) Evan’s Blue 染色液
(88)。取 50 粒種子為一組進行四重覆實驗,種子樣品先以去離子水浸潤 6~8 小時
後,脫去種子外皮後浸潤染色液中,於 25℃避光反應 30 分鐘後,再以去離子水 沖洗二次,每次 10 分鐘。
電導度測定方法是將不同衰化處理之綠豆種子 50 顆,分別置於三角錐瓶內, 再加入預冷的去離子水 250 ml,將樣品存放於 20℃生長箱內 24 小時後,以電導 度計測量其導電度的變化,以分析不同程度的衰化種子,其離子滲漏的情形(83)。 3.3.2.5 乙烯測定 乙烯含量的測定是將種子置於密閉的採血管內 (10 ml),經由不同的衰化條件 處理種子,以針筒抽取 1 ml 的氣體進行氣相層析分析 (Hitachi Ltd., Model 063) (89)。
3.4 mRNA 差異展現法
採用 Boris(90) 改良自 Liang(85) mRNA 差異展現技術,進行篩選與抗綠豆衰化 相關的基因。必須以 mRNA為材料,其概念仿自DNA的 RAPD (random amplified of polymorphic DNA),實驗以 1~3 條 20~25 個核 酸任意組合的寡核 酸引子, 利用 PCR 法分析經衰化處理與未處理綠豆或幼苗間,相關基因表現的差異。以 T 載體選殖有差異的 DNA 帶,經南方轉印及北方轉印的確認,可做為篩選與綠 豆衰化有關基因的探針或是 3’/5’ RACE 之模板。
3.4.1 總 RNA 抽取
總 RNA 的抽取採用 hot phenol 法(92),其方法如下:
1. 綠豆樣品 4 g 以 20 ml 65℃預熱萃取溶液 (100 mM Tris-HCl, pH 7.5 / 100 mM LiCl / 100 mM EDTA, pH 8.0 / 1% SDS / 100 mM β-mercaptoethanol) 加入等體積
phenol,震盪 30 秒。
2. 將樣品置於 65℃水浴槽加熱 5 分鐘,間歇震盪,再加入 1/2 體積 chloroform: isoamyl alcohol = 24:1,震盪 1 分鐘後,15000 rpm 離心 20 分鐘。
15000 rpm 離心 10 分鐘。 4. 加入等體積 chloroform:isoamyl alcohol = 24:1,震盪 1 分鐘後,15000 rpm 離 心 5 分鐘以除去 phenol,取上清。 5. 加入等體積 4 M LiCl 後,置於 -70℃冰箱 1~2 hr 以幫助 RNA 沉澱。 6. 樣品以 15000 rpm 離心 30 分鐘,可得到白色 RNA 樣品沉澱於管底。 7. 取 10 ml 之 2 M LiCl 震盪懸濁 RNA,15000 rpm 離心 10 分鐘。
8. 加入 5 ml 之 0.1% lauro-sarcrosyl 震盪懸濁 RNA,此時 RNA 溶於上清液中。 9. 取 ependoff 加入 1 ml 之 100% EtOH ,40 ìl 之醋酸溶液 (3M CH3COONa, pH
4.3)後,溶解之 RNA 平均加入 ependoff 內,置於 -70℃冰箱過夜。 10. 4℃, 14000 rpm 離心 15 分鐘後,可得白色 RNA 沉澱於管底。
11. 將上清去除,取 80% 酒精清洗白色 RNA 沉澱物,4℃, 14000 rpm 離心 15 分鐘 後真空抽氣凍乾。
12. 將 pellet 溶於 200 ìl 之 0.1% lauro-sarcrosyl 置於-70℃備用,即可得到總 RNA。 13. 以分光光度計測其 A260 / A280比值應達 1.8~2.0 者表示該總 RNA 純度較佳,否則
可以加作 1~2 次 phenol /chloroform (24:1) 與總 RNA 上層液等體積混合,除去蛋 白質的污染。又每一 A260的吸光值表示 RNA 濃度約為 40 ìg/ml。
3.4.2 訊息 RNA 的分離
訊息 RNA (mRNA) 具有 poly (A) +
tail 的特性,可用 Oligo-dT cellulose 從總 RNA 中分離,方法如下:
1. 取 1 g Oligo (dT) cellulose,通入 TES (10 mM Tris-HCl, pH 7.5 / 1 mM EDTA , pH 8.0) 流洗 15 ml,再以 15 ml LTES (10 mM Tris-HCl, pH 7.5 / 1 mM EDTA, pH 8.0) 平衡。
濃度為 0.5 M。
3. 將 RNA 通入已平衡的管柱中,流出液再回加入管柱三次,使 poly (A) + RNA 能完全結合於 Oligo (dT) Cellulose 上。
4. 以 15 ml LTES 流洗,以除去 poly (A)-
RNA。 5. 以 TES 沖提之 mRNA,每 0.4 ml 為一分劃,加入無水酒精 1 ml、醋酸溶液 (3 M CH3COONa, pH 4.3),40 ìl 混合後,置於 -70℃ 30 分鐘後以幫助 mRNA 沉澱。 6. 14000 rpm、4℃離心 30 分鐘後,將樣品以 70% 酒精清洗後,再以 14000 rpm 離心 10 分鐘。 7. 將樣品真空抽氣乾燥後,以緩衝液 (5 mM Tris-HCl, pH 7.5) 回溶,其純度 應達 A260 / A280 >2.0,定量法同總 RNA 定量法。 8. 所得之 mRNA 置 -70℃ 貯存。 3.4.3 cDNA 合成 1. 將 mRNA 於使用前離心以除去不溶物。 2. 依下列方式製備 cDNA 合成反應液︰
Component Sample No RT Control Control RNA
1 to 5 µg total RNA n n - Control RNA - - 1 Random hexamer 2 2 2 DEPC-treated water 10-n 10-n 9 Total 12 12 12 3. 於 70℃ 反應十分鐘後,置於冰浴一分鐘。 4. 再製備以下反應液︰
10X PCR buffer 2 8 25 mM MgCl2 2 8 10 mM dNTP mix 1 4 0.1 M DTT 2 8 Total 7 28 5. 加入以上反應液(7 ìl)於 cDNA 合成反應液中,混合均勻並離心。 6. 25℃ 反應 5 分鐘,每個 PCR 管中加入 1 ìl (200 units) SuperScript II RT 反轉 錄 ,混合均勻並於 25℃反應十分鐘。 7. 42℃ 反應 50 分鐘。 8. 於 70℃ 反應 15 分鐘以終止反應進行,並將樣品置於冰浴冷卻。 9. 每管加入 1 ìl RNase H 以除去 RNA,並於 37℃ 反應 20 分鐘。
10. 將樣品置於 -20℃ 保存,此樣品為 mRNA 差異展現法所用之 cDNA pool。
3.4.4 mRNA 差異展現法
1. 分別合成 C 組(1994-T5 未衰化綠豆浸潤發芽 1 2 小時),及 T 組(標準 衰化條件處理 9 天浸潤發芽 1 2 小時)之 cDNA pool,二者加入經電腦比所 得到的引子,以進行 PCR 反應。Primer 序列如下:
No Name Oligonucleotide sequence(s) 5’→3’ Total Base 1 Heat-Soy-F AGGCTTCCATGGAAAATGGG 20 2 Heat-Soy-R CGTGGCATCGCGTGGAGCGAAGC 23 3 Sen-Ara-F GGCAGCTGCGGTTCAAAC 18 4 Sen-Ara-R CATCGGACATCCGACTAGAG 20 5 Pro-Bra-F GTCAAGCCGCTGTGGGACAG 20 6 Pro-Bra-R TTTAACATTCATATCCATT 19 7 Pro-Soy-F TCTGCTACAAACCCTGCAAG 20 8 Pro-Soy-R GCTGTGATCTCTGCATGTGC 20
其中 No 1~2 為比對黃豆熱休克蛋白質之引子; No 3~4 為比對阿拉伯芥老化相 關基因之引子;No 5~6 為比對青花菜蛋白質水解 之引子;No 7~8 為比對黃豆 蛋白質水解 之引子。
2. 另一方面,我們亦以 OPERON 商業引子 (10 mers),進行 mRNA 差異展現 法,引子序列如下:
No Name Oligonucleotide sequence(s) 5’→3’ Total Base
1 OPA-01 CAGGCCCTTC 10 2 OPA-02 TGCCGAGCTG 10 3 OPA-03 AGTCAGCCAC 10 4 OPA-04 AATCGGGCTG 10 5 OPA-05 AGGGTGCTTG 10 6 OPA-06 GCTCCCTGAC 10 7 OPA-07 GAAACGGGTG 10 8 OPA-08 GTGACGTACG 10 3. mRNA 差異展現法所使用之 PCR 反應液,內含物如下:
Component Control Exp
10 X PCR buffer 5 5
25 mM MgCl2 3 3
2.5 mM dNTP mix 5 5
Glycerol 5 5
Primer (single) 1 1
Taq DNA polymerase 1 1
C-12-h cDNA 5 -
9-d-12-h cDNA - 5
Autoclaved, distilled water 26 26
Total 50 50
5. PCR 反應程式如以下所示: 程式一 94℃反應 3 分鐘,40℃反應 4 分鐘,72℃反應 1 分鐘 程式二 94℃反應 45 秒,40℃反應 1 分鐘,72℃反應 1 分鐘共進行 45 cycle 程式三 72℃反應 10 分鐘 6. PCR 反應後產物以 1% 洋菜膠体電泳進行分析,切離膠體上有差異 的片斷,進行膠體溶離,並加以回收。 3.4.5 DNA 片段之分離純化 1. 將 PCR 產物進行洋菜膠體電泳。 2. 差異之 DNA 片斷以小刀自洋菜膠體割下。
3. 膠體稱重後,加入 3 倍體積之 buffer QG(QIAquick Gel Extraction Kit, QIAGEN)後,置於 50℃水浴 10 分鐘。。
4. 待膠體完全溶解後,將其加入 OIAquick spin column,13000 rpm 離心 1 分鐘。 5. 倒掉濾液,加入 75 µl buffer PE 至管柱中,13000 rpm 離心 1 分鐘。
6. 以 50 ìl buffer EB(10 mM Tris-HCl, pH 8.5)或無菌水加入管柱中,室溫靜 置 1 分鐘後,13000 rpm 離心 1 分鐘將 DNA 溶離。
3.5 T-vector 選殖及定序
1. 將膠體溶離所回收的片斷以 pGEM-T easy vector 進行連接。
2. 其方法為加入 1 ìl vector,3 ìl 回收 DNA,1 ìl ligase 及 5 ìl 10X ligation buffer 於 10℃反應過夜。 3. 取反應過夜的溶液 5 ìl,加入 100 ìl 已製備好的 DH5α competent cell,置於 4℃冰上反應 1.5 小時。 4. 其後進行轉形作用,其方法是將樣品置於 60℃反應 90 秒後,再置於冰上 3 分鐘。 5. 取 1 ml LB 加入樣品內,於 37℃反應 1.5 小時。
6. 將反應後樣品均勻塗佈在 LB / Amp / X-gal 培養基上,37℃隔夜培養,進行 藍白篩選。 7. 挑選白色菌落,以專一性引子進行 colony PCR,colony PCR 反應,PCR 內 含物如下: Component Volume (ìl) 10 X PCR buffer 2.5 25 mM MgCl2 2.5 2.5 mM dNTP mix 2.5
T-vector forward primer 0.5
T-vector reverse primer 0.5
Taq DNA polymerase 0.5
Colony -
Autoclaved, distilled water 16
Total 25 PCR 反應條件如下: 程式一 94℃反應 3 分鐘 程式二 94℃反應 1 分鐘,55℃反應 1 分鐘,72℃反應 1 分鐘共進行 35 cycle 程式三 72℃反應 10 分鐘 8. 選取經 PCR 確認為正相關之菌株,抽取質體進行定序。 9. 定序後之 DNA 核酸序列以 GCG 進行序列分析比對。
3.6 VrRH1 cDNA 全長的選殖
從 mRNA 差異展現法,我們可以得到 1.2 kb 差異片段,我們以 1.2 kb 為模板 設計專一性引子,仿照 3’ / 5’ RACE 概念,選殖 1.2 kb 全長序列。本實驗採用 的試劑組為 CapfinderT M3.6.1 cDNA 合成 1. 加 入 3 ìl mRNA (1 ìg poly A+ mRNA) 、 1 ìl CapSwitch II oligonucleotide 以及 1 ìl CDS / 3’ primer 於離心管內,混合樣品後離心。 2. 樣品置於 72℃反應 2 分鐘後,置於 4℃冷卻 2 分鐘。 3. 將樣品快速離心後加入 2 ìl 5X first-strand buffer、1 ìl DTT (20 mM)、1 ìl dNTP (10 mM)及 1 ìl MMLV (200 units / ìl) 混合後離心。 4. 置於 42℃反應 1 小時後置於冰上終止反應。 5. First-strand cDNA 可保存於 -20℃達三個月。 3.6.2 LD PCR 放大 cDNA 1. 將 PCR 機器於 95℃預熱。 2. 將樣品置於預熱的 PCR 機器內,PCR 內含物如下: Component Volume (ìl) First-strand cDNA 2 Deionized H2O 80 10X KlenTaqnPCR buffer 10 DNTP mix 2
T-vector reverse primer 2
5’PCR primer 2
CDS/3’PCR primer 2
50X Advantage KlenTaq Polymerase Mix 2
Total 100 PCR 反應條件如下: 程式一 95℃反應 1 分鐘 程式二 95℃反應 15 秒,68℃反應 5 分鐘共進行 30 cycle 程式三 72℃反應 10 分鐘 3. 取 5 ìl PCR 產物,以 1.1 % 洋菜膠體電泳進行分析,所合成 ds cDNA 需在 0.5~6 kb 形成模糊的亮帶,甚且大量表現的 mRNAs 會合成大量的 cDNA 而
形成明顯亮帶。
4. 所合成的雙股 cDNA 稱之為 cDNA pool,此 cDNA pool 作為選殖 VrRH1 5’端 及 VrRH1 3’端,PCR 合成所使用之模板 (template),合成之 cDNA 置於 -20 ℃保存。 3.6.3 VrRH1 5’端合成 我們取 1 ìl 已合成之 cDNA 進行 PCR 反應,合成 VrRH1 之 5’端,其 PCR 內 含物如下: Component Volume (ìl) 10 X PCR buffer 5 25 mM MgCl2 5 2.5 mM dNTP mix 5 5’PCR primer 1 VrRH1 reverse primer 1
Taq DNA polymerase 1
cDNA template from CapFinder 1
Autoclaved, distilled water 31
Total 50 PCR 反應條件如下: 程式一 94℃反應 3 分鐘 程式二 94℃反應 1 分鐘,55℃反應 1 分鐘,72℃反應 1 分鐘共進行 35 cycle 程式三 72℃反應 10 分鐘 3.6.3 VrRH1 3’ 端合成 我們取 1 ìl 已合成之 cDNA 進行 PCR 反應,合成 VrRH1 之 3’端,其 PCR 內含 物如下: Component Volume (ìl) 10 X PCR buffer 5 25 mM MgCl2 5
2.5 mM dNTP mix 5
VrRH1 forward primer 1
3’ CDS primer 1
Taq DNA polymerase 1
cDNA template from CapFinder 1
Autoclaved, distilled water 31
Total 50 PCR 反應條件如下: 程式一 94℃反應 3 分鐘 程式二 94℃反應 1 分鐘,65℃反應 1 分鐘,72℃反應 1 分鐘共進行 35 cycle 程式三 72℃反應 10 分鐘 3.6.4 VrRH1 全長合成 我們取 1 ìl 已合成之 cDNA 進行 PCR 反應,所使用的專一性引子為從 VrRH1 5’ 端及 VrRH1 之 3’端得之,其 PCR 內含物如下: Component Volume (ìl) 10 X PCR buffer 5 25 mM MgCl2 5 2.5 mM dNTP mix 5
VrRH1 forward specific primer 1
VrRH1 reverse specific primer 1
Taq DNA polymerase 1
cDNA template from 1
Autoclaved, distilled water 31
Total 50 PCR 反應條件如下: 程式一 94℃反應 3 分鐘 程式二 94℃反應 1 分鐘,55℃反應 1 分鐘,72℃反應 2 分鐘共進行 35 cycle 程式三 72℃反應 10 分鐘 PCR 夾擊出之全長 VrRH1 基因,以 T-vector 選殖及定序,方法同前述,被殖入
T-vector 命名為 pVRH1。
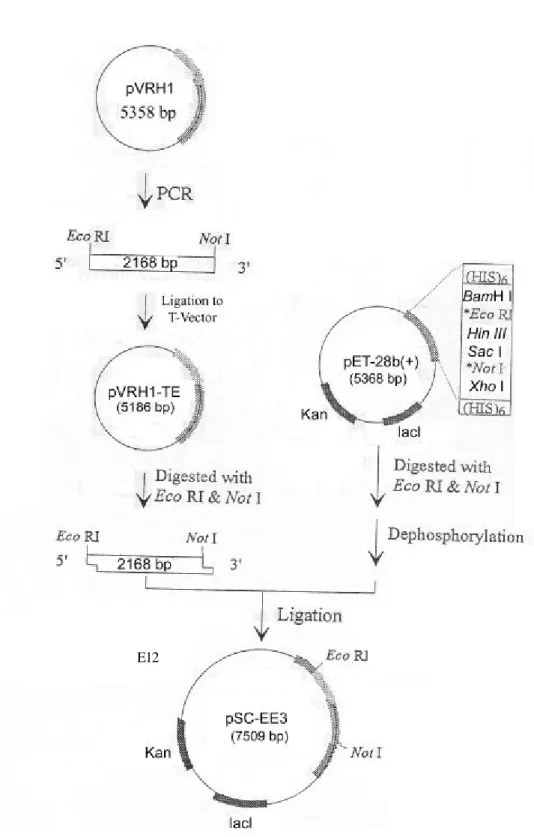
3.7 VrRH1 基因的表現與純化
設計內含 Eco RI 及 Not I 引子夾擊 pVRH1,所得到的 PCR 產物再接入 T-vector 內,經養菌抽取質體分別以 Eco RI 及 Not I 將 insert 切出,此時 insert 因兩端含 有 Eco RI 及 Not I 的限制 切點,我們將表現載體 pET28b(+) 亦以相同的限制 切出缺口,將 insert 與 pET28b(+) 進行接合,可得到 pSCEE3 的重組質體, 操作過程中都分別以限制 或 colony PCR 方法加以確認。
pSCEE3 經轉型作用送入 BL21 中,所得到的表現蛋白質命名為 His-VrRH1, His-VrRH1 因含有 His,因此可藉由鎳離子親合性管柱層析加以純化,我們將 His-VrRH1 蛋白質大量表現 (養菌 1L),集菌溶於一倍的 binding buffer (5 mM imidazole / 500 mM NaCl / 20 mM Tris-HCl, pH7.5 / 1 mM PMSF),以超音波破菌 後加以純化,純化方法如下:
1. 取 5 ml 樹脂以重力沉澱均勻地充填在管柱內,以 15 ml 二次水沖洗樹脂,再 加入 25 ml 50 mM NiSO4,使鎳離子固定在樹脂上,最後以 15 ml 的 binding buffer (5 mM imidazole / 500 mM NaCl / 20 mM Tris-HCl, pH7.5) 平衡管柱。 2. 接者注入約 30 ml 經破菌後的可溶性蛋白質,管柱每分鐘流速為 0.5 ml。 3. 接著先後以 30 ml binding buffer 及 15 ml 的 washing buffer (30 mM imidazole
/ 200 mM NaCl / 20 mM Tris-HCl, pH7.5) 沖洗出與鎳離子樹脂非專一性的蛋 白質,然後以 15 ml elution buffer (300 mM imidazole / 200 mM NaCl / 20 mM Tris-HCl, pH 7.5) 沖提出專一接合在樹脂上的 His-VrRH1。
4. 純化過程皆以 UV 偵測器及自動收集器收集蛋白質。
3.8 His-VrRH1 融合蛋白質活性分析
His-VrRH1 活性分析法,我們是參考文獻製備雙股 RNA 來分析 His-VrRH1 其 helicase 活性(93)。另一方面 ATPase 活性的分析我們參考 Jae-Young 分析
ROK1p,其對於 ATP 水解活性的研究(94)。 3.8.1 雙股 RNA 受質製備 1. 配置以下藥品,以進行 In vitro transcription Cold (ìl) Hot (ìl) 5X buffer 4 4 100 mM DTT 2 2 RNasin 0.5 0.5 pGEM3 1 - pGEM4 - 1 AGU - 4 AGUC (2.5 mM) 4 - DEPC-H2O 7.5 0.1 CTP (100uM) - 2.4 Total 19 14 2. 將上述樣品混合後,於放射線室中先加入 5 ìl *CTP 於 hot tube 中,再分別加 入 1 ìl SP6 polymerase 於兩管中,震盪離心,總體積共 20 ìl。 3. 37℃ 反應 1.5 小時進行 In vitro transcription。 4. 加入 1 ìl DNase I,37℃ 反應 15 分鐘。
5. 加入等體積 phenol / chloroform / isoamyl alcohol = 25: 24: 1,於兩管中,震 盪離心,12000 rpm 5 分鐘。
6. Chroma-spin 10 DEAE column 先以 2000 rpm 離心 3 分鐘,去除 DEPC-H2O 後,保留去除液,換新的載管後,將前 4 步驟所得之上清液通入管柱中,2000 rpm 離心 3 分鐘。
7. 以 liquid scintillation analyzer 計算 hot RNA label 量,方法如下:
a. 取 1 ìl *CTP 以 ddH2O 稀釋 100 倍後,取 1 ìl 滴於濾紙 (Watman GF/C filter paper)上。
b. 將濾紙以鑷子置入含 3 ml cock tail (Nakalai) 之閃爍計數瓶內。
算 RNA 濃度,單管反應時間約 1 分鐘。 Hot value 1 15.4 d. RNA 濃度: × × ×106 = ( n ) fmol / ìl Control value 30000 11 8. Cold 股計算 RNA 量: a. 以分光光度計計算 A260吸光值 b. RNA 濃度:A260吸光值 ×40 ×稀釋倍數 ÷1000 ÷ (324.5×104)×109 = ( n ) fmol / ìl
9. Hot / Cold RNA 比例為 1/10,通常所製備的 hot/cold 等體積加入即可省去計 算量,但必需留 1 ìl hot RNA 為 control。
10. 將 19 ìl 的 hot RNA 與 20 ìl cold-RNA 加入 100 ìl 之 2 X hybridization buffer (40 mM Herpes-KOH, pH 7.6 / 1 M NaCl / 2 mM EDTA / 0.2% SDS)進行 hybridization。 11. 100℃ 水浴加熱 10 分鐘後,置於燒杯中自然冷卻反應過夜。 12. 預鑄 8% (30:1) gel,其濃度如下: 40% Acryl / Bis (29:1) 5 ml 5 X TBE 2.5 ml H2O 17.3 ml APS 175 ìl TEMED 8.75 ìl Total 25 ml 13. 鑄完膠後,其凝膠時間需 2 小時或是置放於室溫下,使其凝膠過夜。 3.8.2 雙股 RNA 受質製備 (continued)
1. 取第一天反應之樣品 200 ìl,加入 50 ìl / 5X loading dye,平均 loading 於 well 內,另加入未 hybridization 之 hot-RNA 為控制組。
2. 以 1X TBE 電泳緩衝液,250 V 進行電泳 1.5 hr。
鐘。
4. 沖片後,將片子與膠片重合,用針頭於雙股 RNA 處戳 4 個孔,以刀片將膠 體割下,置於 15 ml 離心管中,將其搗碎後,加入 3 ml elution buffer (0.5 M (NH4)2OAc, pH 7.0 / 0.1% SDS, 10 mM EDTA, pH 8.0),緩慢震盪 2 小時待其 diffusion。
5. 加入等體積 phenol / chloroform / isoamyl alcohol = 25: 24: 1 於離心管中震 盪一分鐘後,3000 rpm 離心 30 分鐘。
6. 取 8 管 1.5 ml ependoff,內含 0.7 ml 無水酒精,將步驟 5 所得之上清液平均 加入 ependoff 中後,加入 1 ìl glycogen 及 30 ìl幫助沉澱 RNA,置入 -20℃ 30 分鐘。 7. 4℃、12000 rpm 離心 15 分鐘後,可得白色 RNA 沉澱於管底。 8. 將上清去除,以 75% 酒精清洗 pellet,4℃、12000 rpm 離心 5 分鐘後,抽氣 凍乾。 9. 上清溶於 30 ìl 緩衝液 (20 mM Hepes-KOH, pH 7.6) 中,計算雙股 RNA數量。 3.8.2 RNA Helicase 活性分析法 取 0.5 ìg 的 His-VrRH1 純化蛋白質於 20 ìl 的反應混合液中 (20 mM Hepes-KOH, pH 7.0 / 2 mM DTT / 1.5 mM MnCl2 / 2.5 mM ATP / 0.1 mg/ml BSA / 2 units RNasin, 4.4 fmol 雙股 RNA),於 37℃ 反應 1 小時後,加入 5 ìl 5 倍的 RNA loading dye (0.1 M Tris-HCl, pH 7.4 / 20 mM EDTA / 1% SDS / 0.1% bromophenol blue / 0.1% xylene cynal / 50% glycerol)。取 10 ìl 跑 8% native polyacrylamide gel, 跑完膠後乾片並進行放射性顯影,顯影後之底片以 phosphoimager 計算活性。
3.8.3 ATPase 活性分析
取適當酵素量的 His-VrRH1 加入 1 ìl [α-32
P]ATP 或 1 ìl [ã-32P]ATP (3000 Ci / mmol;Amershan) 於 10 ìl 反應液 (20 mM Hepes-KOH, pH 7.0 / 2 mM DTT / 1.5
mM MgCl2),37℃反應 15 分鐘後,加入 0.5 ìl 終止液 (500 mM EDTA) 停止 反應。取出 1 ìl 反應物點在 Silica gel 60 sheet 上,以延展劑 (methyl nitril:H2O =80:20) 進行薄層分析 (Thin layer chromatography, TLC),風乾後進行放射顯 影,顯影後之底片以 phosphoimager 計算活性。。
3.9 蛋白質分析方法
3.9.1 蛋白質定量法
以 Bio-Rad protein Assay Dye Reagent Concentrate,使用前先以去離子水稀釋 5 倍,以 BSA 作標準曲線,可偵測量為 1~20 ìg/ml,測得吸光值可得標準迴歸直 線方程式。同樣地,樣品蛋白質與試劑反應後吸光值,內插標準迴歸直線可估量 樣品含量。 3.9.2 原態膠體電泳法 電泳前先製備以下試劑: A 液:丙烯醯胺液 (T 30%, C 2.6%): 丙烯醯胺 29.2 g Bis 0.8 g 加水至 100 ml。通常加入 5% (w / v) Dowex MR-3 攪拌過夜後再過濾,以免不溶 物干擾膠體凝結,避光保存於 4℃。 B 液:4X 分離膠體緩衝液: Tris (1.5 M) 90.8 g TEMED 1.8 ml 300 ml 水溶解之,以 HCl 調整 pH 值至 8.8,加水至 500 ml。 C 液:4X 焦集膠體緩衝液: Tris (0.5 M) 6.0 g
TEMED 0.4 ml 40 ml 水溶解之,以 HCl 調整 pH 值至 6.8,加水至 100 ml。 3. 10 % APS:取 0.1 g APS 溶於 1 ml 二次水中,4℃保存。 4. 5X 通用電泳緩衝液: Tris (450 mM) 54.5 g Boric acid (400 mM) 24.8 g EDTA.2Na (12.5 mM) 4.7 g 800 ml 水溶解之,以 HCl 調整 pH 值至 8.4,加水至 1000 ml。 5. 追蹤染料:Bromophenol blue 1mg 溶於 10 ml 50% (v/v) 甘油中。 6. 高分子量標準蛋白質組合:其分子量分別為 Thyroglobulin (669
kDa)、Ferritin (440 kDa)、Catalase (232 kDa)、Lactate dehydrogenase (140 kDa) 及 BSA (67 kDa)。 方法: 甲、鑄膠為五組電泳片組合,分離膠濃度為 7.5 %及 10 %,焦集膠體為 4 %, 其配方如下: 膠體 ( % ) 分 離 膠 體 溶 液 焦集膠體溶液 溶液 (ml) 7.5 % 10 % 4 % A 液 5 6.7 2.64 B 液 5 5 - C 液 - - 4.96 水 9.9 8.2 11.8 APS 0.1 0.1 0.4
總體積 20 20 20 乙、電泳:將電泳片組合於電泳槽中,注入 1X 通用電泳緩衝液,每一電泳 槽約須 150 ml,預跑 10 分鐘後,蛋白質樣品加入 2/5 體積的追蹤染料, 以微量針筒注入樣品槽內,以 100 V 進行電泳,等追蹤染料進入分離膠 體後,可調高電壓至 120 V。等追蹤染料跑出膠體後,停止電泳取出膠 片進行染色。 3.9.3 SDS 膠體電泳法 電泳前先製備以下試劑 1. A 液、B 液、C 液、5 % APS、5X 通用電泳緩衝液及追蹤染料同前原態 膠體電泳所述。 2. 2X SDS 膠體電泳樣品緩衝液: Tris (250 mM) 54.5 g β-mercaptoethanol (10 %) 10 ml SDS (4 %) 24.8 g EDTA.2Na (4 mM) 4.7 g 80 ml 水溶解之,以 HCl 調整 pH 值至 6.8,加水至 100 ml。 3. 10 % SDS 溶液。 4. 低分子量標準蛋白質組合:其分子量分別為 Phosphorylase b (94 kDa)、 BSA (67 kDa)、Ovalbumin (43 kDa)、Carbonic anhydrase (30 kDa)、Trypsin inhibitor (20 kDa) 及 α-Lactalbumin (14.4 kDa)。
方法:
1. 鑄膠為五組電泳片組合,分離膠濃度為 10 %及 12.5 %,焦集膠體