Volume 2011, Article ID 523596,13pages doi:10.1155/2011/523596
Research Article
Emodin Induces Apoptotic Death in
Murine Myelomonocytic Leukemia WEHI-3 Cells
In Vitro
and
Enhances Phagocytosis in Leukemia Mice
In Vivo
Yuan-Chang Chang,
1Tung-Yuan Lai,
2, 3Chun-Shu Yu,
4Hung-Yi Chen,
4Jai-Sing Yang,
5Fu-Shin Chueh,
6Chi-Cheng Lu,
7Jo-Hua Chiang,
7Wen-Wen Huang,
8Chia-Yu Ma,
9and Jing-Gung Chung
8, 101School of Chinese Medicine, China Medical University, Taichung 404, Taiwan
2School of Post-Baccalaureate Chinese Medicine, China Medical University, Taichung 404, Taiwan
3Department of Chinese Medicine and Chinese Internal Medicine, China Medical University Hospital, Taichung 404, Taiwan 4School of Pharmacy, China Medical University, Taichung 404, Taiwan
5Department of Pharmacology, China Medical University, Taichung 404, Taiwan
6Department of Health and Nutrition Biotechnology, Asia University, Taichung 412, Taiwan 7Department of Life Sciences, National Chung Hsing University, Taichung 402, Taiwan
8Department of Biological Science and Technology, China Medical University, Taichung 404, Taiwan
9Department of Food and Beverage Management, Technology and Science Institute of Northern Taiwan, Taipei 112, Taiwan 10Department of Biotechnology, Asia University, Taichung 412, Taiwan
Correspondence should be addressed to Jing-Gung Chung,[email protected]
Received 6 January 2011; Revised 28 February 2011; Accepted 3 March 2011
Copyright © 2011 Yuan-Chang Chang et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Emodin is one of major compounds in rhubarb (Rheum palmatum L.), a plant used as herbal medicine in Chinese population. Although many reports have shown that emodin exhibits anticancer activity in many tumor cell types, there is no available information addressing emodin-affected apoptotic responses in the murine leukemia cell line (WEHI-3) and modulation of the immune response in leukemia mice. We investigated that emodin induced cytotoxic effects in vitro and affected WEHI-3 cells in vivo. This study showed that emodin decreased viability and induced DNA fragmentation in WEHI-3 cells. Cells after exposure to emodin for 24 h have shown chromatin condensation and DNA damage. Emodin stimulated the productions of ROS and Ca2+and reduced the level ofΔΨmby flow cytometry. Our results from Western blotting suggest that emodin triggered apoptosis of WEHI-3
cells through the endoplasmic reticulum (ER) stress, caspase cascade-dependent and -independent mitochondrial pathways. In in vivo study, emodin enhanced the levels of B cells and monocytes, and it also reduced the weights of liver and spleen compared with leukemia mice. Emodin promoted phagocytic activity by monocytes and macrophages in comparison to the leukemia mice group. In conclusions, emodin induced apoptotic death in murine leukemia WEHI-3 cells and enhanced phagocytosis in the leukemia animal model.
1. Introduction
Emodin (1,3,8-trihydroxy-6-methylanthraquinone) is one of the major compound in the root of rhubarb (Rheum
palma-tum L.) [1,2] and possesses immunosuppressive, anticancer, antiinflammatory, antiatherosclerotic, vasorelaxant, and vas-orelaxant effects [3–5]. Numerous reports have shown that emodin has antiproliferative effects on many kinds of cancer cell lines such as HER-2/neu-overexpressing breast cancer
[6,7], leukemia [8,9], hepatoma [10], and lung [11] cancer cells. Also, emodin-triggered apoptosis is mediated through the caspase- and mitochondria-dependent pathways in prox-imal tubular epithelial HK-2 cells [12]. Emodin enhanced gefitinib-induced cytotoxicity via Rad51 downregulation and ERK1/2 inactivation in human breast cancer BCap-37 cells [13]. Therefore, emodin has now been proposed as a potential agent in the management of tumors [14]. In our laboratory, we also found that emodin induced apoptosis
in human tongue squamous cancer SCC-4 cells through reactive oxygen species (ROS) and mitochondria-dependent pathways [15], but it has cytotoxic and protective effects in rat C6 glioma cells, and Mdr1a and nuclear factor kappaB play important roles in cell survival [16].
Leukemia is one of the major causes of deaths in the human population. In Taiwan, about 4.0 per 100,000 people die each year of leukemia and it is the 11th most common malignancy based on the report of the Department of Health,
Taiwan in 2009 [17]. In the clinical therapy, the major
strategies for patients with leukemia including bone marrow transplant, radiotherapy, and chemotherapy [18,19]. How-ever, the cure rate and side effects are still unsatisfied, and to find new agent for leukemia patients is urgent. Numerous clinical drugs for cancer patients are obtained from natural products. Although various studies of biological activities on emodin have been carried out, regarding the molecular mechanisms of emodin-acted the cell death in vitro and promotion of immune responses in animal model in vivo are still undefined. Therefore, we investigated the effects of emodin on growth inhibition and apoptotic cell death of murine WEHI-3 leukemia cells in vitro, and modulation of immune responses in leukemia mice model in vivo.
2. Material and Methods
2.1. Reagents and Antibodies. Dimethyl sulfoxide (DMSO), N-acetylcysteine (NAC), propidium iodide (PI), Triton
X-100, and RNase A were obtained from Sigma-Aldrich Corp. (St. Louis, MO, USA). The pan-caspase inhibitor (Z-VAD-FMK), 3 inhibitor (Z-DEVD-(Z-VAD-FMK), and caspase-9 inhibitor (Z-LEHD-FMK) were purchased from R&D Systems (Minneapolis, Menn, USA). All primary antibodies were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, Calif, USA). The peroxidase-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology, Inc. Enhanced chemiluminescence (ECL) kit and Western blotting reagents were purchased from Pierce Chemical Co. (Rockford, Ill, USA).
2.2. In Vitro Studies
2.2.1. Murine Leukemia Cell Line. WEHI-3 murine
myelo-monocytic leukemia cell line was purchased from the Food Industry Research and Development Institute (Hsinchu, Taiwan). Cells were seeded in 75-cm2cell culture flasks and
maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine 100 U/mL
penicillin, and 100μg/mL streptomycin at 37◦C under a
humidified 5% CO2atmosphere.
2.2.2. Cell Growth Inhibition Assay. The viable WEHI-3 cells
were assessed by the MTT assay. About 1×104cells/100μL
per well plated in 96-well plates were treated with emodin
at 0, 25, 50, 100, and 150μM for 24 and 48 h. MTT
solution (Sigma-Aldrich Corp., 5 mg/mL) was prepared and a volume of 10μL was individually added to each well for 4-h
incubation [20]. MTT is reduced to form purple formazan
a, b a, b, c a, b, c a, b, c a, b, c, d a, b a 48 24 (h) 0 20 40 60 80 100 120 140 Cell vi abilit y (% o f cont rol) 0μM 25μM 50μM 100μM 150μM (a) Emodin Marker 0 50 100 150 (μM) 300 500 700 1000 1500 2000 3000 (bp) (b)
Figure 1: Emodin affected the percentage of viable cells and DNA fragmentation in WEHI-3 cells. Cells (1×104cells/well; 96-well plates) were plated in RPMI 1640 medium + 10% fetal bovine serum (FBS) with 0, 25, 50, 100, and 150μM of emodin for 24 and 48 h. The cells were collected by centrifugation and the viable cells were determined by using the MTT assay (a). Cells were treated with 0, 50, and 100μM of emodin for 24 h, and then DNA was isolated for DNA gel electrophoresis (b) as described in Section2. Columns, mean of three determinations; bars, SD. a,P < .05 shows significantly different when compared with DMSO-treated control; b, c, and d,P < .05 indicates significantly different compared with 25, 50, and 100μM emodin-treated groups, respectively (one-way ANOVA followed by Bonferroni’s test for multiple comparisons).
product by the mitochondrial dehydrogenases of viable cells. The MTT-purple formazan productions were dissolved in DMSO and then were measured by absorbance at 570 nm in an ELISA plate reader as described previously [20].
2.2.3. DNA Laddering Fragmentation. Approximately 2 ×
105cells/well of WEHI-3 cells were grown in 12-well plates
and treated with 0, 50, 100, and 150μM of emodin for 24 h.
(A) (B) (C) (D) 100 50 25 0 Emodin (μM) 0 2 4 6 8 10 Fluor esc enc e int ensit y (fold o f cont rol) ∗ ∗ ∗ ∗ (a) Control 200x 100μM 200x 50μM 200x 100 50 0 Emodin (μM) 0 1 2 3 4 C o met length (fold of co nt ro l) ∗ ∗ ∗ ∗ ∗ ∗ (b)
Figure 2: Effects of emodin on apoptosis and DNA damage in WEHI-3 cells by using DAPI staining and Comet assay. Cells (2×105cells/well) in 12-well plate were incubated with 0, 25, 50, and 100μM emodin for 24 h and apoptosis was determined using DAPI staining (a). Data represent mean±SD of at least three experiments. Cells were treated with 0, 50, and 100μM emodin for 24 h and then were harvested for the examination of DNA damage using the Comet assay (b) as described in Section2. Comet tail length was calculated, quantified and expressed in mean±S.D for at least three replicates.∗Significantly different compared with DMSO-treated control, P < .05, and∗∗∗significantly
different from the control sample at P < .001.
and then were measured in 1.5% agarose gel electrophoresis, followed to photograph after being stained with ethidium bromide (Sigma-Aldrich Corp.) under UV illumination as described previously [21].
2.2.4. DAPI Staining for Apoptosis. Cells (2×105cells/well)
were seeded in 12-well plates and emodin was individually
added to the cells at final concentrations at 0, 50, and 100μM
for 24 h. Cells were harvested, washed with PBS, and fixed with 4% paraformaldehyde. The fixed cells were then washed with PBS and stained with 4,6-diamidino-2-phenylindole
(DAPI, 1μg/mL; Invitrogen) for 30 min in the dark. Cells
were then examined and determined under a fluorescent microscope, photographed and apoptotic cells identified [22,23].
3 h 1 h 6 h 12 h 0 h 104 103 102 101 100 ROS 0 20 40 60 80 100 Co u n ts 12 6 3 1 0 Time of incubation (h) 0 20 40 60 80 100 120 R O Sp ro d u ct io n( % ) ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ (a) 3 h 24 h 6 h 12 h 0 h 104 103 102 101 100 Fluo-3/AM 0 30 60 90 120 150 Co u n ts 24 12 6 3 0 Time of incubation (h) 0 10 20 30 40 50 In tr ac ellular C a 2+ re le as e (% ) ∗ ∗ ∗ ∗ (b) 0 h 1 h 6 h 3 h 12 h 104 103 102 101 100 MMP 0 40 80 120 160 200 Co u n ts 12 6 3 1 0 Time of incubation (h) 0 20 40 60 80 100 120 Loss of ΔΨ m (% of co nt ro l) ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ (c)
Figure 3: Emodin affected the levels of ROS, Ca2+andΔΨ
min WEHI-3 cells. Cells were cultured in 100μM emodin for 0, 1, 3, 6, 12 or
24 h. Cells were harvested and resuspended in DCFH-DA for determining the changes in ROS (a), in Fluo-3/AM for staining of Ca2+(b) and in DiOC6for determiningΔΨm(c) as described in Section2.∗P < .05 and∗∗∗P < .001 were considered significant when compared with
DMSO-treated control.
2.2.5. Comet Assay for DNA Damage. The Comet assay was
followed the procedures of Wang et al. with some modifi-cations [24]. Cells (2×105cells/well) in 12-well plates were
incubated with 0, 25, 50, and 100μM of emodin for 24 h.
Cells were harvested for the examination of DNA damage
using the Comet assay. Comets of cells on slides acquired DNA damage by using the CometScore Freeware analysis (TriTek Corporation, Sumerduck, VA, USA). Comet tail length was calculated, quantified, and expressed in mean±
Caspase-9 Caspase-8
Caspase-3
Control Emodin
Emodin + pan-caspase inhibitor 0 0.2 0.4 0.6 0.8 1 1.2 1.4 Caspases acti vi ty (O .D .405) ∗ ∗ ∗ ∗ ∗ ∗ (a) Emodin NAC Caspase-9 inhibitor Caspase-3 inhibitor ∗ ∗ ∗∗ ∗ ∗ ∗ ∗ ∗ − − − − − + − − − − + − − − − + + − − − + + − − + − + − + − − + 0 20 40 60 80 100 120 140 Cell vi abilit y (% of co nt ro l) (b)
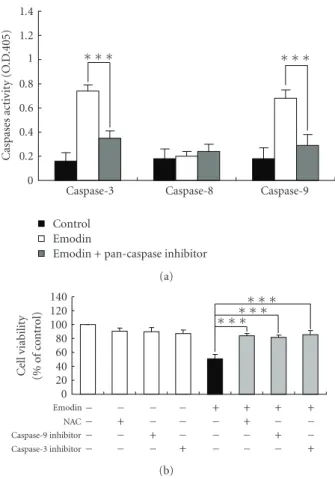
Figure 4: Emodin stimulated the activities of caspase-3 and -9 of WEHI-3 cells. Cells were seeded in RPMI 1640 medium + 10% FBS with pretreatment with NAC, inhibitors of caspase-9 and -3 or a pan-caspase inhibitor, and then were exposed to 100μM of emodin for 24 h. Cells were determined the caspase-3, -8, and -9 activity (a) and percentage of viable cells (b) as described in Section 2. Each experiment was done with triple sets, and columns, mean of three determinations; bars, SD.∗P < .05 and∗∗∗P < .001 were considered significant different as compared to the DMSO-treated control group.
2.2.6. Detection of Reactive Oxygen Species (ROS), Ca2+ Pro-duction Levels and Mitochondrial Membrane Potential (ΔΨm). Cells (2×105cells/mL) in RPMI 1640 medium were treated
with 100μM of emodin for 0, 1, 3, 6, 12, or 24 h. The cells
were harvested, washed twice with PBS, and then resus-pended in 500μL of 2,7-dichlorodihydrofluorescein diacetate
(10μM) (DCFH-DA) for determining the changes in ROS,
in 500μL of Fluo-3/AM (2.5 μg/mL) for staining of Ca2+
and in 500μL of DiOC6(500 nmol/L) for determiningΔΨm. Cells then were individually incubated at 37◦C for 30 min and were analyzed by flow cytometry (Becton Dickinson FACSCalibur, San Jose, Calif, USA) and BD CellQuest Pro software [21,23,25].
2.2.7. Determinations for Caspase-3, -8 and -9 Activity. Cells
(1×106cells/mL) in RPMI 1640 medium in 10-cm dishes
were exposed to 100μM of emodin for 24 h or pretreated
with a pan-caspase inhibitor (10μM, Z-VAD-FMK) for 3 h.
The cells were harvested and lysed in lysis buffer (50 mM Tris-HCl (pH 7.4), 1 mM EDTA, 10 mM EGTA, 10 mM digi-tonin and 2 mM DTT). The cell lysates (50μg proteins) were
incubated with caspase-3, -9, and -8 specific substrates (Ac-DEVD-pNA, Ac-LEHD-pNA, and Ac-IETD-pNA) (R&D systems Inc., Minneapolis, MN, USA) for 1 h at 37◦C. The caspases activities were determined by measuring the release of pNA at OD405 as previously described [26, 27]. Cells
were also pretreated with the caspase-9 inhibitor (10μM,
Z-IETD-FMK), caspase-3 inhibitor (10μM, Z-DEVD-FMK)
(R&D Systems) and NAC, and then were treated with 100μM emodin for 24-h exposure. Cells were harvest for
determination of viability as previously described [28,29].
2.2.8. Western Blotting for Examining the Apoptosis-Associated Protein Levels. Cells (2 × 105 cells/mL) in RPMI 1640
medium were treated with 100μM of emodin for 0, 6, 12,
and 24 h, isolated cells from each treatment were lysed and the protein levels were determined as described previously [15,25] for determining apoptosis-associated proteins levels such as caspase-3, -7 and -9, PARP, cytochrome c,
Apaf-1, AIF, Endo G, GADD153, GRP78, ATF-6α, Bcl-2, Bcl-xL,
Bax and Bad. All samples were separated by sodium dodecyl sulfate polyacrylamide (SDS-PAGE) gel electrophoresis as described previously [15,25]. Quantification of band density was determined using NIH Image J software.
2.3. In Vivo Studies
2.3.1. BALB/c Mice. Fifty male BALB/c mice, each was
ap-proximately 22–28 g in body weight at 8 weeks of age were purchased from the Laboratory Animal Center, College of Medicine, National Taiwan University (Taipei, Taiwan). These animals were maintained at 25◦C on a 12-h light/dark cycle in the animal center of the China Medical University followed the animal guideline as previously described [30, 31].
2.3.2. Emodin Treatment. Fifty mice were randomly divided
into 5 groups (10 animals per group). Group I was control (normal animal) and Group II was only treated olive oil. Group III was intraperitoneally (i.p.) injected with WEHI-3 cells (1×105cells) and the olive oil treatment only as negative control. Group IV was i.p. injected with WEHI-3 cells (1×
105cells) and orally treated with emodin (5 mg/kg) in olive oil. Group V was i.p. injected with WEHI-3 cells (1×105cells) and oral treatment with emodin (10 mg/kg) in olive oil [8,32]. Emodin (5 and 10 mg/kg) was administered by oral gauge in 100μL of olive oil. Animals were inoculated with
WEHI-3 cells for 2 weeks of incubation, and subsequently leukemia mice were administrated daily for 2 weeks before being weighed and sacrificed.
2.3.3. Blood Collection and Immunofluorescence Staining.
After emodin treatment for 2 weeks, one mL of blood was collected from each mouse of examined groups. The
indi-vidual samples were added 1 × Pharm Lyse lysing buffer
Emodin (100μM) (h) 24 12 6 0 Caspase-3 (32/17 kDa) Active form (Active form) 2.9 2.5 2.1 1 Caspase-7 (35/20 kDa) Active form (Active form) 1.9 1.9 1.6 1 Caspase-9 (45/35 kDa) Active form (Active form) 2.9 4.2 3.8 1 Cleavage fragment 3.9 2 2.4 1 PARP (116/85 kDa) 1 0.9 0.9 1 Actin (43 kDa) (a) Emodin (100μM) (h) 24 12 6 0 Cytochrome c (15 kDa) 2.8 2.4 1.8 1 Apaf-1 (130 kDa) 2.5 3.5 1 1 AIF (67 kDa) 3.8 3.6 3.5 1 2.4 3.2 2.6 1 Endo G (35 kDa) 1 1.2 1.1 1 Actin (43 kDa) (b) Emodin (100μM) (h) 24 12 6 0 GADD153 (30 kDa) 2.4 4.5 3.2 1 GRP78 (78 kDa) 3.4 1.2 0.8 1 ATF-6α (90 kDa) 2.6 2.9 4.8 1 3 3.9 4.4 1 Caspase-12 (55 kDa) 1 1 1 1 Actin (43 kDa) (c) Emodin (100μM) (h) 24 12 6 0 Bcl-2 (26 kDa) 0.4 0.3 0.8 1 Bcl-xL (30 kDa) 0.2 1.5 3.2 1 Bax (23 kDa) 3.8 2.5 1.6 1 2.6 2.9 4.8 1 Bad (25 kDa) 0.9 1 0.9 1 GAPDH (37 kDa) Anti-apoptosis Pro-apoptosis (d)
Figure 5: Emodin affected the apoptosis-associated protein levels in WEHI-3 cells. Cells were treated with emodin at 100 μM for 0, 6, 12, and 24 h, and then the total proteins were prepared and determined as described in Section2. The levels of associated proteins expressions ((a): caspase-3, -7, and -9 and PARP; (b): cytochrome c, Apaf-1, AIF and Endo G; (c): GADD153, GRP78, ATF-6α and caspase-12; (d): Bcl-2, Bcl-xL, Bax, and Bad) were estimated by Western blotting as described in Materials and Methods.
blood cells and then centrifuged at 1000 ×g at 4◦C for
15 min. Isolated white blood cells were stained with antiCD3fluorescein isothiocyanate (FITC), CD11bFITC, -CD19-phycoerythrin (PE), and -Mac-3-PE antibodies (BD Pharmingen, San Diego, CA, USA) for measuring the cell surface markers of T cell (CD3), B cell (CD19), monocytes, and macrophages (CD11b and Mac-3, resp.) and then were determined the cell marker levels by flow cytometry as previously described [30,33].
2.3.4. Phagocytic Activity of Monocytes and Macrophages.
Leukocytes from the mice were collected for determining the phagocytosis by using a PHAGOTEST kit (Glycotope
Biotechnology GmbH/Orpegen Pharma, Heidelberg, Ger-many) as previously described [30,31]. Cells were isolated from peripheral blood mononuclear cells (PBMC), and peritoneal cavity of control and emodin-treated animals. Isolated cells were individually incubated with opsonised FITC-labeled E. coli (20μL) for 1 h at 37◦C according to the manufacturer’s instruction. After incubation, an ice-cold quenching solution (100μL) was added to stop the reaction
then and DNA content of the monocytes/macrophages for cell cycle analysis. Cells were prepared then were analyzed by flow cytometery. Fluorescence data were collected on 10,000 cells by flow cytometry and analyzed using the BD CELLQUEST Pro software [30,31].
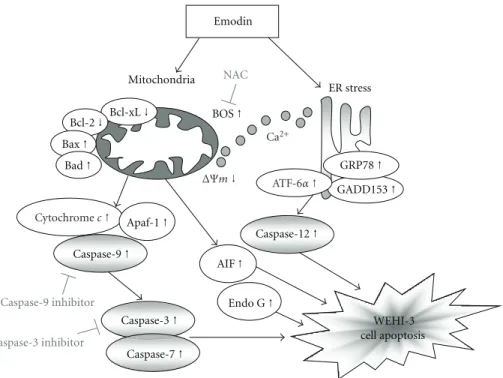
Emodin Mitochondria ER stress NAC Caspase-9 inhibitor Caspase-3 inhibitor WEHI-3 cell apoptosis Bcl-xL ↓ Bcl-2 ↓ Bax ↑ BOS ↑ GRP78 ↑ GADD153 ↑ Caspase-12 ↑ Bad ↑ Apaf-1 ↑ Caspase-9 ↑ Caspase-3 ↑ Endo G ↑ AIF ↑ Caspase-7 ↑ ΔΨm↓ Cytochrome c↑ ATF-6α↑ Ca2+
Figure 6: The proposed mechanisms of emodin-induced apoptosis in WEHI-3 cells. The flow chart shows that emodin induced apoptosis through the ER stress, mitochondria-, and caspase-3-dependent signaling pathways in murine leukemia WEHI-3 cells in vitro.
2.4. Statistical Analyses. Data were expressed as mean±SD and differences between control and emodin-treated groups were analyzed by one-way ANOVA followed by Bonferroni’s test for multiple comparisons or Dunnett’s test.∗P < .05 and ∗∗∗P < .001 were considered significant.
3. Results
3.1. Emodin Induced Cytotoxic Effects and DNA Fragmenta-tion in WEHI-3 Cells. The doses of emodin (50 to 150μM)
are required to induce cell death in WEHI-3 cells for 24-h treatment and t24-he percentage of viable cells decrease was
41–63% (Figure 1(a)). However, emodin (25–150μM) for
48-h treatment decrease viable cells and the results show 19–82% (Figure1(a)). Herein, emodin in the concentration
as low as 25μM initiated cell death of WEHI-3 cells and
the IC50 is 100μM (Figure 1(a)). DNA gel electrophoresis
assay indicated that 100–150μM of emodin-induced DNA
fragmentation in WEHI-3 cells after exposure for 24 h (Figure1(b)).
3.2. Emodin Induced Apoptosis and DNA Damage in WEHI-3 Cells. To investigate the mechanism of emodin-induced
leukemic cell death, the effect of emodin on DNA damage
was evaluated. The effects of emodin on DNA integrity were further analyzed using the DAPI staining and Comet assay. Emodin-induced chromatin condensation (an apoptotic characteristic) in a dose-dependent response (Figure2(a)). Also, emodin triggered DNA damage after 24-h treatment and quantification of the number of leukemic WEHI-3 cells
displaying a Comet tail strongly increased after emodin treatment (Figure2(b)).
3.3. Emodin Altered the Levels of ROS, Ca2+ and ΔΨ m in WEHI-3 Cells. Cells treated with 100μM emodin were
deter-mined by flow cytometric assays and results are shown in Fig-ures3(a),3(b), and3(c), which indicated that emodin pro-moted the levels of ROS (Figure3(a)) and Ca2+(Figure3(b)),
but decreased the level ofΔΨm(Figure3(c)). To investigate emodin-induced cell death is through the disruption of mito-chondrial respiratory chain, leading to ROS accumulation and cellular damage. An increase in intracellular fluorescence after DCFH-DA loading showed the reversal of ROS accu-mulation (Figure3(a)). In order to elucidate the mechanism of emodin-induced mitochondria-dependent apoptotic cell death, the effects of emodin on the levels of intracellular Ca2+
andΔΨmwere analyzed. These data indicated that
emodin-induced apoptosis is possibly mediated through alteration of mitochondrial permeability transition in WEHI-3 cells in
vitro.
3.4. Emodin Affected Activities of Caspase-3 and -9 of WEHI-3 Cells. In order to evaluate the roles of caspase-mediated
pathways in emodin-induced apoptotic death of WEHI-3 cells, cells were pretreated with a pan-caspase inhibitor (Z-VAD-FMK), and then exposed to emodin. We found that emodin promoted the activities of caspase-3 and -9, but it did not affect that of caspase-8 (Figure4(a)). Pretreatment with Z-VAD-FMK could decrease emodin-stimulated the activities of caspase-3 and -9 in WEHI-3 cells (Figure4(a)). Furthermore, cells were individually preincubated with
WEHI-3/ emodin 10 mg/kg WEHI-3/ emodin 5 mg/kg WEHI-3 Olive oil Control 0 10 20 30 40 50 Bod y w eig ht (g) ∗ ∗ (a) WEHI-3/ emodin 10 mg/kg WEHI-3/ emodin 5 mg/kg WEHI-3 Olive oil Control 0 0.1 0.2 0.3 0.4 0.5 Spleen w eig h t (g) ∗ ∗ (b) WEHI-3/ emodin 10 mg/kg WEHI-3/ emodin 5 mg/kg WEHI-3 Olive oil Control 0 1 2 3 4 Li ve r w eig h t (g) ∗ (c)
Figure 7: Emodin affected the weights of the leukemia mice which were treated with without or with emodin 2 weeks. BALB/c mice were intraperitoneally injected with WEHI-3 cells (1×105cells/100μL) in PBS for 2 weeks and/or treated with emodin once daily by oral administration for 14 days. Blood was collected and animals were sacrificed for examinations of weights of body (a) spleen (b) and liver (c) tissues, and then were individually weighed. Each point is the mean±SD and similar results were observed in at least three independent experiments (n=10) followed by one-way ANOVA followed by Dunnett’s test.
an antioxidant scavenger (NAC), caspase-9 inhibitor (Z-IETD-FMK), and caspase-3 inhibitor (Z-DEVD-FMK), which led to increase emodin-reduced the percentage of viable WEHI-3 cells compared to the emodin-treated cells only (Figure4(b)). These data suggest that emodin triggered apoptosis of WEHI-3 cells through the ROS and caspase-dependent pathways.
3.5. Emodin Altered the Apoptosis-Associated Protein Levels in WEHI-3 Cells. Cells were exposure to 100μM of emodin for
0, 6, 12, and 24 h and then the total protein were prepared and determined by Western blotting analysis. These results are presented in Figure 5((a): caspase-3, -7 and -9, PARP; (b): cytochrome c, Apaf-1, AIF, Endo G; (c): GADD153,
GRP78, ATF-6α, caspase-12; (d): Bcl-2, Bcl-xL, Bax, and
Bad), which indicated that the levels of caspase-3, -7 and -9, PARP (Figure5(a)), cytochrome c, Apaf-1, AIF, Endo G
(Figure 5(b)), GADD153, GRP78, ATF-6α, caspase-12
(Figure 5(c)), Bax, and Bad (Figure 5(d)) were increased,
while the levels of Bcl-2 and Bcl-xL (Figure 5(d)) were
decreased and these effects may lead to cell apoptosis. Overall, the proposed possible signal pathways for emodin-induced apoptosis are shown in Figure6.
3.6. Emodin Affected the Weight of BALB/c Mice after Injec-tion with WEHI-3 Cells and/or Exposure to Emodin for 2 Weeks. The BALB/c mice after injection with WEHI-3 cells
were treated with or without emodin (5 and 10 mg/kg). Results indicate that emodin increased the body weight of
mice (Figure 7(a)) but decreased the weights of spleen
(Figure7(b)) and liver (Figure 7(c)) when compared with the leukemia mice group.
3.7. Emodin Affected the Cell Markers of White Blood Cells from Leukemia BALB/c Mice. The leukemia mice were
WEHI-3/ emodin 10 mg/kg WEHI-3/ emodin 5 mg/kg WEHI-3 Olive oil Control 0 10 20 30 40 50 CD3 (%) N.S. N.S. (a) WEHI-3/ emodin 10 mg/kg WEHI-3/ emodin 5 mg/kg WEHI-3 Olive oil Control 0 10 20 30 40 50 CD19 (%) ∗ (b) WEHI-3/ emodin 10 mg/kg WEHI-3/ emodin 5 mg/kg WEHI-3 Olive oil Control 0 10 20 30 40 50 CD11b (%) ∗ (c) WEHI-3/ emodin 10 mg/kg WEHI-3/ emodin 5 mg/kg WEHI-3 Olive oil Control 0 5 10 15 20 M ac-3 (%) ∗ ∗ (d)
Figure 8: Emodin affected the cell markers of white blood cells from leukemia BALB/c mice. The leukemia mice were orally treated without and with emodin (5 and 10 mg/kg) in olive oil for 14 days. Blood was collected from individual animals and was analyzed for surface cell markers by flow cytometry as described in Section2. Each point is the mean±SD and similar results were observed in at least three independent samples (n=10) followed by one-way ANOVA followed by Dunnett’s test. N.S.=Not Significant (P > .05).
10 mg/kg) for 14 days. Blood was collected individually from animals of each group and was analyzed for cell markers by flow cytometry. The results are shown in Figures 8(a), 8(b),8(c), and8(d). Emodin did not affect the level of CD3
surface marker (Figure 8(a)); however, emodin increased
the levels of CD19 (Figure 8(b)) and CD11b (Figure8(c)) in emodin-treated groups and it also decreased the level of Mac-3 marker after emodin at 5 and 10 mg/kg treatment (Figure8(d)) in comparison to leukemia mice group.
3.8. Emodin Affected on Phagocytosis by Monocytes and Ma-crophages from BALB/c Mice. Animals were injected with
WEHI-3 cells for 2 weeks and then treated without or with emodin for 2 weeks. Leukocytes were collected from PBMC and the peritoneal cavity and were analyzed for phagocytosis of monocytes and macrophages by flow cytometry. Results
shown in Figures9(a)and 9(b)indicate that emodin
pro-moted the phagocytosis from PBMC and peritoneal cavity of leukemia mice.
4. Discussion
The major differences between leukemic cells and tumors
are that leukemic cells capable of circulating and having access to various organs through interaction with activated vascular cells [34]. However, the subsets of leukemic cells may also adhere to vascular cells for establishing perivascular infiltrates then may be endowed with a unique mechanism of resistance to chemotherapy. It is well documented that circulating and vascular-adherent leukemic cells require cytoskeletal stability for maintaining mitochondrial and cel-lular function to avoid cell death [35]. In this study, we show that various doses of emodin selectively induced apoptosis of murine WEHI-3 leukemic cells by caspase-dependent response as well as ROS-mediated signal pathways for leading to cell death. Moreover, we also found that emodin-triggered apoptotic death is involved in the unfolded protein response (endoplasmic reticulum stress) in WEHI-3 cells. The molecular mechanisms of emodin-caused cell death are
WEHI-3/ emodin 10 mg/kg WEHI-3/ emodin 5 mg/kg WEHI-3 Olive oil Control 0 10 20 30 40 50 Phagocy totic acti vi ty (%) ∗ (a) WEHI-3/ emodin 10 mg/kg WEHI-3/ emodin 5 mg/kg WEHI-3 Olive oil Control 0 5 10 15 20 25 30 Phagocy totic acti vi ty (%) ∗ ∗ ∗ ∗ (b)
Figure 9: Emodin affected on phagocytosis by monocytes and macrophages from leukemia BALB/c mice. Mice were injected with WEHI-3 cells (1×105cells/100μL) in PBS for 2 weeks and treated without or with emodin for 2 weeks. Leukocytes were collected from PBMC (a) and peritoneal cavity (b) from animals and were analyzed for phagocytosis by flow cytometry as described in Section2. Each point is the mean±SD and similar results were observed in at least three independent samples (n=10), and∗P < .05 and∗∗∗P < .001 (n=10) were shown significant followed by one-way ANOVA followed by Dunnett’s test.
complex and most likely mediated through recruitment of caspase-dependent and -independent pathways.
In leukemia cells, various concentrations of emodin have been shown to result in caspase activations and apoptotic cell death [9,36]. Emodin also induced G2/M phase arrest and subsequent cell death in leukemia cells [9]. However, our data showed that emodin impaired leukemic cell survival by caspases activation and in part through ROS production, Ca2+release and the disruption ofΔΨ
mand releases of some proapoptotic factors including cytochrome c, AIF and Endo G. Mitochondrial dysfunction might result in the release of cytochrome c, AIF and Endo G in emodin-treated WEHI-3 cells as can be seen in Figure5.
Mitochondrial membrane potential disruption and G2/M arrest have previously been described in leukemia cells upon treatment with emodin or aloe-emodin and rhein from rhubarb (Da-huang) [8,9,37,38]. In the present study, our data suggest that emodin induced leukemic cell death by an apoptotic pathway that is distinct from conventional other compounds agents. We also examined whether emodin-induced mitochondrial damage promoted the release of
cytochrmoe c, AIF and Endo G (Figure 5(b)). AIF and
Endo G that are ubiquitously expressed flavoproteins, which might play a critical role in caspase-independent apoptosis [39,40]. It was reported that AIF is similar to cytochrome
c normally localized to the mitochondrial intermembrane
space and released in response to apoptotic stimuli [41]. Our results also showed that emodin promoted the level of Bax, and decreased the level of Bcl-2 (Figure5(c)), and it is well documented that the ratio of Bax/Bcl-2 involved the dysfunction of mitochondria, resulting in cell apoptosis [42]. Bcl-2, an upstream effector molecule in the apoptotic pathway, has been recognized to be a potent suppressor of apoptosis [43], and most cancers generally overexpress Bcl-2 [44,45]. In the present study, we observed that emodin
significantly downregulated Bcl-2 protein and up-regulated
the level of Bax protein in WEHI-3 cells (Figure 5(d)),
suggesting that the involvement of an intrinsic apoptotic pathway is mediated in emodin-induced apoptosis in WEHI-3 cells.
Our results also showed that emodin promoted ROS
and Ca2+ production in WEHI-3 cells (Figure3). We also
used DAPI staining and Comet assay to show that emodin
induced DNA damage in WEHI-3 cells (Figure 4). As the
increased ROS accumulation and DNA damage have been identified as mediators or initiators of apoptosis in certain condition [46]. These data indicated that emodin-induced
cell death in WEHI-3 cells is mediated via loss of ΔΨm,
leading to intracellular ROS and DNA damage then followed by caspase-3 activation and the releases of AIF and Endo G for causing apoptosis. We further showed that these events lead to cell death via caspase-dependent or -independent mitochondrial apoptosis with DNA fragmentation. Taken together, these data indicate that emodin might induce cell death in part through a caspase-dependent as well as in part through a caspase-independent (mitochondria-dependent led to AIF and Endo G and ER stress) apoptotic pathway by accumulation of ROS as a result of the disruption of the mitochondria in WEHI-3 cells as shown in Figure6.
Emodin induced apoptosis in human promyeloleukemic HL-60 cells accompanied by activation of caspase cascade but
independent ROS production [9]. However, other reports
also showed that emodin induced apoptosis in human lung adenocarcinoma cells through a ROS-dependent
mitochon-drial signaling pathway [11]. Our previous studies also
showed that emodin induced apoptosis in SCC-4 cells through the production of ROS and
mitochondria-depend-ent pathways [15]. Currently, we showed that emodin
induced apoptosis in murine leukemia WEHI-3 cells through ROS production, caspase-3 and mitochondria-dependent
pathways. Therefore, it is raising the possibility that emodin may have some chemotherapeutic chance for human leu-kemia. However, the effects of emodin on leukemia in vivo provided no clear information.
Other reports [47] and our previous studies [48,49] also showed the in vivo model through the mice intraperitoneally injected with WEHI-3 cells is well established. This mice model has been demonstrated as an ideal system for the study of potential therapeutic drugs (ATRA, aclacinomycin A, IL-6, G-CSF, and vitamin D3) which could induce in
vitro differentiation of WEHI-3 cells in monocytic and
granulocytic lineages [50–52]. Therefore, the purpose of this
study was to examine the effects of emodin on WEHI-3
cells in BALB/c mice in vivo. Herein, our results showed that emodin inhibited spleen leukemia tumor growth in a WEHI-3 leukemia murine model. We observed that the size of the spleens decreased the emodin-treated leukemia groups. These observations were also seen in liver tissues, and there was a significant difference between the control and
emodin-treated groups (Figure 7). Emodin also promoted
the phagocytosis by monocytes and macrophages in PBMC and peritoneal cavity of WEHI-3 leukemia mice in vivo (Figure9).
In summary, emodin provokes apoptosis in mice leuke-mia WEHI-3 cells in vitro and tends to inhibit leukeleuke-mia mice through stimulating phagocytosis in vivo. Our study is the first report and the in vitro the linkages of apoptotic cell death and the in vivo phagocytic activity of macrophages or monocytes were indeed needed for further investigation.
Acknowledgment
This work was supported by Grant DOH100-TD-C-111-005 from Taiwan Department of Health, China Medical Univer-sity Hospital Cancer Research Center of Excellence.
References
[1] J. W. Liang, S. L. Hsiu, H. C. Huang, and P. D. Lee-Chao, “HPLC analysis of emodin in serum, herbs and Chinese herbal prescriptions,” Journal of Food and Drug Analysis, vol. 1, no. 3, pp. 251–257, 1993.
[2] F. Yang, T. Zhang, G. Tian, H. Cao, Q. Liu, and Y. Ito, “Prepara-tive isolation and purification of hydroxyanthraquinones from Rheum officinale Baill by high-speed counter-current chro-matography using pH-modulated stepwise elution,” Journal of Chromatography A, vol. 858, no. 1, pp. 103–107, 1999. [3] H. C. Huang, S. H. Chu, and P. D. L. Chao, “Vasorelaxants
from Chinese herbs, emodin and scoparone, possess immuno-suppressive properties,” European Journal of Pharmacology, vol. 198, no. 2-3, pp. 211–213, 1991.
[4] X. M. Zhou and Q. H. Chen, “Biochemical study of Chinese rhubarb. XXII. Inhibitory effect of anthraquinone derivatives on Na+-K+-ATPase of the rabbit renal medulla and their diuretic action,” Acta Pharmaceutica Sinica, vol. 23, no. 1, pp. 17–20, 1988.
[5] M. Koyama, T. Ross Kelly, and K. A. Watanabe, “Novel type of potential anticancer agents derived from chrysophanol and emodin. Some structure-activity relationship studies,” Journal of Medicinal Chemistry, vol. 31, no. 2, pp. 283–284, 1988.
[6] L. Zhang, Y. K. Lau, L. Xi et al., “Tyrosine kinase inhibitors, emodin and its derivative repress HER-2/neu-induced cellular transformation and metastasis-associated properties,” Onco-gene, vol. 16, no. 22, pp. 2855–2863, 1998.
[7] L. Zhang, Y. K. Lau, W. Xia, G. N. Hortobagyi, and M. C. Hung, “Tyrosine kinase inhibitor emodin suppresses growth of HER-2/neu- overexpressing breast cancer cells in athymic mice and sensitizes these cells to the inhibitory effect of paclitaxel,” Clinical Cancer Research, vol. 5, no. 2, pp. 343–353, 1999.
[8] W. Chun-Guang, Y. Jun-Qing, L. Bei-Zhong et al., “Anti-tumor activity of emodin against human chronic myelocytic leukemia K562 cell lines in vitro and in vivo,” European Journal of Pharmacology, vol. 627, no. 1–3, pp. 33–41, 2010.
[9] Y. C. Chen, S. C. Shen, W. R. Lee et al., “Emodin induces apop-tosis in human promyeloleukemic HL-60 cells accompanied by activation of caspase 3 cascade but independent of reactive oxygen species production,” Biochemical Pharmacology, vol. 64, no. 12, pp. 1713–1724, 2002.
[10] D. E. Shieh, Y. Y. Chen, M. H. Yen, L. C. Chiang, and C. C. Lin, “Emodin-induced apoptosis through p53-dependent pathway in human hepatoma cells,” Life Sciences, vol. 74, no. 18, pp. 2279–2290, 2004.
[11] Y. U. T. Su, H. L. Chang, S. K. Shyue, and S. L. Hsu, “Emodin induces apoptosis in human lung adenocarcinoma cells through a reactive oxygen species-dependent mitochondrial signaling pathway,” Biochemical Pharmacology, vol. 70, no. 2, pp. 229–241, 2005.
[12] C. Wang, X. Wu, M. Chen et al., “Emodin induces apoptosis through caspase 3-dependent pathway in HK-2 cells,” Toxicol-ogy, vol. 231, no. 2-3, pp. 120–128, 2007.
[13] R. S. Chen, J. Y. Jhan, Y. J. Su et al., “Emodin enhances gefitinib-induced cytotoxicity via Rad51 downregulation and ERK1/2 inactivation,” Experimental Cell Research, vol. 315, no. 15, pp. 2658–2672, 2009.
[14] N. J. Lee, J. H. Choi, B. S. Koo et al., “Antimutagenicity and cytotoxicity of the constituents from the aerial parts of Rumex acetosa,” Biological and Pharmaceutical Bulletin, vol. 28, no. 11, pp. 2158–2161, 2005.
[15] S. Y. Lin, W. W. Lai, C. C. Ho et al., “Eniodin induces apoptosis of human tongue squamous cancer SCC-4 cells through reactive oxygen species and mitochondria-dependent pathways,” Anticancer Research, vol. 29, no. 1, pp. 327–335, 2009.
[16] T. C. Kuo, J. S. Yang, M. W. Lin et al., “Emodin has cytotoxic and protective effects in rat C6 glioma cells: roles of Mdr1a and nuclear factorκB in cell survival,” Journal of Pharmacology and Experimental Therapeutics, vol. 330, no. 3, pp. 736–744, 2009. [17] J.-P. Lin, J.-S. Yang, J.-J. Lin et al., “Rutin inhibits human
leukemia tumor growth in a murine xenograft model in vivo,” Environmental Toxicology, 2011.
[18] K. C. Nau and W. D. Lewis, “Multiple myeloma: diagnosis and treatment,” American Family Physician, vol. 78, no. 7, pp. 853– 859, 2008.
[19] A. K. Fotoohi, Y. G. Assaraf, A. Moshfegh et al., “Gene expres-sion profiling of leukemia T-cells resistant to methotrexate and 7-hydroxymethotrexate reveals alterations that preserve intracellular levels of folate and nucleotide biosynthesis,” Biochemical Pharmacology, vol. 77, no. 8, pp. 1410–1417, 2009. [20] Y. U. H. Chang, J. S. Yang, S. C. Kuo, and J. G. Chung, “Induction of mitotic arrest and apoptosis by a novel syn-thetic quinolone analogue, CWC-8, via intrinsic and extrinsic apoptotic pathways in human osteogenic sarcoma U-2 OS
cells,” Anticancer Research, vol. 29, no. 8, pp. 3139–3148, 2009.
[21] J. S. Yang, G. W. Chen, TE. C. Hsia et al., “Diallyl disulfide induces apoptosis in human colon cancer cell line (COLO 205) through the induction of reactive oxygen species, endo-plasmic reticulum stress, caspases casade and mitochondrial-dependent pathways,” Food and Chemical Toxicology, vol. 47, no. 1, pp. 171–179, 2009.
[22] C.-C. Lu, J.-S. Yang, A.-C. Huang et al., “Chrysophanol induces necrosis through the production of ROS and alter-ation of ATP levels in J5 human liver cancer cells,” Molecular Nutrition and Food Research, vol. 54, no. 7, pp. 967–976, 2010. [23] J.-H. Chiang, J.-S. Yang, C.-Y. Ma et al., “Danthron, an anthraquinone derivative, induces DNA damage and caspase cascades-mediated apoptosis in SNU-1 human gastric cancer cells through mitochondrial permeability transition pores and Bax-triggered pathways,” Chemical Research in Toxicology, vol. 24, no. 1, pp. 20–29, 2011.
[24] S. C. Wang, J. G. Chung, C. H. Chen, and S. C. Chen, “2- and 4-Aminobiphenyls induce oxidative DNA damage in human hepatoma (Hep G2) cells via different mechanisms,” Mutation Research, vol. 593, no. 1-2, pp. 9–21, 2006.
[25] H. F. Lu, H. L. Wang, Y. Y. Chuang et al., “Danthron induced apoptosis through mitochondria- and caspase-3-dependent pathways in human brain glioblastoma multiforms GBM 8401 cells,” Neurochemical Research, vol. 35, no. 3, pp. 390–398, 2010.
[26] S. S. Lin, H. P. Huang, J. S. Yang et al., “DNA damage and endoplasmic reticulum stress mediated curcumin-induced cell cycle arrest and apoptosis in human lung carcinoma A-549 cells through the activation caspases cascade- and mitochondrial-dependent pathway,” Cancer Letters, vol. 272, no. 1, pp. 77–90, 2008.
[27] J. P. Lin, J. S. Yang, C. C. Lu et al., “Rutin inhibits the proliferation of murine leukemia WEHI-3 cells in vivo and promotes immune response in vivo,” Leukemia Research, vol. 33, no. 6, pp. 823–828, 2009.
[28] W. U. Ping-Ping, K. Sheng-Chu, W. W. Huang et al., “(-)-epigallocatechin gallate induced apoptosis in human adrenal cancer NCI-H295 cells through caspase-dependent and caspase-independent pathway,” Anticancer Research, vol. 29, no. 4, pp. 1435–1442, 2009.
[29] J.-S. Yang, M.-J. Hour, W.-W. Huang, K.-L. Lin, S.-C. Kuo, and J.-G. Chung, “MJ-29 inhibits tubulin polymerization, induces mitotic arrest, and triggers apoptosis via cyclin-dependent kinase 1-mediated Bcl-2 phosphorylation in human leukemia U937 cells,” Journal of Pharmacology and Experimental Thera-peutics, vol. 334, no. 2, pp. 477–488, 2010.
[30] J. S. Yang, C. C. Wu, C. L. Kuo et al., “Solannm lyratum extract affected immune response in normal and leukemia murine animal in vivo,” Human and Experimental Toxicology, vol. 29, no. 5, pp. 359–367, 2010.
[31] T. W. Tan, Y. T. Lin, J. S. Yang et al., “A. cantoniensis inhibits the proliferation of murine leukemia WEHI-3 cells in vivo and promotes immunoresponses in vivo,” In Vivo, vol. 23, no. 4, pp. 561–566, 2009.
[32] Y. Jing, J. Yang, Y. Wang et al., “Alteration of subcellular redox equilibrium and the consequent oxidative modification of nuclear factor κB are critical for anticancer cytotoxicity by emodin, a reactive oxygen species-producing agent,” Free Radical Biology and Medicine, vol. 40, no. 12, pp. 2183–2197, 2006.
[33] M. R. Loken, C. I. Green, and D. A. Wells, “Immunofluo-rescence of surface markers,” in Flow Cytometry: A Practical
Approach, M. G. Ormerod, Ed., pp. 61–82, Oxford University Press, Oxford, UK, 2000.
[34] I. Petit, M. A. Karajannis, L. Vincent et al., “The microtubule-targeting agent CA4P regresses leukemic xenografts by dis-rupting interaction with vascular cells and mitochondrial-dependent cell death,” Blood, vol. 111, no. 4, pp. 1951–1961, 2008.
[35] G. Kroemer, L. Galluzzi, and C. Brenner, “Mitochondrial membrane permeabilization in cell death,” Physiological Reviews, vol. 87, no. 1, pp. 99–163, 2007.
[36] H. E. Y. Zheng, J. D. Hu, Z. H. Zheng et al., “Emodin induces leukemic HL-60 cells apoptosis probably by inhibiting Akt signal pathway,” Yao Xue Xue Bao, vol. 42, no. 11, pp. 1142– 1146, 2007.
[37] H. C. Chen, W. T. Hsieh, W. C. Chang, and J. G. Chung, “Aloe-emodin induced in vitro G2/M arrest of cell cycle in human promyelocytic leukemia HL-60 cells,” Food and Chemical Toxicology, vol. 42, no. 8, pp. 1251–1257, 2004. [38] S. Lin, M. Fujii, and DE. X. Hou, “Rhein induces
apopto-sis in HL-60 cells via reactive oxygen species-independent mitochondrial death pathway,” Archives of Biochemistry and Biophysics, vol. 418, no. 2, pp. 99–107, 2003.
[39] E. Daugas, D. Nochy, L. Ravagnan et al., “Apoptosis-inducing factor (AIF): a ubiquitous mitochondrial oxidoreductase involved in apoptosis,” FEBS Letters, vol. 476, no. 3, pp. 118– 123, 2000.
[40] S. A. Lipton and E. Bossy-Wetzel, “Dueling activities of AIF in cell death versus survival: DNA binding and redox activity,” Cell, vol. 111, no. 2, pp. 147–150, 2002.
[41] S. A. Susin, H. K. Lorenzo, N. Zamzami et al., “Molecular characterization of mitochodrial apoptosis-inducing factor,” Nature, vol. 397, no. 6718, pp. 441–446, 1999.
[42] A. Gross, J. Jockel, M. C. Wei, and S. J. Korsmeyer, “Enforced dimerization of BAX results in its translocation, mitochon-drial dysfunction and apoptosis,” EMBO Journal, vol. 17, no. 14, pp. 3878–3885, 1998.
[43] D. M. Hockenbery, Z. N. Oltvai, X. M. Yin, C. L. Milliman, and S. J. Korsmeyer, “Bcl-2 functions in an antioxidant pathway to prevent apoptosis,” Cell, vol. 75, no. 2, pp. 241–251, 1993. [44] J. C. Reed, “Regulation of apoptosis by bcl-2 family proteins
and its role in cancer and chemoresistance,” Current Opinion in Oncology, vol. 7, no. 6, pp. 541–546, 1995.
[45] K. Revelos, C. Petraki, A. Gregorakis, A. Scorilas, P. Papanasta-siou, and M. Koutsilieris, “Immunohistochemical expression of Bcl2 is an independent predictor of time-to-biochemical failure in patients with clinically localized prostate cancer following radical prostatectomy,” Anticancer Research, vol. 25, no. 4, pp. 3123–3134, 2005.
[46] W. Ding, L. G. Hudson, and KE. J. Liu, “Inorganic arsenic compounds cause oxidative damage to DNA and protein by inducing ROS and RNS generation in human keratinocytes,” Molecular and Cellular Biochemistry, vol. 279, no. 1-2, pp. 105– 112, 2005.
[47] Q. He and X. Na, “The effects and mechanisms of a novel 2-aminosteroid on murine WEHI-3B leukemia cells in vitro and in vivo,” Leukemia Research, vol. 25, no. 6, pp. 455–461, 2001. [48] J. S. Yang, L. F. Kok, YU. H. Lin et al., “Diallyl disulfide inhibits WEHI-3 leukemia cells in vivo,” Anticancer Research, vol. 26, no. 1 A, pp. 219–225, 2006.
[49] FU. S. Yu, C. C. Wu, C. T. Chen et al., “Diallyl sulfide inhibits murine WEHI-3 leukemia cells in BALB/c mice in vitro and in vivo,” Human and Experimental Toxicology, vol. 28, no. 12, pp. 785–790, 2009.
[50] A. W. Burgess and D. Metcalf, “Characterization of a serum factor stimulating the differentiation of myelomonocytic leu-kemic cells,” International Journal of Cancer, vol. 26, no. 5, pp. 647–654, 1980.
[51] D. Metcalf, “Actions and interactions of G-CSF, LIF, and IL-6 on normal and leukemic murine cells,” Leukemia, vol. 3, no. 5, pp. 349–355, 1989.
[52] J. Li and A. C. Sartorelli, “Synergistic induction of the differentiation of WEHI-3B D+ myelomonocytic leukemia cells by retinoic acid and granulocyte colony-stimulating factor,” Leukemia Research, vol. 16, no. 6-7, pp. 571–576, 1992.