國立交通大學應用化學研究所
碩士論文
Institute of Applied Chemistry
National Chiao-Tung University
Master Thesis
交替式共聚高分子聚醯胺型聚氨酯內超分子
識別系統之研究
The Study on the Supramolecular Recognition within a
Poly(amide urethane) System
蔡佳佑
Jia-You Tsai
指導教授:張豐志 博士
Advisor: Feng-Chih Chang, Ph.D.
中華民國九十九年六月
交替式共聚高分子聚醯胺型聚氨酯內超分子識別
系統之研究
The Study on the Supramolecular Recognition within a
Poly(amide urethane) System
研 究 生:蔡佳佑 Student: Jia-You Tsai
指導教授:張豐志 Advisor: Feng-Chih Chang
國 立 交 通 大 學
應 用 化 學 系
碩 士 論 文
A Thesis
Submitted to Department of Applied Chemistry College of Science
National Chiao Tung University in Partial Fulfillment of the Requirements
for the Degree of Master
in
Applied Chemistry
June 2010
Hsinchu, Taiwan, Republic of China
Acknowledgement
在這即將要脫離學生身分,進入人生旅程另ㄧ階段的這一刻,許多的回憶不 斷地輪流湧上心頭...時光荏冉,兩年的時間猶如一場夢般,很快就過去了。 但是這夢境,卻是如此的真實又無法忘懷。從考上交大和剛來到新竹的抗拒,慢 慢到接受並習慣,真的是很意外,也讓我再次與當初從台大畢業一般,相當不捨。 然而,來到交大後奇蹟而幸運地進入了夢寐以求的實驗室,張豐志老師的 Polymer Research Center,也開啟了我這奇幻之旅最美的一頁... 感謝恩師張豐志老師及實驗室學長們在這兩年內的細心指導,並於研究過程 中給予我在實驗上的訓練、觀念的灌輸及參考文獻查閱方面的要點,使得此篇 論 文能夠順利完成。每當遇到事情時,你總是用鼓勵學生而不責備學生的方式,讓 我體會到您對學生的愛護與諄諄教誨。很謝謝您提供良好了如此良好的學習環境、 資源與空間,讓我可以充分的在研究上發揮,並且順利完成研究。每當我有問題 困惑時,學長們總是在適當的時機下指正,將我導入正確的研究方向,並且又能 適時地從旁提攜教誨,不斷給予正面鼓勵,給予獨立研究空間,而解決問題的方 式與危機處理,也同時讓我了解到積極思考的重要性,致使我心智及處事上更為 積極負責,在此致上最崇高的敬意。 感謝口試委員郭紹偉老師、王志逢老師、陳建光老師在口試時的建言與觀念 討論,使我更能了解實驗本身的意義及需要補強之處,讓我的論文能更佳完整豐 富。 感謝學長姐英傑、智嘉、倩婷、婉君、didi、你胖、阿發、宜弘、狗弟、小 豬、業昇、佳樺、Sandy、仁智,及同學阿罵、嘉蔚、唱邱、雅萍與學弟妹Alex、 建忠、修哥、可風、咪咪在研究期間內,對於學業實驗研究方面的切磋或日常生 活中問題分享與解惑,讓我用積極的態 度面對任何實驗或生活上的問題與挑戰, 使我這兩年的生活更加精采。特別要感謝漢清學長於我在NDL研究奈米壓印時的 指點,以及宜弘和你胖學長在相當多理論和實驗實作方面的指導和協助,特別是 在同幅中心打小角的那晚,讓我覺得永生難忘。在這一路上的歡笑、淚水,以及 很多知識及人生經驗都讓我倍感珍貴。希望大家珍惜這得來不易的緣份並保持連 絡,相信以後大家出社會,還是有機會遇到與再合作的。 還有在新竹課後唯一的娛樂就是音樂,感謝MB樂團的每一份子,團長變態、 吉他手小何、大P、bass手與安、KB手阿珮、阿ㄆ以及鼓手賴彥,因為有了你們, 讓我在開心時有人分享,實驗不順難過及悲傷時再度有了動力。大家一定會在人 生的路上再度重相逢的! 最後要感謝我親愛的家人爸爸、媽媽各方面的支持,大學同學思齊和昭博一 路上的互相鼓勵,還有求學路上親朋好友們的栽培愛護,讓我無後顧之憂地在研 究上努力;特別謝謝女友小葉的支持、體諒與包容,在我這兩年求學路程中給予 勉勵與關懷,使我順利完成學業,邁向人生嶄新的階段。 佳佑 2010 年 6 月Outline of Contents
Page Acknowledgments Outline of Contents І List of Tables Ⅴ List of Schemes ⅥList of Figures VIII
Abstract (in English) XIV
Abstract (in Chinese) XVI
Chpater1 Introduction 1
1.1 An Overview on Polyurethane 1
1-1.1 The Development and Utilities of Polyurethane 1
1-1.2 Chemistry of Polyurethanes 5
1-1.3 The Structure and Properties of Polyurethanes 8
1-1.4 The Microstructure and Morphology of Polyurethanes 11
1-2 The Crystalline Behavior in Polymers (Thermal Properties
of PU)
15
1-3 Hydrogen Bonding within Polymers 18
2-1 Introduction to Natural Nucleobases and its Applications in
Supramolecular
24
2-2 Versatile Hydrogen-Bonding Motifs Through Nucleobase-
pairing
27
2-2.1 Nature and Stability of Hydrogen Bonds 27
2-2.2 Hydrogen Bonding of Nucleobase-pairing Allows
for Versatility
30
2-3 An overview on Supramolecules 36
2-3.1 Basic Principle of Supramolecules 36
2-3.2 Basic Principle of Recognition in Supramolecules 37
2-3.3 An overview of Supramolecular Materials 40
2-3.4 Supramolecular Chemistry 42
2-3.5 Supramolecular Polymerization 44
2-4 Nucleobase Hydrogen Bonding as Applications in Polymer
Material Systems
46
2-4.1 Nucleobases in Polymeric and Material Systems 46
2-4.2 Chain-End Nucleobase Modified Monomers and
Polymers
48
2-4.4 Supramolecular Block copolymers 58
2-5 Motivation 63
Chapter3 Experimental Section 65
3-1 Materials 65
3-1.1Material Sources 65 3-1.2 Purification of Solvents 67
3-2 Synthesis of Diamidepyridine Diacid (Compound 1) 68
3-3 Syntheses of Poly(amide urethane), (PAU) 70
3-4 Synthesis of 1-Hexadecyluracil (U-16) 73
3-5 Preparation of Complexes 74
3-6 Characterizations 75
3-6.1 Thermogravimetric Analysis (TGA) 75
3-6.2 Differential Scanning Calorimetry (DSC) 75
3-6.3 Infrared Spectroscopy (FTIR) 76
3-6.4 Gel Permeation Chromatography (GPC) 76
3-6.5 NMR Spectroscopy 76
3-6.6 Transmission Electron Microscopy (TEM) 77
3-6.7 Atomic force microscopy (AFM) 78
3-6.9 Wide-Angle X-ray Scattering (WAXS) 79
Chapter 4 Results and Discussion 80
4-1The complementary interaction within the PAU/U16 blends 82
4-2 Thermal Analyses 90
4-3 Small angle X-ray scattering and Wide angle X-ray
scattering
96
4-4 TEM Observation of the Morphology 102
4-5 Atomic Force Microscopy Analysis 106
Chapter 5 Conclusion 110
Chapter 6 References 112
List of Tables
Tab le 1-1. Development of the significant research on polyurethane in history
2
Tab le 2-1. The length of hydrogen bonds 28
Tab le 2-2. Some selected non-covalent interaction energies useful in supramolecular chemistry
44
Tab le 3-1. The elementary analysis data of Compounds 1 68
Tab le 3-2. The GPC data of PAU 71
Tab le 3-3. Intrinsic viscosity of PAU dissolved in DMSO (0.1 g/10 ml) obtained from Ostwald viscometer at 25 oC.
List of Schemes
Scheme 1-1. Mechanism of Urethane Formation 5
Scheme 1-2. Basic reactions of isocyanate with different reactants 6
Scheme 1-3. Basic reaction scheme for urethane formation. 8 Scheme 1-4. Schematic of the PU chains studied in this work. Soft and
hard segments have different lengths and length distributions. The hard segment consist of 4,4'-diphenylmethane diisocyanate (MDI) and the chain extender 1,4-butanediol (BD) whereas the soft segment consist of original diol segments.
10
Scheme 1-5. The hard, soft segments and urethane groups in the backbone of PU (R2 may be used as chain extender)
10
Scheme 1-6. Physical crosslink among hard segment and soft segment in PU
11
Scheme 1-7. Schematic model for the morphological changes that occur during DSC scans of polyurethane elastomers: (a) below the microphase mixing transition temperature; (b) between the microphase mixing temperature and the melting temperature; and (c) above the melting temperature. The microcrystalline hard-segment domains are indicated
17
Scheme 1-8. An example of intermolecular hydrogen bonding in a self-assembled dimer complex reported by Meijer and coworkers
20
Scheme 1-9. Intramolecular hydrogen bonding in acetylacetone helps stabilize the enol tautomer
Scheme 1-10 Carboxylic acids often form dimers in vapor phase 20 Scheme 1-11 Examples of hydrogen bond donating (donors) and hydrogen
bond accepting groups (acceptors)
21
Scheme 1-12 Hydrogen bonding between guanine and cytosine, one of two types of base pairs in DNA.
21
Scheme 2-1. Stepwise Construction of π-Stacked Dimers from Monomers for Homostack of 1k
26
Scheme 2-2. Synthesis and structures of the homoditopic (A-A, B-B) nucleobase terminated bis(phenylethynyl)-benzene monomers. (BP1aBP and BP1bBP). BP=nucleobase derivative.
52
Scheme 2-3. Synthesis of the supramolecular telechelic macromonomers
CPbz43CPbz, GCbz43GCbz, AAn43AAn, T43T, BIP43BIP and
H43H.
53
Scheme 3-1. Synthesis and chemical structure of Compound 1. 68
Scheme 3-2. Syntheses and chemical structures of PAU. 70
Scheme 3-3. Syntheses and chemical structures of U16. 73
Scheme 4-1. Schematic representation of structures of PAU and U16 with the complex process.
81
Scheme 4-2 Some different modes of hydrogen-bonding motifs within PAU/U16 complexes.
85
Scheme 4-3. Graphical representations of (a) the physical cross-linked structure formed.from PAU/U16 complexes in the bulk state and (b) the transition of the lamellar structures of various PAU/U16 complexes in bulk state.
List of Figures
Figure 1-1. Hydrogen bonding within hard segments of PU 11
Figure 1-2. Hard domains (HD) and soft domains (SD) of TPUs with (a) a low hardsegment content where HDs are isolated; and (b) a high hard segment content where HDs are interconnected.
13
Figure 1-3. The microstructure within PU and hydrogen bonding among hard segments
22
Figure 1-4. The hydrogen bonding within diamidepyridine 22
Figure 1-5. Schematic of the hydrogen-bonding in MDI-butanediol hard segment ,as proposed by Bonart et al. The staggering of the chains leads to planes in this projection at 30° to the perpendicular as indicated
23
Figure 2-1. Some of the non-covalent interactions which are present in the nucleobase guanine and the nucleotide deoxyguanosine monophosphate.
25
Figure 2-2. Hydrogen bonding in DNA base pairs 29
Figure 2-3. Selected multiple hydrogen bonded complexes with high associationconstants
29
Figure 2-4 The canonical Watson-Crick hydrogen bonding motifs. 33
Figure 2-5 Some common non Watson–Crick (non-traditional) base-pairing modes
34
Figure 2-6 Some Examples of base-triplets as a combination of different forms of base-pair binding modes.
35
G-quartet, and (c) G-quadruplex formed by stacking of G-quartets around a column of cations.
Figure 2-8 The route from "Molecular Chemistry" to "Supramolecular Chemistry."
36
Figure 2-9 Supramolecules form through multiple hydrogen bonds. 38
Figure 2-10 large ring system form via π- π interaction and crosslink between rings.
39
Figure 2-11 Cartoon representations of polymer architectures made using covalent and non-covalent links between building blocks.
42
Figure 2-12 (a) Supramolecular polymeric material based on a low molecular weight compound equipped with two ureido-pyrimidinone (UPy) units. (b) Schematic picture of the underlying polymeric network. (c) Schematic picture of the self-complementary UPy dimer.
42
Figure 2-13 Modes of dimerization of the UPy functional group via quadruple hydrogen bonding: keto tautomer (a) and enol tautomer (b).
47
Figure 2-14 Schematic representation of two different types of supramolecular polymers which are formed by the association of monomers with complementary end groups. (a) Self assembly of a heteroditopic monomer to yield a (AB)n
supramolecular polymer and (b) self assembly of two complementary homoditopic monomers to yield a (AA-BB)n
supramolecular polymer. The dynamic nature of these polymers allows them to break and recombine in response to changes in
the environment.
Figure 2-15 Self-assembly of monomers with different but complementary endgroups resulting in an (AB)n copolymer.
51
Figure 2-16 (a) Nucleobase terminated bolaamphiphiles and (b) a possible hydrogen bond scheme for the heteroassembly formed from an equimolar mixture of 1 and 2.
51
Figure 2-17 Schematic model of the AAn43AAnassembly as it transitions at 90 ºC from a linear system to a gel-like material. The schematic shows the segregation of the nucleobase hard segments [blue disks] connected by chains of poly(tetrahydrofuran)s [green lines].
54
Figure 2-18 A cartoon depicting multi-step self-assembly via metalcoordination based cross-linking and polymer functionalization via hydrogen-bonding.
57
Figure 2-19 (a) Schematic illustration of noncovalent polymer cross-linking of copolymer 2a using a complementary bifunctional cross-linker 2b. (b) Schematic representation of the diacyldiaminopyridine – and thymine-based random copolymers, 2a and 2c, as assembled in three dimensional vesicles.
58
Figure 2-20 Block copolymers constitute an important class of soft material capable of self-assembling into nanoscale microdomains with well-defined geometries. The cylindrical domains thus formed have been found to pack almost exclusively in hexagonal lattice.
Figure 2-21 Schematic illustration of the possible arrangements of comb blocks in the comb-coil copolymers: (a) monolayer arrangement in linear architecture; (b) double-layer arrangement in linear architecture; (c) monolayer arrangement in heteroarm star architecture; (d) doublelayer arrangement in heteroarm star architecture.
61
Figure 2-22 Schematics and morphologies of the interactions in PS-block-P4VP(PDP)1.0
62
Figure 2-23 One of the potential scenarios to construct hierarchically self-assembled polymeric structures. Construction units of different sizes allow a natural selection of different self-assembled length scales
62
Figure 2-24 Sketch of the formation of a stoichiometric polyelectrolyte/surfactant complex.
64
Figure 2-25 Schematic representation of the structure of the ionic complex. 64
Figure 3-1. The (a) 1H NMR and (b) 13C NMR spectra of Compound 1. 69
Figure 3-2. The 1H NMR spectra of PAU. 71
Figure 3-3. The Fourier Transform Infrared spectra of poly (amide urethane).
72
Figure 3-4. The 1H NMR spectra of U16 73
Figure 4-1. Variable temperature FT-IR spectra recorded in the 1550-1800 cm-1 region (C=O stretching region) of Poly (amide urethane) [PAU].
86
Figure 4-2. FTIR spectra of PAU/U16 blends incorporating various amount of U16 in the presence of bulk state and recorded at room
temperature in the 1600-1800 cm-1 region.
Figure 4-3. Variable temperature FT-IR spectra recorded in the 3000-3500 cm-1 region of Poly (amide urethane) [PAU].
88
Figure 4-4. FTIR spectra of PAU/U16 blends incorporating various contents in the presence of bulk state and recorded at room temperature in the 3140-3400 cm-1 region.
89
Figure 4-5. Thermal Gravitivity Analysis (TGA) curves of PUA/U16 blends of different compositions.
94
Figure 4-6. DSC thermograms of (a) PUA/U16 blends of different compositions. (b) PAU ranges from 40°C to 150°C shows the second Tg.
95
Figure 4-7. DSC curves indicate the change in melting points (Tm) of
PUA/U16 blends in the presence of various amounts of U16.
96
Figure 4-8. The SAXS patterns indicate that both PAU and other PAU/U16 blends possess lamella structures.
99
Figure 4-9. Representative synchrotron small angle X-ray scattering (SAXS) data as a function of the scattering vector (q) for a series of poly(amide urethane)/ 1-Hexadecyluracil (PAU/U16) blends having different PAU to U16 weight ratios.
100
Figure 4-10. Representative wide angle X-ray diffraction (WAXD) patterns for PAU/U16 complexes with different weight ratios. The magnitude of the scattering vactor is given by q=(4π/λ) sinθ, where 2θ is the scattering angle and λ=1.54Å. Arrows are a guide for eyes.
101
of the pure PAU stained with RuO4. The dark region (matrix)
corresponds to the phase of hard segments—PAU; the white region corresponds to the excluded PEG soft segment. The scale bar represents 100 nm.
Figure 4-12. Transmission electron micrograph of the cryo-microtomed film of the PAU/U16 (10/1) blend stained with RuO4. The dark
region (matrix) corresponds to the phase of hard segments— PAU; the white region corresponds to the excluded PEG soft segment. The scale bar represents 100 nm.
106
Figure 4-13. AFM 2D height and phase images of different ratio of blends: (a)(b) PUA1 and (c)(d) 60:1 (e)(f) 10:1 with scale of 2× 2 μm2.
109
Figure 4-14. AFM 3D image was used in the investigation of the morphology of different ratio of blends (a) PUA1 and (b) 60:1 (c) 10:1 with scale of 2× 2 μm2.
The Study on the Supramolecular Recognition
within a Poly(amide urethane) System
Student: Jia-You Tsai Advisor: Dr. Feng-Chih Chang
Institute of Applied Chemistry
National Chiao Tung University
30050 Hsinchu, Taiwan
ABSTRACT
Self-assembled polymers such as block copolymers are usually formed through covalent linkages in conventional polymer chemistry, including bonds connecting monomer units and attaching functional groups to the polymer backbone. Recently, novel structural organizations of self-assembled polymers formed through highly directional and sufficiently strong non-covalent host-guest pairs have attracted great attention. These new polymers utilize non-covalent multiple-hydrogen-bonding interactions similar to those found in bio-molecules such as protein, DNA, and RNA to direct and modulate their 3-D topology. In addition, the moderately strong and highly directional multiple-hydrogen-bonding interactions within these new generation polymers also result in unique physical properties, such as high specificity, controlled affinity, and reversibility. In previous studies, the study on the complementary nature and its effect on material properties can be broadly classified into side chain and chain ends types. Until now, studying and controlling the microstructures within supramolecular polymers with different hydrogen-bonding
motifs still remain as a challenging task. In this study, the amphiphilic alkylated nucleobase, hexadecyluracil (U16) was incorporated into poly(amide urethane) which was synthesized ourselves with self-complementary group and several hydrogen bonding motifs for the study on the heterodimer recognition behavior within the alternative polymer. Biocomplementary PAU/U16 supramolecular complexes formed in dilute DMF through molecular recognition, that is, the hydrogen bonding between the diaminopyridine (DAP) groups of the PAU and the Uracil (U) of U16. Moreover, FTIR, DSC, WAXD, SAXS, and TEM analysis provided furthur detail into the nature of self-assembly of these systems. The effect of the heterodimer recognition on the microphase separation was investigated, revealing that the heterodimer recognition led the poly(amide urethane) to possessing the “plug and play” behavior even the heterodimer recognition coincided with several other hydrogen bonding motifs. The period and morphlogies of PAU/U16 complexes can be rationally tuned by the amount of U16. Upon the adding of U16, the lamellar structure within long-range lamellar changes from bilayer to bilayer with interclated and further to monolayer.
交替式共聚高分子聚醯胺型聚氨酯內超分子
識別系統之研究
學生: 蔡佳佑 指導教授:張豐志 教授
國立交通大學應用化學研究所 碩士班
摘 要
傳統高分子化學中,自組裝型高分子(self-assembled polymers)如嵌段式共聚 高分子常經由共價鍵方式將單體-單體間及單體與高分子主鏈上之對應官能基間 相互以連結。近年來,藉由具高度方向性及主客配對之非共價鍵方式形成自組裝 型高分子之新穎結構已逐漸獲得重視。此種作用力常見於生物體內許多蛋白質、 DNA、RNA及3D立體架構,因此近年來向自然界取材之仿生材料發展相當蓬勃。 此類新型高分子中適當強度及具方向性之多點式氫鍵作用力導致了許多特殊的 物理性質。近期對於互補性單元導入高分子材料中的研究,可簡略分類為側鏈修 飾及鏈尾修飾等兩種。如果我們可以控制多重氫鍵的鍵結能力,就可以間接改變 超分子的型態。然而,藉由不同之多點式氫鍵作用力調控超分子中之微結構及研 究直到現在仍相當具有挑戰性。於此篇研究中,我們將雙重極性(amphiphilic)之 長碳鏈核鹼基,導入具有自身互補性基團及多種氫鍵作用力之聚醯胺型聚氨酯高 分子中,用以研究在交替式高分子中異雙聚體(heterodimer)分子辯識的行為以及 在導入之後微相分離的變化。研究結果顯示,即使在系統中仍有其它種類氫鍵的 存在,異雙聚體的分子辯識使得聚醯胺型聚氨酯高分子呈現「熱插式」(plug and play)的行為。藉由U16混掺比例的不同,可使得整個高分子系統的型態有所改變 並得以調控。隨著U16加入的量增加,鏈與鏈間之次級層狀結構由原本雙層之結 構變成深入彼此之相互交錯狀態,並進而變成單層結構。1
Chapter1
Introduction
1-1 An Overview on Polyurehtane1-1.1 The Development and Utilities of Polyurethane
Polyurethanes(PUR), also known as Segmented polyurethanes(SPU or PUs),
are widely used in thermal plastic elastomers, owing to their excellent mechanical and
chemical characteristics. Polyurethane(PU) was earliest found by Bayer and
Farbenindustric. Segmented polyurethanes (PUs), which comprise hard and soft
segments, are a major consumer plastic material with an annual production capacity
worldwide of nearly 12 million tons (2007). Segmented polyurethane elastomers are linear block copolymers of -(HS)n- type (H, hard segment; S, soft segment), or so
called alternative copolymer, whose versatile physical properties are generally attributed to their microphase-separation structure. Soft segment means the region where polyol located and mainly consisted of polyester or polyether with 500~4000 molecular weight; hard segment indicates regimes contain no polyol but NCO end group and chain extenders,hard segments are also with 500~4000 molecular weight.
Opposite to other consumer plastics they are no polymerization but condensation
polymers produced in a stepwise polyaddition process without the elimination of side
products. Polyurethanes are generally produced from one or more polyhydroxyl
compounds and one or more polyisocyanates. Due to the numerous compounds on
both sides of the reaction the type, properties, and fields of application of the resulting
polyurethanes are without any limit. Otherwise, the raw materials have been produced
in several steps from crude oil (or, more recently, also renewable resources), thus,
they constitute high commercial value.
2
Time (Period) Research Topic References
1849 Isocyanate reaction with an alcohol Wurtz, A. Ann 71, 326, 1849
1937 I. G. Farbenindustrie applies for first
polyurethane patent German Patent 728, 981
1938 Firest U.S. payent award to Rinke, et al. U.S. Patent 2, 511, 544
1942
DuPont receives patents for reaction of polyisocyanates with glycols, diamine, and polyesters
1942 Introduction of Igamid U, and Ugamid UL in Germany
1943 Vulkollan polyester-based elastomers introduced in Europe
1945 Allies recognize German industry and create Farbenfabriken
1954 Patent on Lycra spandex elastomeric
fiber awarded U.S. Patent 2, 692, 893
1954 Bayer and Monsanto Co. form Mobay Chemical Co.
1955 Patent on Estane thermoplastic elastomers
awarded U.S. Patent 2, 871, 218
1955 Union Carbide developes first one-shot foam
1956 Teracol 30-PTMEG introduced Bulletin HR-11 DuPont 1959 First use of chlorofluorocarbons as
blowing agents
1971 Patent on medical-grade
polyurethane-silicone elastomer U.S. Patent 3, 562, 352 1993 Patent on aliphatic biostable polyurethane
elastomer U.S. Patent 5, 254, 662
Tab le 1-1. Development of the significant research on polyurethane in history.[4]
Segmented polyurethanes are generally composed of polyether or polyester soft segments and urethane based hard segments along the polymer backbone giving rise to a microphase-separated morphology caused by the usual poor compatibility
3
between both segments. The unique properties of these polymers are directly related to their two phasemicrostructure, in which the hard domains act as a reinforcing filler and as a thermally reversible cross-link.[1] Such polyurethanes have been widely used industrially due to their versatile properties[2] but, due to their low degradability in nature, many research efforts have been put on biodegradable polyurethanes.[3]
Environmental concerns are also related to the use of organic solvents during the processing and even during the use of polymers in applications such as coatings, adhesives, fibers among others. Polyurethanes derived from water dispersions can overcome this type of problem by replacing more toxic solvents with water. Waterborne poly(urethane-urea)s, WPUUs, are multiblock copolymers consisting of alternating the soft and hard segments. WPUUs not only conform to environmental emission legislation, but also reduce the consumptions of both cost and energy.[4] Therefore, they are widely used as the elastomer, coating, adhesive, and nanocomposite.[5–8]
In the last years, as interest in polymers for biomedical devices is increasing, biocompatible and biodegradable polymer precursors are used to synthesise segmented thermoplastic polyurethane elastomers (STPU) which can be visualized as possible candidates for many applications as vascular prostheses, soft tissue adhesives, pericardial patches, and in tissue engineering cancellous bone substitutes.[9,10]
Shape memory polyurethanes (SMPU) with phaseseparated morphology are composed of soft and hard segments, in which the former mostly comprises the reversible phase to fix the temporary shape, while the latter forms the fixed phase to memorize the permanent shape. In the thermal sensitive shape memory function, most of the temporary deformation can be fixed well after cooling to a low temperature (Tl)
under a certain temperature Ts (melting point (Tms) or glass transition temperature
4
recovered from the temporary shape with the stimulus of heating to a high temperature (Th) above Ts. In heating scan, such a sharp recovery usually happens in
the vicinity of Ts. This unique feature made this type of material arouse lot of research interest from both academia and industry in recent decades. In the linear segmented SMPU system, the strong intermolecular force (physical cross-linking) among hard segments results from the hydrogen bonding and the high polarity due to the presence of urethane and urea units. The shape memory effect (SME) of SMPU therefore is significantly influenced by the hard-segment content and the moiety of its molecular structure. In addition, it is reported that the soft-segment content, its molecular weight, conformation, and morphological structure play significant roles on SME. Therefore, these parameters are paramount in controlling the shape memory properties in general.
5
1-1.2 Chemistry of Polyurethanes
Isocyanates can undergo a broad spectrum of chemical reactions and are of great importance for contemporary polymer chemistry. Isocyanates can, for instance, be polymerized along the nitrogen-carbon double bond producing substituted polyamides. By far the most important reaction, however, is the reaction between an isocyanate and an alcohol forming an urethane bond. Also the reaction with an amine forming an urea bond is widely used. The basic reaction of polyurethanes is the addition of an H-active compound to an isocyanate compound. The reaction is belong to nucleophilic addition and the polyol is attached to the carbon atom on the isocyanate(see eq. 1-1). The more detail mechanism(Scheme 1-1) discovered by H. Herlinger is a six centered ring mechanism in which a proton is shifted from the H-active compound to the nitrogen atom of the isocyanate[11]:
(1-1)
Scheme 1-1. Mechanism of Urethane Formation
There are also different reactions between H-active compound and isocyanates (RNCO) and listed as below:
6
Scheme 1-2. Basic reactions of isocyanate with different reactants
At the initial stage of reaction or under the condition of excess isocyanates, urethane, urea, and amide may lead to side reactions such as route e and f in Scheme
1-2. These side reactions give rise to branching and crosslinking in polymers. In
general, the relative reaction rate of isocyanate and H-active compound is listed as follow:
aliphatic
NH2>aromatic NH2>primary OH>H2O>secondary OH>tertiary OH >phenolic OH>R-COOH>RNHCONHR>RNHCOR>RNHCOOR.
Polyurethanes belong to the class of polycondensation polymers. There are two common ways to synthesize polyurethane: the first one is let bischloroformates react
7
with diamine; the other one and also popular method is by the reaction between diisocyanate and dihydroxy(diol) groups.[12]
(I) The source of bischloroformates is by the reaction between phosgene and diol or bisphenol(1-1), and then react with diamine or bisphenol as the following: (a) bischloroformates react with diamine to form polyurethanes(1-2):
(1-1)
(1-2)
(b) or sometimes by the reaction of biscarbanyl chlorides and bisphenol A:
(1-3)
(II) In general, the synthesis of polyurethane is achieve by the polymerization
diiocyanate, diol and chain extender. There are also two types of reactions to complete the goal: "One-shot process" and "Prepolymer process." The former one means let all the reactants complete the reaction in only one process; and the latter is let excess diisocyanate react with diol and forms prepolymer with NCO end group first, then add
8
chain extender to increase the molecular weight of (pre)polymers. The alternative copolymers which contain soft and hard segments is elastic, so these elastomers also called TPU(thermal elastomer of polyurethane).
Scheme 1-3. Basic reaction scheme for urethane formation. [12] 1-1.3 The Structure and Properties of Polyurethanes
Segmented polyurethane elastomers are alternative copolymer or so-called linear block copolymers of -(HS)n- type (H, hard segment; S, soft segment) with Hard
and Soft segments on the backbone(Scheme 1-4).[13] The ‘‘soft segment’’ typically is a polar polyol region, such as polyether or polyester, with 500~4000 molecular weight and a glass transition temperature below ambient, which imparts elastomeric character to the polymer. The ‘‘hard segment’’ indicates regimes contains the highly polar urethane and urea linkages, typically with 500~4000 molecular weight and a glass transition above room temperature. Hard segment may also include and chain extenders(Scheme 1-5)[14] and NCO end groups. The soft segments behaves like rubbers at room temperature, and hard segments usually is semi-crystalline or glass state. Because of the hydrogen bonding, coulombic force and segmental polarity difference, these two segment types tend to phase separate in the bulk. The hard domains act as physical crosslinkers and reinforcing fillers. The variable physical properties of PUs depend strongly on the degree of phase separation and the cohesion of the hard domains.
9
intramolecular and intermolecular hydrogen bonding of hard segments(Scheme 1-6). The content of such kind of physical crosslink structure plays an important role of several properties of polyurethane. For example, the modulus of polyurethane material mainly depends on the ratio of hard segment content to soft segment content as a result of the amount of physical crosslink. The more hard segment content, the more physical crosslinking inside the PU, thus the modulus of rigidity is larger. On the contrary, the more soft segment content, the modulus of PU material is smaller.[15-17] The structure of the physical crosslink within PAU applied in this paper was shown in Figure1-1.
These kinds of physical crosslink structure mentioned above are composed of varies types of intramolecular and intermolecular hydrogen bonds and exist in two kinds of forms: (1)intermolecular urethane-urethane group hydrogen bonding (2) urethane-polyol hydrogen bonding forms owing to the dispersion of hard segments in soft segments.
In general, urethane-urethane hydrogen bond is stronger in the whole PU structure because the polarity of urethane is higher than polyol segments and thus make the urethane group in hard segments attract each other. Specific interaction called inter-urethane hydrogen bonding also forms as the driving force of phase separation(segregation) due to the aggregation of hard segments led by hydrogen bonding. Therefore, the polarity of structure composition and the strength of hydrogen bonding within the PU have a big influence on the microphase separation which can be detected and known by X-ray diffraction(WAXS and SAXS).
The degree of microphase separation within PU varies due to the unevenly distributed(unhomogeneous) hard segments and soft segments then results in multiple transition temperatures. In general, there are three glass transition temperatures found in DSC or DMA scanning[17]:
10
(1) The first phase transition point is below 0℃ and mainly comes from the glass transition temperature of soft segment.
(2) Another phase transition temperature found generally in the range of 30℃~90℃ is higher than room temperature and mainly comes from the glass transition temperature of hard segments
(3) The rest endothermic peak locates between two glass transition temperature mentioned above and mainly bring about from the heat destruction of hydrogen bonding within hard and soft segments.
Scheme 1-4. Schematic of the PU chains studied in this work. Soft and hard segments
have different lengths and length distributions. The hard segment consist of 4,4'-diphenylmethane diisocyanate (MDI) and the chain extender 1,4-butanediol (BD) whereas the soft segment consist of original diol segments.
Scheme 1-5. The hard, soft segments and urethane groups in the backbone of PU
11
Scheme 1-6. Physical crosslink among hard segment and soft segment in PU
R N H C O O R O C O N H R N H C O O R N H C O O R O C O N H R N H C O O
: soft segment :hard segment
Figure 1-1. Hydrogen bonding within hard segments of PU 1-1.4 The Microstructure and Morphology of Polyurethanes
Segmented polyurethanes (PUs) have a great application potential due to their wide range of mechanical properties. Depending on the specific end use, PUs can be designed to possess either elastic or rigid mechanical characteristics, depending on the macromolecular structure.[18–20] Thus, considerable effort has been made to
12
understand structure–property relationships of segmented PUs. In order to tailor mechanical properties, the macromolecules of segmented PUs are built up of two types of blocks.[21–24] One block type, the so-called hard segment, is glassy (or crystalline) as it is below its Tg (or Tm) at the use temperature. The other soft block is
flexible and rubbery, as it is above its corresponding Tg at the use temperature. The
hard segments due to their thermodynamic incompatibility with the soft segments usually associate to form small domains that serve as physical cross-links and reinforcement sites; hence they limit flow of the other phase, which is made up by the soft segments.
The domain size of segmented PU is so small at the scale of 10nm and hardly observed by TEM. Thus, the literature on the TEM morphology of PU is seldom rarely published. Meier calculated the morphology of block copolymer of PU varies with the increasing of volume fraction by thermodynamics. Cooper et. al[41] put much effort on PB based PU and both SAXS and HVEM was utilized on the research. Hard segment distributed in soft segments with a dislike cylinder shape when PB based PUs with 31% hard-segment content (HSC)(Volume fraction: 27%) within is discovered. Also, for a 42~67% HSC PU(volume fraction: 35~61%), hard segments displays a lamella like structure. When HSC is higher than 75% (volume fraction: 68%), hard-segment domain was continuous phase and soft-segment domain shows dislike shapes. Therefore, For thermoplastic and solvent-polymerized PUs with 50% hard-segment content, a lamellar domain structure[24–26] or bicontinuous phase separation[27] is usually assumed. For PU samples with 30% HSC, a dispersed hard-segment domain structure is expected to be having spherical, disclike,[28–30] or cylindrical[31] domain shapes.
The degree of phase separation in PUs depends on the types of diisocyanate and polyol employed to produce the PU, CE(chain extender) nature, type of
13
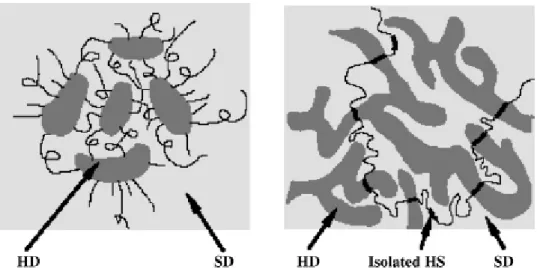
polyfunctional compound used in the crosslinking process, NCO:OH ratio, sizes of HSs and SSs, method of synthesis, etc. Figure 1-2. shows the HD (hard domain) and SD(soft domain) structures of TPUs at low and high hard segment content. The domain size may range from 3 to 20 nm and depends on the reactants used as well as the polymerization conditions.
Figure 1-2. Hard domains (HD) and soft domains (SD) of TPUs with (a) a low hard
segment content where HDs are isolated; and (b) a high hard segment content where HDs are interconnected.[32,33,34]
In real case, thermoset PUs are formed by using either one or a combination of the following:
• using polyols with functionalities greater than 2,
• substitution of a trifunctional hydroxyl compound in place of the normal glycol CE, • using isocyanates with functionalities greater than 2,
• using NCO:OH ratios greater than 1, and
• introduction of a crosslinker into the HS, SS or CE.
On the other hand, the mechanical stability of PUs are low at high temperatures,[35,36] and their initial temperature of degradation is typically around 200°C, which is near their melting temperature.[36–39] Therefore, the mechanical characteristics and thermal stability are improved by altering the structure of the
14
segments, such as by incorporating into the hard or the soft segment an aromatic amide,[39] an aromatic imide,[37] or a silicone group.[38,39] Although the hard and soft segments of PUs influence the stability of thermal degradation, the morphology of the domains that consist of hard segments also, importantly, affects the mechanical stability in practical application.
Polyurethanes can be obtained in both amorphous and semicrystalline forms.[40–43] It was found that the morphology of semicrystalline PUs is much more complicated for polymers obtained by the so-called cast reactive method (CRM) as compared with the thermoplastic procedure.[44–48] In semicrystalline PUs obtained by CRM, multi-phase separation may occur on two levels of structural organization: of the order of tens of micrometers (micro level), corresponding to micro-phase separation (hard segment-rich spherulites, hard segment-rich globules); and of the order of tens of nanometers (nano level), associatedwith nano-phase separation (hard segment-rich fibrils and lamellae, and domain structure).[43,49–51]
There is a discussion in the literature regarding the nature of the morphological features observed by transmission electron microscopy (TEM) that exist outside hard-segment rich spherulites and globules.[51,52] The observations are not complete due to limitations of TEM and additional investigations are necessary to solve this issue. The atomic force microscopy (AFM)[53–55] has been used to study polymer morphology with great success (see, e.g., melting and crystallization of poly(ethylene oxide) in real-time,[56] the spherulitic morphology of isotactic polypropylene,[57] the lamellar organization of melt-crystallized bisotactic polypropylene,[58] the lamellar morphology of polyethylene and polyoxymethylene (POM), microfibrils of oriented POM,[59] etc.). In the work nowadays, the morphology of various semicrystalline polyetherurethanes is discussed, based on new, complementary AFM and TEM studies.
15
1-2 The Crystalline Behavior in Polymers (Thermal Properties of PU)
If crystalline polymers contact with specific solid when they heat to melt then
cool down and crystallized, it forms a large amount of nuclei on the interface, and therefore make crystals form latter only grow toward the direction perpendicular to the interface and finally at the interior of polymers because of overcrowded in the space. This kind of columnar crystal is named as transcrystal, which is a result of heterogeneous nucleation. The grow behavior of transcrystal in polymers was earliest found in the interface between templates of hot-embossing manufacturing. After a series of research, it was found out the density and surface energy are higher in the transcrystal region than that in bulk polymers and thus enhance the mechanical properties and bonding strength in polymers.[60,61] This phenomenon has readily been utilized as an application in the reinforcement of the adhesion on polymer surfaces[62]. In recent years, the development of thermoplastic nanocomposite is prosperous, and the discussion over crystalline behavior on the surface between matrix and reinforcement material is popular. XRD analysis technique and the variation of DSC curve are both common methods to realize the detail structure and crystalline behavior in nanocompsite materials.
Seymour and Cooper[63] discovered there are three melting endothermic peaks(T1、T2、T3) in the higher temperature region of DSC diagram and respectively
responsible for different order of arrangements within hard segments (Scheme 1-7). The existence of multiple endotherms has been documented in several studies of the thermal behavior of segmented polyurethane block copolymer.[63-65] Multiple endotherms are as a result of partially amorphous and partially crystalline, or so-called semi-crytalline behavior inside the polymer microstructure because no existence of totally crystalline polymer in reality. Actually, the morphology in crystalline polymers contain amorphous, short-range order, long range order and
16
crystalline regions. In general, three distinct endotherms are observed in differential scanning calorimeter (DSC) experiments.[63-66] The lowest temperature endotherm (T1)
is observed at temperatures ca. 20-30℃ higher than the annealing temperature and has been attributed to a local restructuring of hard-segment units(short-range order of unknown nature) within the hard microdomains. An intermediate temperature endotherm (T2), found generally in the range 120-200 ℃, has been associated with the
destruction of long-range order of an unspecified nature or the onset of microphase mixing of the hard and soft microphases. A higher temperature endotherm (T3)
observed above 200 °C is generally ascribed to the melting of microcrystalline regions within hard microdomains (hard segments).
As for soft segments in polyurethanes, the constrain of hard segments on both side ends of soft segments has a tremendous influence on the crystalline behavior of soft segments due to the stereo structure and continuous circumstance of hard segments. In general, the ability of crystallization of soft segments decrease as the increasing of specific volume(surface area/volume, S/V). Although the specific volume(S/V) of block copolymer varies with morphology, the value of S/V decreases with the increasing of molecular chain length for all kinds of morphologies. Therefore, it is concluded that the chain length of soft segments in both styrenic block copolymers or linear copolymers should be long enough to induce crystalline behavior.
17
Scheme 1-7. Schematic model for the morphological changes that occur during DSC
scans of polyurethane elastomers: (a) below the microphase mixing transition temperature; (b) between the microphase mixing temperature and the melting temperature; and (c) above the melting temperature. The microcrystalline hard-segment domains are indicated
18
1-3 Hydrogen Bonding within Polymers
A hydrogen bond is the attractive interaction of a hydrogen atom with an electronegative atom, like nitrogen, oxygen or fluorine (thus the name "hydrogen bond", which must not be confused with a covalent bond to hydrogen). The hydrogen must be covalently bonded to another electronegative atom to create the bond. A hydrogen atom attached to a relatively electronegative atom is a hydrogen bond donor.[67] This electronegative atom is usually fluorine, oxygen, or nitrogen. An electronegative atom such as fluorine, oxygen, or nitrogen is a hydrogen bond acceptor, regardless of whether it is bonded to a hydrogen atom or not (Scheme 1-11). An example of a hydrogen bond donor is ethanol, which has a hydrogen bonded to oxygen; an example of a hydrogen bond acceptor which does not have a hydrogen atom bonded to it is the oxygen atom on diethyl ether.
In general, the strength of hydrogen bonding is (ca. 1~10 kcal/mol) is weaker than covalent or ionic bonds(50 kcal/mol), but far stronger than a van der Waals interaction(0.2 kcal/mol). Hydrogen bonds break and reform continuously because of thermal motions in molecules. Polymers contains polar functional groups, such as hydroxyl group, carbonyl group, urethane group and amide groups forms physically hydrogen bonding within or between polymers. The hydrogen bonds in polymers can occur between molecules (intermolecularly, self-association, Scheme 1-8)[68], or within different parts of a single molecule (intramolecularly, inter-association,
Scheme 1-9). In general, the hydrogen bonding in polymer forms dimers in dilute
polymer solution but timers in concentrated solution (Scheme 1-10). Hydrogen bonding forms among different functional groups of molecules is called intermolecular hydrogen bond. Also, the hydrogen bonding among molecules make the charge density of electron cloud rearrange and also the total energy of electromagnetic radiation altered with the strength of interaction. The change of
19
interaction can be detected and measured by FT-IR spectrum at suitable frequency. Many polymers are strengthened by hydrogen bonds in their main chains. Among the synthetic polymers, the best known example is nylon, where hydrogen bonds occur in the repeat unit and play a major role in crystallization of the material. The bonds occur between carbonyl and amine groups in the amide repeat unit. They effectively link adjacent chains to create crystals, which help reinforce the material. The effect is greatest in aramid fibre, where hydrogen bonds stabilize the linear chains laterally. The chain axes are aligned along the fibre axis, making the fibres extremely stiff and strong. Hydrogen bonds are also important in the structure of cellulose and derived polymers in its many different forms in nature, such as wood and natural fibres such as cotton and flax.
Hydrogen bonding also plays an important role in determining the three-dimensional structures adopted by proteins and nucleic bases. In these macromolecules, bonding between parts of the same macromolecule cause it to fold into a specific shape, which helps determine the molecule's physiological or biochemical role. The double helical structure of DNA, for example, is due largely to hydrogen bonding between the base pairs (Scheme 1-12), which link one complementary strand to the other and enable replication. Figure 1-4 also shows the hydrogen bonding between base pairs in the compound utilized in this study.
In the secondary structure of proteins, hydrogen bonds form between the backbone oxygens and amide hydrogens. When the spacing of the amino acid residues participating in a hydrogen bond occurs regularly between positions i and i + 4, an alpha helix is formed. When the spacing is less, between positions i and i + 3, then a 310 helix is formed. When two strands are joined by hydrogen bonds involving
alternating residues on each participating strand, a beta sheet is formed. Hydrogen bonds also play a part in forming the tertiary structure of protein through interaction
20
of R-groups.
Scheme 1-8. An example of intermolecular hydrogen bonding in a self-assembled
dimer complex reported by Meijer and coworkers.[67]
Scheme 1-9. Intramolecular hydrogen bonding in acetylacetone helps stabilize the
enol tautomer
21
Scheme 1-11. Examples of hydrogen bond donating (donors) and hydrogen
bond accepting groups (acceptors)
Scheme 1-12. Hydrogen bonding between guanine and cytosine, one of two types of
22
Figure 1-3. The microstructure within PU and hydrogen bonding among hard segments[69] N N H N H O O N H N HN O O
23
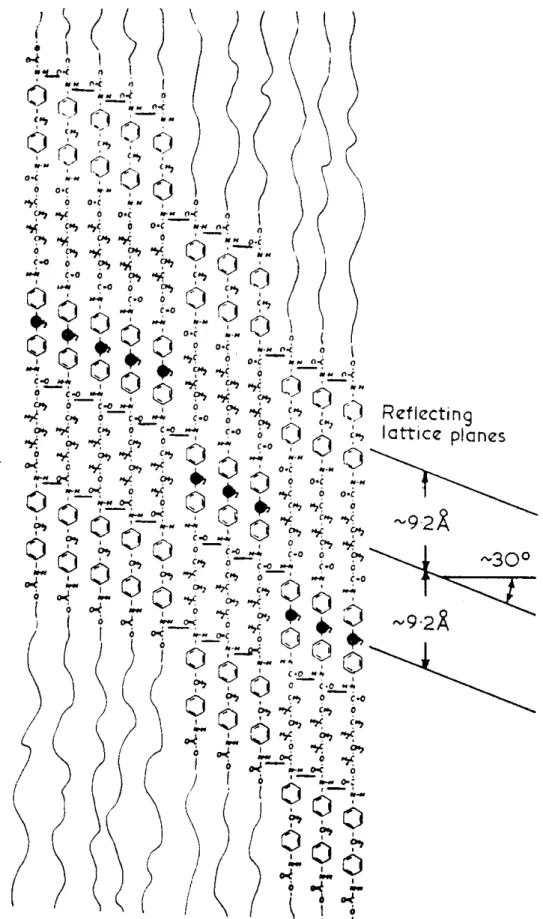
Figure 1-5. Schematic of the hydrogen-bonding in MDI-butanediol hard segment ,as
proposed by Bonart et al. The staggering of the chains leads to planes in this projection at 30° to the perpendicular as indicated[70-71].
24
Chapter 2
Literature Review and Motivation
2-1 Introduction to Natural Nucleobases and its Applications in Supramolecular
The five main natural nucleobases adenine, cytosine, guanine, thymine and uracil are involved in the self-assembly of one of nature’s most interesting and intriguing biopolymers, namely the nucleic acids DNA and RNA. As such, these nucleobases have held a fascination to researchers in a diverse range of fields. With the growth in the field of supramolecular chemistry and consequently a better understanding of how molecules interact with each other, more and more information is emerging on the complex supramolecular behaviour of these nucleobases.
Nature has excelled at utilizing supramolecular chemistry to store, transmit and replicate information in a challenging environment with a limited number of structural units. DNA and DNA-like materials offer the opportunity of preparing controlled self-assembled architectures. The interaction between two DNA strands is primarily mediated by four nucleobases adenine (A), thymine (T), guanine (G) and cytosine (C). The two anti-parallel strands of DNA are held together by A-T and C-G base-pairs to form the famous double helix. While the selectivity of these base-pair interactions is controlled mainly by hydrogen bonding, both π−π stacking and hydrophobic effects also play a role in stabilizing the resulting structure. Utilization of the common nucleobases in supramolecular chemistry offers the flexibility of exploiting four different binding units A, C, G, and T (or U), all of which offer different binding characteristics. A limiting factor, however, in the utilization of single base-pairing interactions lies in the fact that in polar solvents, in which functionalized purine (A, G) and pyrimidine (C, T) derived systems are generally most soluble, the energies of the complementary base-pair interactions are small. Biological systems have overcome this problem by utilizing a combination of non-covalent interactions, in addition to
25
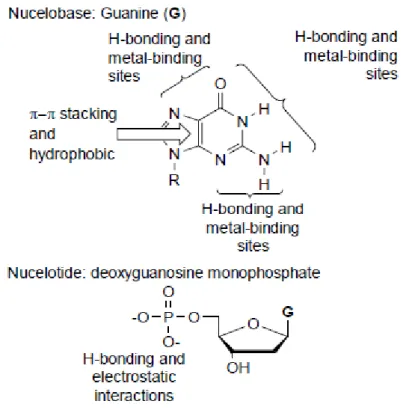
multiple base-pairs, to contribute toward the formation of desired complexes. Figure.
2-1, as an example, shows some of the possible non-covalent interactions of guanine
and its corresponding nucleotide deoxyguanosine monophosphate which could be utilized in supramolecular systems. All nucleobases offer a similar plethora of potential non-covalent interactions, and so a multiple site binding approach, where the weak base-pairing interactions are complemented by additional binding forces, adds an extra dimension to the use of nucleobase recognition in supramolecular chemistry. Furthermore, nucleotides, which also contain a charged sugar phosphate, expand on these molecules repertoire of non-covalent bonds by including electrostatic interactions.
Figure 2-1. Some of the non-covalent interactions which are present in the
nucleobase guanine and the nucleotide deoxyguanosine monophosphate.
Supramolecular self-assembly has attracted great attention because of the novel structural organizations formed through highly complementary molecular recognition events.[72,73] Nature has excelled at utilizing supramolecular chemistry to
26
store, transmit, and replicate information in a challenging environment with a limited number of structural units such as DNA and RNA offering the opportunity of preparing controlled self-assembled architectures.[74] These noncovalent interactions containing materials exhibit unique physical properties, such as high specificity, controlled affinity, and reversible, selective, self-healing, and spontaneous self-assembly behavior.[75] Recently, several supramolecular structures have been incorporated into polymers5 to form novel materials with features of conventional polymers and reversibility in the bonding between monomer units.[77] On the other hand, chemists have not only started to learn how to mimic natural use of noncovalent chemistry in polymer science but also how we might be able to harness these interesting biomolecules to construct complex nanostructures and materials in the foundation of supramolecular polymer science. In this chapter, the discussion of the scope and limitations of supramolecular chemistry and nature of hydrogen bonds in polymeric materials with an emphasis on properties of the resulting materials will be presented. In addition, the design principles and the methodology of supramolecular materials in particular multi-functionalization indicated in the published literatures will also be discussed.
Scheme 2-1. Stepwise Construction of π-Stacked Dimers from Monomers for
27
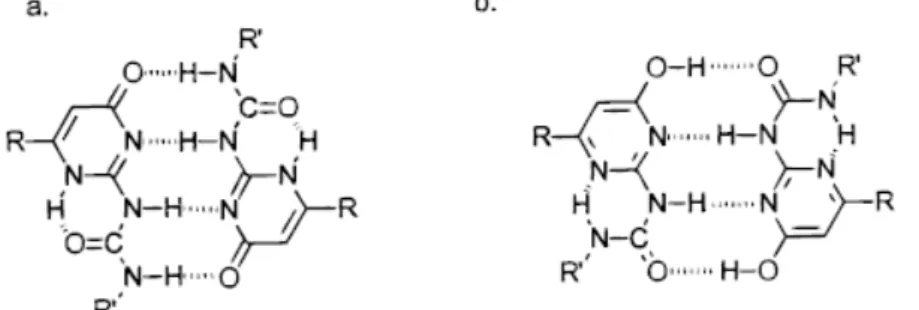
2-2 Versatile Hydrogen-Bonding Motifs Through Nucleobase-pairing 2-2.1 Nature and Stability of Hydrogen Bonds
Hydrogen bonds between neutral organic molecules play an important role in determining the three-dimensional structure of chemical and biological systems as a consequence of their specificity and directionality. The most familiar hydrogen bond motifs are the nucleobases found in DNA and RNA. Hydrogen bonds connect atoms X and Y which have electronegativities larger than that of hydrogen, such as C, N, O, F, P, S, Cl, Se, Br, and I. The XH group is generally referred to the “proton donor” (D) and the Y atom is called the “proton acceptor”. The strength of a hydrogen bond increases with the increase in the dipole moment of the X-H bond and the electron lone pair on atom Y (Table 2-1). Thus, the strongest hydrogen bonds formed between atoms N, O, and F acting as X and Y. Recently, it have been reported that C-H also acts as a donor and hydrogen bonds involve an interaction between a partially positive hydrogen atom and the electrons of unsaturated double or triple bonds.78
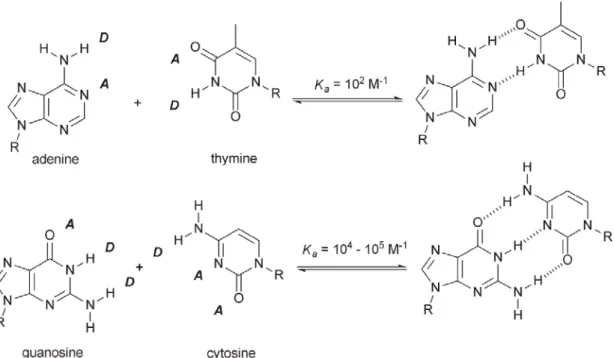
The stability of any hydrogen-bond assembled complex will be strongly influenced by the number of hydrogen-bonds – in general, two hydrogen-bonds will be stronger than one hydrogen-bond, three hydrogen-bonds will be stronger than two, and so on. This can be seen for DNA base pairs where the Guanine-Cytosine (G-C, Ka= 104-105 M-1) complex is two to three orders of magnitude morestable than the
Adenine-Thymine (A-T, Ka= 102 M-1) complex (Figure 2-2). In addition, this
suggests that multiple hydrogen-bonding systems should posses relatively higher Ka
attributed to four (or more) attractive secondary interactions (Figure 2-3). Indeed, the association constants based on this system were reported as values of more than 104-107 M-1.21,22 In summary, among various multiple hydrogen bonded modules, the association constant increases in the order, but the complexity does not refer to the objects directly. Compared to the DNA types, the systems, G-C and A-T, are
28
relatively easy to be synthesized through molecular design. For the molecular assembly, the trouble should be avoided in the synthesis of a modular component, as possible.
Table 2-1: The length of hydrogen bonds[104]
29
Figure 2-2 Hydrogen bonding in DNA base pairs.
Figure 2-3 Selected multiple hydrogen bonded complexes with high association
30
2-2.2 Hydrogen Bonding of Nucleobase-pairing Allows for Versatility
Before discussing the various structural architectures that synthetic chemists can prepare using nucleobases, a review of the base-pairing characteristics of the natural nucleobases is appropriate. This is because, in order to comprehend better the plethora of synthetic structures that can be constructed through nucleobase interactions, a summary of the various modes of hydrogen-bonding between nucleic acids is in need. Also, from a design perspective the advantages of base-pair derived systems must be recognized and exploited while the disadvantages must likewise be noted and addressed. There are two major nucleobase binding motifs present in nucleic acids, the adenine-thymine, AT (or adenine-uracil, AU in RNA) and guaninecytosine, GC. These nucleobase-pairs interact via 2 or 3 hydrogen bonds, respectively (Figure 2-4). The Watson–Crick motif (Figure 2-4), found in a range of DNA- and RNA-containing structures, is the most widely recognized hydrogen-bonding interaction in Nature. This canonical motif is defined by the pairing of guanosine with cytidine and adenosine with either thymidine or uridine. The guanosine–cytidine (GC) couple (Ka ~104–105 M-1 in CDCl3)[81] is stabilized by a
three-point hydrogen-bonding interaction, while the adenosine–thymidine (AT or AU) grouping (Ka ≈ 102 M-1 in CDCl3)[82] contains a two-point hydrogen-bonding mode. Thus, based solely on the strength of association, the GC couple represents a stronger base-pairing motif. It is therefore more attractive for incorporation as a recognition ‘‘subunit’’ into new structures. For this reason, GC binding interactions have been popular and widely used. However, there are many examples where the AT (or AU) Watson–Crick motif has been used with good effect to stabilize a number of elegant supramolecular structures. Both types of ensembles are covered in the research of our laboratory. However, while this Watson-Crick base-pairing is dominant within nucleic acids and prevalent within DNA-duplex, it is important to note that the
31
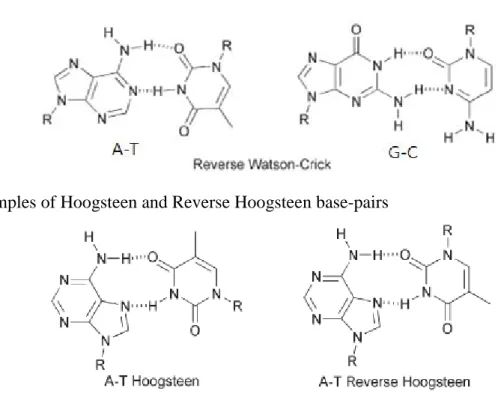
nucleobases are not exclusive in their binding behaviour. Other hydrogen-bonding motifs are available and expand the possibility for the creation of different structural networks even though the Watson–Crick mode of bonding is prevalent in natural systems.[83] There are 28 possible base-pairing motifs that involve at least two hydrogen bonds which can be formed between the four common nucleobases.[85] These include reverse Watson-Crick (Figure 2-5a), Hoogsteen (Figure 2-5b) or ‘wobble’ (or mismatched) base-pairs (Figure 2-5c), and various homo dimers (self-pairing). Special attention needs to be paid to the Hoogsteen[84] mode of bonding, which is another mode that we have exploited for the development of new, synthetic self-assembled ensembles. Hoogsteen interactions occur on the opposite face, between the C6–N7, of the purine nucleosides. Along with Hoogsteen interactions, other non-traditional base-pairs (see Figure 2-5) are found extensively in various DNA and RNA structures. In addition, these modes are also present in protein–DNA and drug–DNA interactions. These, and other nucleobase binding modes, can play important roles in any nucleobase self-assembly process which is controlled by its hydrogen bonding, especially but not exclusively when the interaction is not constrained by the geometry of the double helix. The conformation of the sugars with respect to a base-pair (i.e., cis or trans conformation with regard to the sugar on the complementary nucleobase) can also lead to variant basepairing modes. For example, the ‘reverse’ base-pairing mode is defined by a trans or antiparallel conformation of the two sugar moieties. Such a conformation can lead to reverse Watson–Crick (see
Figure 2-5a) and reverse Hoogsteen (Figure 2-5b) base-pairing modes. Other
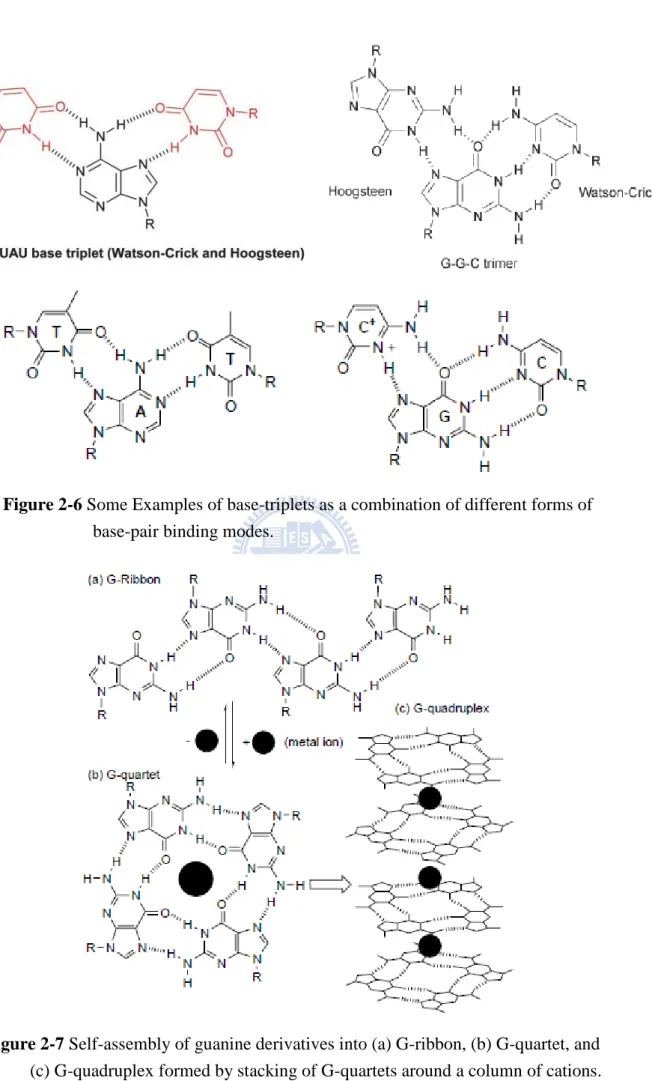
dimeric binding modes are also possible due to tautomerism and ionization of nucleobases, but these are far less prevalent. In addition to base dimerization, individual nucleobases can also form trimers and other higher order oligomers. Furthermore, the purines, A and G, also contain two major hydrogen bonding sites,
32
the Watson-Crick and the Hoogsteen sites. Thus, in addition to being able to form 1:1 complexes through either of these two sites, both of these nucleobases can form 2:1 aggregates (or base-triplets) with an appropriate partner (Figure 2-6). Such complexes are involved in the formation of triple helix DNA.[86] In the case of the C+:G:C base-triplet, the N3 of one of the cytosines needs to be protonated in order to allow the donation of a hydrogen bond to the N7 of guanine (Figure 2-6). Furthermore, in addition to interacting with other nucleobases, each of the nucleobases can also homo-dimerize(Figure 2-5d). In the case of purines, A and G, larger aggregates are also possible on account of the two binding sites. Guanine in particular is known to self aggregate[87] into tapes and macrocycles aided by its relatively high association constant for dimerization (KGG ca. 102-104 M-1 in CDCl3,
c.f. KAA ca. < 5 M-1 in CDCl3).[88,89]
In fact, in addition to the complementary interactions, all of the nucleobases can homodimerize, albeit with greatly reduced binding constants (KTT = 3.5 M-1, KAA
= 2.4 M-1, KCC = 40, KGG = 102-104M-1).Therefore, monomer units containing single
nucleobases for binding generally exhibit degrees of interaction too low to form polymers through the MSOA mechanism in solution. However, of the natural nucleobases, homo-oligomers of guanine derivatives are perhaps the most diverse. The presence of two complementary hydrogen bonding arrays, a donor/donor site comprised of the N1H amide and N2H amine and an acceptor/acceptor site comprised of the O6 and N7 functionalities, allows it to oligomerize via hydrogen bonding. This supramolecular arrangement combined with the strength of the interaction (KGG ca.
102-104 M-1 in CDCl3), makes it the most studied homo-nucleobase interaction.
Guanine derivatives can utilize hydrogen bonding to self-assemble into either linear tapes[90] or macrocycles. Figure 2-7 shows an example of a guaninebased linear tape that self–assembles through Hoogsteen interactions. The other possibility is for the
33
self-assembly to result in macrocycles. In fact, guanine is well known to self-assemble into hydrogen-bonded cyclic tetramers, the so-called G-quartet, in the presence of metal ions (Figure 2-7).
On the other hand, non Watson–Crick binding modes, such as the Hoogsteen motif, can be exploited to assemble architectures that are not possible to access via simple Watson–Crick base pairing. Further, the aforementioned binding modes can be used in conjunction with other intermolecular forces to prepare synthetic molecular cages and supramolecular polymers. Indeed, a number of supramolecular architectures assembled through non Watson–Crick base-pairing interactions will be highlighted in some examples within this article.
34
(a) Reverse Watson-Crick mode
(b) Examples of Hoogsteen and Reverse Hoogsteen base-pairs
(c) Two Wobble (Mismatched) base-pairs and an Reverse Wobble base-pair
(d) Self-dimerization (homo-dimerization)
35
Figure 2-6 Some Examples of base-triplets as a combination of different forms of
base-pair binding modes.
Figure 2-7 Self-assembly of guanine derivatives into (a) G-ribbon, (b) G-quartet, and
36
2-3 An overview on Supramolecules 2-3.1 Basic Principle of Supramolecules
"Molecular Chemistry" and "Supramolecular Chemistry" is two famous topic in recent researches. Research related to molecular chemistry is mainly focus on the interaction between molecules; and supramolecular chemistry, earliest investigate by J.M. Lehn[91-92] and also called chemistry superior than molecular, is a research field study on non-covalent bonding among molecules.The formation of supramolecules is mainly due to the intermolecular interaction and thus make two or more chemical compounds combine with each other to form highly complex but ordered structures. (see Figure 2-8). These specific supramolecules not only owns the characteristics of original monomers but also with new forming structures and physical properties. Supramolecules forms mainly via self-assembly and molecular recognition. The novel sturctures and properties provide fancy ways of thinking and challenges on the molecular design and applications in real products, therefore, supramolecules fix everyone' eyes on and developed prosperously in recent decades.
Figure 2-8. The route from "Molecular Chemistry" to "Supramolecular Chemistry."
In fact, supramolecules exist not only in artificial systems but do also exist in living organisms and human bodies. For example, highly complex and effective
![Table 2-1: The length of hydrogen bonds [104]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8094780.164814/47.892.126.784.280.835/table-the-length-of-hydrogen-bonds.webp)