Poly(aryl ether oxadiazole)s
FANG-IY WU, CHING-FONG SHUDepartment of Applied Chemistry, National Chiao Tung University, Hsin-Chu 30035, Taiwan (Republic of China)
Received 10 June 2001; accepted 20 August 2001
ABSTRACT: A new AB2 monomer was synthesized for use in the preparation of a hyperbranched poly(aryl ether oxadiazole) with terminal phenol functionality. The AB2 monomer contains two phenolic groups and a single aryl fluoride group that is activated toward nucleophilic displacement by the attached oxadiazole ring. The nucleophilic substitution of the fluoride with the phenolate groups led to the formation of an ether linkage. Subsequently, a hyperbranched poly(aryl ether oxadiazole) having approxi-mately a 44% degree of branching, as determined by a combination of model compound studies and1H NMR, was obtained. The terminal phenolic groups underwent facile functionalization, furnishing hyperbranched polymers with a variety of functional chain ends. The nature of the chain-end groups had a significant influence on the physical properties of the polymers, such as the glass-transition temperature and their solubility.© 2001 John Wiley & Sons, Inc. J Polym Sci Part A: Polym Chem 39: 3851–3860, 2001
Keywords: hyperbranched; poly(aryl ether oxadiazole); AB2monomer
INTRODUCTION
In view of their unique highly branched structure, which would be expected to confer some unusual properties, hyperbranched polymers have been the subject of considerable interest in recent years.1,2 Although such polymers can be conve-niently prepared via the one-pot polymerization of ABn-type monomers, they maintain many of the architectural features and properties found in their more perfectly defined dendrimer counter-parts3that are built up via step-by-step synthetic sequences.4,5 The one-step synthesis allows hy-perbranched polymers to be more readily avail-able as well as their preparation on a large scale for potential applications. These attractive fea-tures have led to the development of novel syn-thetic routes for the preparation of such poly-mers.1,2
Poly(aryl ether)s represent a class of high-per-formance polymers that possess high thermal sta-bility and good mechanical properties.6 It has been demonstrated that aromatic nucleophilic substitution reactions between activated aryl ha-lide monomers and bisphenolates lead to the for-mation of linear poly(aryl ether)s.7 Electron-with-drawing groups such as ketones, sulfones, and some heterocycles, which can serve to stabilize the anionic intermediate, are frequently used as activating groups to facilitate the aromatic nu-cleophilic substitution in the synthesis of these types of polymers.7–11 Recently, these synthetic strategies have been extended to the preparation of hyperbranched poly(aryl ether)s in the one-step polymerization of AB2monomers that contain ac-tivating moieties such as sulfone, ketone, and het-erocyclic rings.12,13
It is well known that the high thermal stability and specific properties of aromatic poly(1,3,4-ox-adiazole)s are largely due to the 1,3,4-oxadiazole ring.6(b) In this article, we report on a new AB
2 monomer that can be used in the one-pot
prepa-Correspondence to: C.-F. Shu (E-mail: [email protected])
Journal of Polymer Science: Part A: Polymer Chemistry, Vol. 39, 3851–3860 (2001) © 2001 John Wiley & Sons, Inc.
ration of a hyperbranched poly(aryl ether oxadia-zole) with terminal phenolic groups. The AB2 monomer contains an aryl fluoride and two phe-nolic groups connected by a 1,3,4-oxadiazole ring. Activated by the oxadiazole moiety, the nucleo-philic substitution of the fluoride with a phenolic group results in ether linkage, subsequently lead-ing to the formation of the correspondlead-ing hyper-branched poly(aryl ether oxadiazole). The phe-nolic terminal units of this polymer were modified by reaction with several end-capping agents. The effect of the nature of the chain-end groups on the solubility and glass-transition temperature of the hyperbranched polymers was also examined.
EXPERIMENTAL General Directions
Anhydrous K2CO3 was ground into fine powder and dried at 120 °C under vacuum. Anhydrous tetrahydrofuran (THF) was distilled from a so-dium diphenyl ketyl solution just prior to use. Diisopropyl azodicarboxylate (DIAD) and other starting materials and reagents were used as ob-tained from the suppliers. NMR spectra were re-corded on a Varian Unity 300-MHz or a Bruker-DRX 300-MHz spectrometer, and the solvent peak served as the internal standard. DSC was performed on a Seiko SSC 5200 DSC unit using a heating/cooling rate of 10 °C min⫺1. Samples were scanned from 25 to 330 °C and then cooled to 25 °C and again scanned for the second time from 25 to 330 °C. The glass-transition temperature was determined from the second heating scan. Ther-mogravimetric analyses (TGAs) were conducted on a Seiko TG/DTA 200 instrument. The thermal stabilities of the samples were determined in ni-trogen by measuring weight loss while heating at a rate of 10 °C min⫺1. Size exclusion chromatog-raphy (SEC) was carried out on a Waters chro-matographer, interfaced with a Waters 410 differ-ential refractometer. Three 5-m Waters Styragel columns (300⫻ 7.8 mm) connected in series in the decreasing order of pore size (105, 104, and 103 Å) were used with dimethylformamide (DMF)/0.05 M LiBr as the eluent, and poly(methyl methacry-late) standard samples were used for calibration. Mass spectra were obtained on a JEOL JMS-SX/SX 102A mass spectrometer.
3,5-Dimethoxyphenyltetrazole (1)
A mixture of 3,5-dimethoxybenzonitrile (4.60 g, 28.2 mmol), sodium azide (3.00 g, 46.2 mmol), and
ammonium chloride (2.47 g, 46.2 mmol) in DMF (35 mL) was heated at 120 °C for 9 h. After cool-ing, the resulting mixture was poured into water (500 mL) and neutralized with 1 N HCl. The resulting precipitate was collected by filtration, washed with water, and dried to give 1 as a white solid (5.54 g, 95.3%).
1H NMR [dimethyl sulfoxide (DMSO-d 6)]: ␦ 3.82 (s, 6H), 6.70 (t, 1H, J⫽ 2.4 Hz), 7.20 (d, 2H, J⫽ 2.4 Hz). 13C NMR (DMSO-d 6):␦ 55.5, 102.9, 104.8, 125.7, 155.2, 161.0. High-resolution mass spectrometry (HRMS) [M⫹]: 206.0811. Calcd. 206.0803 for C9H10O2N4. 2-(3,5-Dimethoxyphenyl)-5-(4-fluorophenyl)-1,3,4-oxadiazole (2)
To a solution of 1 (4.56 g, 22.1 mmol) in pyridine (10 mL), 4-fluorobenzoyl chloride (3.17 mL, 26.5 mmol) was added dropwise. The reaction mixture was refluxed for 1.5 h and then poured into water (500 mL). The precipitate was filtered off, washed with water, and dried in vacuo to give 2 as a white solid (6.21 g, 93.7%). 1H NMR (CDCl 3):␦ 3.82 (s, 6H), 6.55 (t, 1H, J ⫽ 2.3 Hz), 7.16 (dd, 2H, J ⫽ 8.9, 8.9 Hz), 7.17 (d, 2H, J⫽ 2.3 Hz), 8.07 (dd, 2H, J ⫽ 8.9, 5.2 Hz).13C NMR (CDCl3):␦ 55.7, 104.2, 104.7, 116.4 (d, JCOF ⫽ 22 Hz), 120.2 (d, JCOF⫽ 3 Hz), 125.3, 129.2 (d, JCOF⫽ 9 Hz), 161.2, 163.8, 164.6, 164.8 (d, JCOF ⫽ 252 Hz). HRMS [M⫹]: 300.0904. Calcd. 300.0910 for C16H13O3N2F. 5-[5-(4-Fluorophenyl)-1,3,4-oxadiazol-2-yl]-1,3-benzenediol (3)
A mixture of 2 (2.40 g, 8.0 mmol) and pyridine hydrochloride (6.5 g, 56 mmol) was heated at 205 °C for 1.5 h. After cooling to 80 °C, water (50 mL) was slowly added to the reaction mixture. The precipitate was collected by filtration, washed with water, and purified by column chromatogra-phy (hexane/EtOAc 3:1) to give 3 as a white solid (1.86 g, 85.7%). 1H NMR (DMSO-d 6):␦ 6.45 (t, 1H, J ⫽ 2.2 Hz), 6.96 (d, 2H, J⫽ 2.2 Hz), 7.46 (dd, 2H, J ⫽ 8.9, 8.9 Hz), 8.13 (dd, 2H, J ⫽ 8.9, 5.4 Hz). 13C NMR (DMSO-d6): ␦ 104.6, 106.1, 116.7 (d, JCOF ⫽ 22 Hz), 120.1 (d, JCOF⫽ 3 Hz), 124.5, 129.3 (d, JCOF ⫽ 9 Hz), 159.1, 163.1, 164.1 (d, JCOF⫽ 250 Hz), 164.2. HRMS [M⫹]: 272.0594. Calcd. 272.0597 for C14H9O3N2F.
2-4-[3,5-Di(tert-butyl)phenoxy]phenyl-5-(3,5-dimethoxyphenyl)-1,3,4-oxadiazole (4)
A mixture of 2 (1.50 g, 5.0 mmol), 3,5-di-tert-butyl phenol (1.55 g, 7.5 mmol), K2CO3 (0.69 g, 5.0 mmol), benzene (1.5 mL), and N-methylpyrroli-done (NMP) (4 mL) was heated at 120 °C. The water that was formed during the reaction was removed by azeotropic distillation and collected in a Dean–Stark trap. After 5 h, the remaining ben-zene in the reaction mixture was removed by dis-tillation. The reaction mixture was then heated at 160 °C for 5 h, cooled, poured into water (200 mL), and extracted with EtOAc (3 ⫻ 100 mL). The combined extracts were dried over Na2SO4, and the solvent was removed in vacuo. The product was purified by column chromatography (hexane/ EtOAc 2:1) to give 4 as a white solid (2.19 g, 90.1%). 1H NMR (CDCl 3):␦ 1.30 (s, 18H), 3.85 (s, 6H), 6.60 (t, 1H, J⫽ 2.3 Hz), 6.92 (d, 2H, J ⫽ 1.7 Hz), 7.07 (d, 2H, J⫽ 8.9 Hz), 7.23–7.24 (m, 3H), 8.06 (d, 2H, J ⫽ 8.9 Hz). 13C NMR (CDCl 3): ␦ 31.3, 53.0, 55.6, 104.1, 104.6, 114.4, 117.7, 117.8, 118.6, 125.4, 128.8, 153.1, 154.9, 161.1, 161.3, 164.2, 164. HRMS [M⫹]: 486.2527. Calcd. 486.2518 for C30H34O4N2. 5-(5-4-[3,5-Di(tert-butyl)phenoxy]phenyl-1,3,4-oxadiazol-2-yl)-1,3-benzenediol (5)
A mixture of 4 (1.00 g, 2.13 mmol) and pyridine hydrochloride (3.00 g, 25 mmol) was heated at 210 °C for 5 h. After cooling to 80 °C, water (50 mL) was slowly added to the reaction mixture, and the resulting mixture was extracted with EtOAc (3 ⫻ 40 mL). The combined extracts were dried over Na2SO4, and the solvent was removed in vacuo. The product was purified by column chromatog-raphy (hexane/EtOAc 2:1) to give 5 as a white solid (0.49 g, 51.9%). 1H NMR (DMSO-d 6): ␦ 1.27 (s, 18H), 6.44 (t, 1H, J⫽ 2.2 Hz), 6.94–6.95 (m, 4H), 7.14 (d, 2H, J ⫽ 8.8 Hz), 7.27 (t, 1H, J ⫽ 1.6 Hz), 8.06 (d, 2H, J ⫽ 8.8 Hz), 9.78 (s, 2H). 13C NMR (DMSO-d 6): ␦ 31.1, 34.7, 104.5, 106.0, 114.1, 117.6, 117.9, 118.3, 124.6, 128.8, 152.8, 154.5, 159.1, 160.4, 163.5, 163.9. HRMS [M⫹ ⫹ H]: 459.2291. Calcd. 459.2283 for C28H31O4N2.
Synthesis of Model Compounds 6 and 7
A mixture of 5 (0.40 g, 0.87 mmol), 2 (0.26 g, 0.87 mmol), K2CO3 (0.12 g, 0.87 mmol), benzene (1
mL), and NMP (2 mL) was heated at 120 °C. The water that was formed during the reaction was removed by azeotropic distillation and collected in a Dean–Stark trap. After 5 h, the remaining ben-zene in the reaction mixture was removed by dis-tillation. The reaction mixture was then heated at 155 °C for 6 h. The resulting mixture was poured into water (50 mL), neutralized with 1 N HCl, and extracted with EtOAc (3⫻ 30 mL). The combined extracts were dried over Na2SO4, and the solvent was removed in vacuo. The products were purified by column chromatography (CHCl3). Compound 6: 1H NMR (DMSO-d 6): ␦ 1.26 (s, 18H), 3.84 (s, 6H), 6.73– 6.76 (m, 2H), 6.93 (d, 2H, J⫽ 1.6 Hz), 7.11 (d, 2H, J ⫽ 8.8 Hz), 7.22 (d, 2H, J ⫽ 2.3 Hz), 7.24–7.26 (m, 2H), 7.29 (d, 2H, J ⫽ 8.9 Hz), 7.35 (t, 1H, J ⫽ 1.8 Hz), 8.08 (d, 2H, J ⫽ 8.8 Hz), 8.18 (d, 2H, J ⫽ 8.8 Hz), 10.35 (s, 1H). 13C NMR (DMSO-d 6): ␦ 31.0, 34.7, 55.6, 103.9, 104.4, 108.0, 109.5, 109.8, 114.1, 117.4, 117.8, 118.3, 118.6, 119.1, 124.9, 125.7, 128.9, 129.1, 152.8, 154.5, 157.2, 159.3, 159.7, 160.5, 161.0, 163.2, 163.6, 163.7, 163.8. HRMS [M⫹ ⫹ H]: 739.3143. Calcd. 739.3131 for C44H43O7N4. Com-pound 7: 1H NMR (DMSO-d 6): ␦ 1.26 (s, 18H), 3.84 (s, 12H), 6.73 (t, 2H, J⫽ 2.3 Hz), 6.91 (d, 2H, J⫽ 1.7 Hz), 7.08 (d, 2H, J ⫽ 8.9 Hz), 7.17 (t, 1H, J ⫽ 2.2 Hz), 7.19 (d, 4H, J ⫽ 2.3 Hz), 7.24 (t, 1H, J⫽ 1.7 Hz), 7.36 (d, 4H, J ⫽ 8.8 Hz), 7.63 (d, 2H, J⫽ 2.2 Hz), 8.08 (d, 2H, J ⫽ 8.9 Hz), 8.18 (d, 4H, J⫽ 8.8 Hz).1H NMR (CDCl 3):␦ 1.29 (s, 18H), 3.86 (s, 12H), 6.62 (t, 2H, J⫽ 2.3 Hz), 6.88 (d, 2H, J⫽ 1.7 Hz), 6.96 (t, 1H, J ⫽ 2.2 Hz), 7.05 (d, 2H, J⫽ 8.7 Hz), 7.20 (d, 4H, J ⫽ 8.8 Hz), 7.23–7.25 (m, 5H), 7.62 (d, 2H, J⫽ 2.2 Hz), 8.02 (d, 2H, J ⫽ 8.9 Hz), 8.15 (d, 4H, J ⫽ 8.8 Hz). 13C NMR (DMSO-d6):␦ 31.1, 34.7, 55.6, 103.9, 104.5, 113.0, 114.0, 114.1, 117.2, 117.8, 118.3, 119.0, 119.3, 124.9, 126.6, 129.1, 129.2, 152.8, 154.4, 157.7, 159.0, 160.6, 161.0, 162.6, 163.6, 163.8, 164.2. HRMS [M⫹⫹ H]: 1019.3967. Calcd. 1019.3979 for C60H55O10N6.
Synthesis of Model Compound 8
To a solution of 6 (52 mg, 135mol), PPh3 (106 mg, 405 mol), methanol (13 mg, 405 mol) in anhydrous THF (1 mL), and DIAD (62 mg, 405 mol) were added dropwise under nitrogen. The reaction mixture was stirred at 25 °C overnight, and the product was purified by preparative thin-layer chromatography (TLC) (EtOAc:CHCl3 1:4) to give 8 as a white solid.
1H NMR (CDCl 3):␦ 1.29 (s, 18H), 3.86 (s, 6H), 3.88 (s, 3H), 6.61 (t, 1H, J⫽ 2.3 Hz), 6.79 (t, 1H, J⫽ 2.3 Hz), 6.90 (d, 2H, J ⫽ 1.6 Hz), 7.06 (d, 2H, J⫽ 8.9 Hz), 7.17 (d, 2H, J ⫽ 8.9 Hz), 7.23–7.25 (m, 3H), 7.39 (dd, 1H, J⫽ 2.1, 1.4 Hz), 7.49 (dd, 1H, J⫽ 2.2, 1.3 Hz), 8.04 (d, 2H, J ⫽ 8.9 Hz), 8.12 (d, 2H, J ⫽ 8.8 Hz). 13C NMR (CDCl 3): ␦ 31.3, 35.0, 55.7, 55.9, 104.2, 104.7, 107.8, 109.3, 110.3, 114.4, 117.5, 117.7, 118.6, 118.8, 119.1, 125.4, 126.3, 128.8, 129.0, 153.1, 154.8, 157.5, 159.8, 161.2, 161.4, 161.5, 163.6, 164.1, 164.4, 164.6. HRMS [M⫹ ⫹ H]: 753.3287. Calcd. 753.3288 for C45H45O7N4.
Preparation of Hyperbranched Poly(aryl ether oxadiazole) (P1)
A mixture of 3 (2.00 g, 7.35 mmol), K2CO3(2.03 g, 14.7 mmol), benzene (4 mL), and NMP (11 mL) was heated at 120 °C. The water that was formed during the reaction was removed by azeotropic distillation and collected in a Dean–Stark trap. After 5 h, the remaining benzene in the reaction mixture was removed by distillation. The reaction mixture was then heated at 160 °C for 13 h. The resulting mixture was poured into a solution of water (200 mL) and methanol (200 mL) and neu-tralized with 1 N HCl. The polymer was collected by filtration and purified by precipitation from DMF into methanol to give P1 (1.56 g, 84.1%).
1H NMR (DMSO-d
6):␦ 6.40–7.65 (m, 5H), 7.96 (br, 2H), 9.75 (br), 10.23 (br).
Preparation of Hyperbranched Poly(aryl ether oxadiazole) (P2)
To a solution of P1 (210 mg, 0.83 mmol), ethanol (81 mg, 2.5 mmol) and PPh3(656 mg, 2.5 mmol) in anhydrous DMF (4 mL) and DIAD (506 mg, 2.50 mmol) were added dropwise under nitrogen. The reaction mixture was stirred at 25 °C for 2 days and then added to a solution of water (25 mL) and methanol (25 mL). The collected polymer was pu-rified by precipitation from CHCl3into methanol to give P2 (189 mg, 81.9%).
1H NMR (CDCl
3):␦ 1.25 (br, 3H), 4.05 (br, 2H), 6.56 – 6.90 (m, 1H), 7.13 (br, 2H), 7.34 –7.56 (m, 2H), 8.06 (br, 2H).
Preparation of Hyperbranched Poly(aryl ether oxadiazole) (P3)
P3 was prepared from P1 and 1-hexanol using
the same procedure as was used for P2 (168 mg, 74.1%).
1H NMR (CDCl
3):␦ 0.87 (br, 3H), 1.31–1.77 (m, 8H), 3.98 (br, 2H), 6.58 – 6.92 (m, 1H), 7.15 (br, 2H), 7.30 –7.62 (m, 2H), 8.09 (br, 2H).
Preparation of Hyperbranched Poly(aryl ether oxadiazole) (P4)
To a solution of P1 (70 mg, 0.28 mmol) and Et3N (113 mg, 1.12 mmol) in anhydrous DMF (4 mL), acetyl chloride (88 mg, 1.12 mmol) was added dropwise under nitrogen. The reaction mixture was stirred at 25 °C for 2 days and then added to a solution of water (20 mL) and methanol (20 mL). The collected polymer was purified by precipita-tion from CHCl3into methanol to give P4 (71 mg, 86.8%).
1
H NMR (CDCl3):␦ 2.30 (br, 3H), 6.97 (br, 1H), 7.15 (br, 2H), 7.61 (br, 2H), 8.07 (br, 2H).
Preparation of Hyperbranched Poly(aryl ether oxadiazole) (P5)
To a solution of P1 (50 mg, 0.20 mmol), 4-(dim-ethylamino)pyridine (97 mg, 0.80 mmol) in anhy-drous DMF (3.0 mL) and hexanoic anhydride (171 mg, 0.80 mmol) were added dropwise under nitro-gen. The mixture was stirred at 25 °C for 2 days and then added to a solution of water (20 mL) and methanol (20 mL). The collected polymer was pu-rified by precipitation from CHCl3into methanol to give P5 (56 mg, 80.3%).
1
H NMR (CDCl3):␦ 0.89 (br, 3H), 1.35 (br, 4H), 1.73 (br, 2H), 2.56 (br, 2H), 6.93–7.22 (m, 3H), 7.60 –7.75 (m, 2H), 8.10 (br, 2H).
RESULTS AND DISCUSSION
Synthesis and Characterization of the AB2
Monomer
The synthesis of the AB2oxadiazole monomer 3 is outlined in Scheme 1. The oxadiazole derivatives have usually been synthesized according to one of the following two synthetic routes: (1) by ring closure of dihydrazides with dehydrating agents such as phosphorous oxychloride14 and (2) from the reaction of tetrazole with an acid chloride followed by intramolecular ring transformation.15 The relatively high yields and facile workup pro-cedures render the tetrazole route attractive for the preparation of pure oxadiazole derivatives.16 Commercially available 3,5-dimethoxybenzoni-trile was treated with sodium azide to give
tetra-zole 1 that was transformed into oxadiatetra-zole de-rivative 2 by reaction with 4-fluorobenzoyl chlo-ride. The subsequent demethylation of 2 with pyridine hydrochloride produced diol monomer 3 containing an activated aryl fluoride suitable for nucleophilic substitution. All compounds were characterized by1H NMR,13C NMR, and HRMS. NMR has been used as an indicator for the ability of potential monomers to undergo nucleo-philic displacement of the aryl fluoride.17The19F NMR chemical shift was the most sensitive probe for the reactivity of nucleophilic substitution of aryl fluorides, with a span of 9 ppm between the most activated monomer, 4,⬘-difluorophenyl sul-fone (⫺104.28 ppm), and nonactivated fluoroben-zene (⫺112.77 ppm).17 Figure 1 shows the 19F NMR of oxadiazole monomer 3 along with that of 4,4⬘-difluorophenyl sulfone and fluorobenzene. The19F NMR chemical shift of 3 (⫺107.20 ppm) indicates a downfield shift that is closer to 4,4 ⬘-difluorophenyl sulfone than that of fluorobenzene. The magnitude of the downfield shift is compara-ble to other polymerizacompara-ble fluoro-substituted
monomers that are activated by heterocyclic rings.17The19F NMR data suggest that the fluoro group in monomer 3 is, in all likelihood, undergo-ing aromatic nucleophilic substitution.
Model Reaction
To demonstrate the feasibility of the oxadiazole-activated aryl ether synthesis, the reaction of po-tassium 3,5-di-tert-butylphenoxide with 2 in a so-lution of NMP and benzene was examined as a model reaction for the polymerization of monomer
3 (Scheme 2). Water, generated during phenoxide
formation, was removed in the form of a benzene azeotrope during the initial stage of the reaction and, subsequently, the remaining benzene was removed from the system. The reaction mixture was then heated at 160 °C for 5 h, and the progress of the reaction was monitored by TLC. The crude product was purified by column chro-matography to give compound 4 in quantitative yields. The model reaction reveals that the aryl fluoride at the 2-position of the oxadiazole ring is cleanly substituted by a phenoxide, and this transformation is suitable for use in a polymer-ization reaction.
Synthesis and General Properties of
Hyperbranched Poly(aryl ether oxazole) P1
As shown in Scheme 3, the one-step polymeriza-tion of monomer 3 was carried out using a proce-Scheme 1
Figure 1. 19F NMR spectra in DMSO-d
6of a mixture of (a) 4,4⬘-difluorophenyl sulfone, (b) monomer 3, and (c) fluorobenzene.
Scheme 2
dure similar to that described for the model reac-tion. The nucleophilic substitution of the fluoride with the phenolic groups, activated by the oxadia-zole moiety, led to the formation of an ether link-age and, subsequently, to the hyperbranched poly-(aryl ether oxadiazole) P1 with terminal phenolic groups. A high molecular weight polymer was pro-duced within 12 h as judged by a pronounced in-crease in viscosity. The result of the one-step poly-merization of monomer 3 is summarized in Table I. The molecular weight of P1 was determined by SEC analysis in DMF solution calibrated against linear poly(methyl methacrylate) standards. Because of the highly irregular, branched nature of hyper-branched macromolecules, SEC analysis does not provide an accurate measurement of molecular weight and tends to underestimate the true molec-ular weight.18 Figure 2 shows the progression of molecular weight with reaction time for P1. There is an increasing gap in the growth of number-aver-age molecular weight (Mn) and weight-average mo-lecular weight (Mw), leading to broad molecular weight distributions at higher conversions. This ob-servation is consistent with previous reports of other hyperbranched polymers and is in agreement with Flory’s predictions on the molecular distribu-tion behavior for highly branched systems.19 The glass-transition temperature (Tg) of the hyper-branched poly(aryl ether oxadiazole) was deter-mined by DSC. The Tgvalue for P1 was observed at 286 °C. TGA was used to measure thermal stability.
P1 had a high thermal stability with a 5% weight
loss observed at 419 °C, followed by an additional 5% weight loss at 456 °C.
Degree of Branching
The degree of branching (DB), defined as the sum of dendritic and terminal units versus total units (linear, dendritic, and terminal units), is a typical characteristic frequently used to evaluate the ir-regularity of the structure of hyperbranched poly-mers.20 A combination technique of model
com-Scheme 4 Table I. Data of the One-Step Polymerization of
Monomer 3 Reaction Time (h) Mn a M w a M w/Mn 2.5 3,200 3,400 1.06 4.0 3,600 3,900 1.08 5.5 4,200 4,900 1.17 7.0 4,600 5,600 1.22 8.5 5,800 8,400 1.45 10.0 7,000 11,500 1.64 11.5 7,900 14,600 1.85 13.0 8,800 19,800 2.25 14.5 11,000 39,400 3.58
aDetermined by SEC on the basis of poly(methyl
methac-rylate) standards. Figure 2.analysis versus reaction time plot for the polymeriza-Molecular weight as determined by SEC
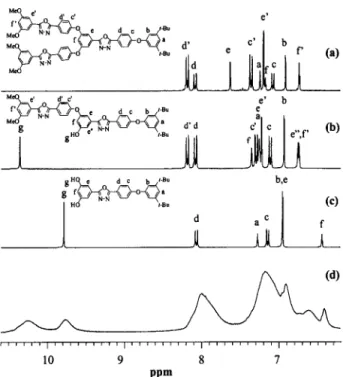
pound studies and NMR spectroscopy has been used to quantify the different subunits that ap-pear in the hyperbranched polymer and subse-quently to determine its DB.20The model reaction performed to determine the DB of P1 is given in Scheme 4. An equimolar reaction of compounds 2 and 5, the demethylated form of 4, was conducted under experimental conditions that were similar to those used earlier for the polymerization of monomer 3. The model compounds were sepa-rated from the reaction mixture by preparative TLC and characterized by 1H NMR, 13C NMR, and HRMS. Figure 3 depicts the1H NMR spectra of compounds 5, 6, and 7 that resemble the ter-minal unit, the linear unit, and the dendritic unit, respectively. The peak assignments were based on the peak positions of compounds 4 and 5 as well as the auxiliary of 2D (H, H) and (C, H)-correlated NMR spectroscopy. Figure 3 also shows the 1H NMR spectrum of the hyper-branched poly(aryl ether oxadiazole) P1. The peaks that are associated with the aromatic pro-tons of P1 are not well resolved, whereas reso-nances due to hydroxyl protons appear at signif-icantly different positions, 9.75 and 10.23 ppm, respectively. A good correlation is observed in the comparison of the1H NMR spectrum of P1 with that of model compounds 5 and 6. The resonance
at 9.75 ppm is assigned to hydroxyl protons of the terminal subunits, whereas the resonance at 10.23 ppm is assigned to the hydroxyl proton of the linear subunits. The relative integrations of the resonances at 9.75 and 10.23 ppm are 100 and 79, respectively, allowing the relative percentage of each subunit to be determined. According to the theoretical prediction, the number of terminal units is equal to the number of dendritic units for an AB2-type hyperbranched polymer possessing high molecular weight.19,20The DB is given by20
DB⫽ D⫹ T
D⫹ L ⫹ T ⬵ 2T 2T⫹ L
where D, L, and T represent the fractions of den-dritic, linear, and terminal units, respectively. On the basis of this formula, the DB of the hyper-branched poly(aryl ether oxadiazole) P1 was de-termined to be 44% based on the relative integra-tion of the hydroxyl protons. The DB is lower than the statistical value of 50%, expected for a ran-dom AB2 polycondensation.21 The preference of linear product in the polycondensation reaction may result from steric hindrance because of the two hydroxyl groups of the AB2monomer that are arranged in a metaorientation on the benzene ring.
By a similar rationale, the DB of the ether derivative P2 (vide infra) that had three well-resolved signals in the region of 6.45– 6.98 ppm was also evaluated. Figure 4 represents the 1H NMR spectra of model compounds 4 (terminal), 8 (linear), and 7 (dendritic) as well as polymer P2. A comparison of the 1H NMR spectra of these model compounds with that of P2 allows the res-onances corresponding to the dendritic, linear, and terminal subunits of the hyperbranched poly-mer to be identified. The resonances at 6.56, 6.72, and 6.90 ppm are assigned to the proton of the terminal, linear, and dendritic subunits with rel-ative integrations of 23.2, 54.1, and 22.7, respec-tively. The integration of the peak assigned to the terminal units is approximately equal to that of the peak assigned to the dendritic units. This result is consistent with the theoretical prediction that the number of terminal units should be equal to the number of dendritic units for a high molec-ular weight AB2-type hyperbranched polymer. On the basis of the integration ratio of these protons, the DB of P2 was determined to be 46%, which is in good agreement with the DB of P1.
Figure 3. 1H NMR spectra in DMSO-d
6 of model compounds (a) 7, (b) 6, and (c) 5 as compared with (d) the hyperbranched poly(aryl ether oxadiazole) P1.
Chemical Modification of Hyperbranched Poly(aryl ether oxadiazole) P1
Hyperbranched polymers are characterized by their large number of chain-end groups. As shown in Scheme 5, different functional groups could be introduced into P1 via reactions of the terminal phenolic groups. Using the Mitsunobu reaction,22 the phenolic groups of P1 were converted into ether groups to yield the ether derivatives P2 and
P3. P1 could also be acrylated with an acid
chlo-ride or acid anhydchlo-ride to give the corresponding ester derivatives P4 and P5, respectively. These derivatives contain alkyl chain ends exhibiting 1H NMR peaks that are well separated from the peaks associated with the aromatic units. The conversion of the end-capping reaction was calcu-lated by comparing the integration ratio of the protons attributed to the alkyl end groups versus those from the aromatic units. For all the afore-mentioned modification reactions, the use of ex-cess reagents resulted in a nearly complete (95– 100%) functionalization, indicating that the hy-droxyl groups at the chain ends are readily accessible to reagents in solution.
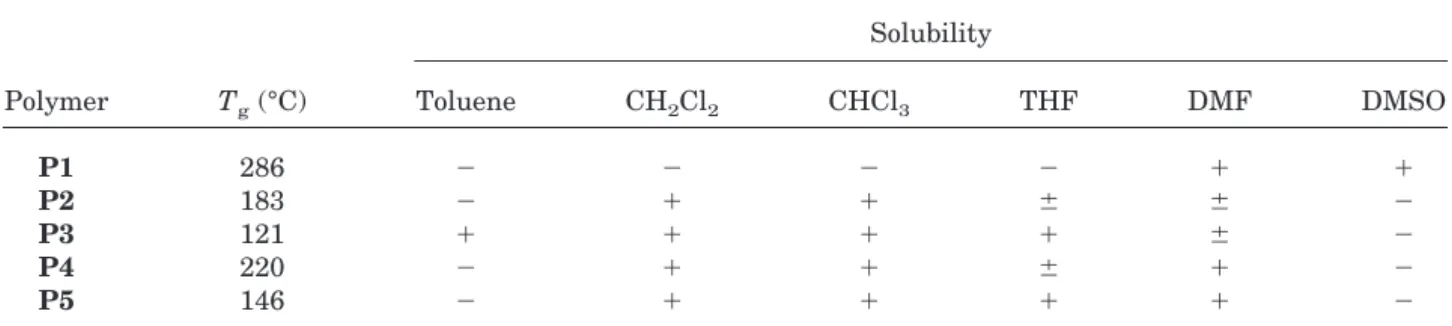
The nature of the end groups influences the physical and chemical properties of the
hyper-branched polymers.23Table II summarizes the T g and solubility of polymers P1–P5. It is known that, for hyperbranched polymers, the transition from the polar hydroxyl function to nonpolar ali-phatic end groups results in a decrease in Tg because of the reduction in the extent of intermo-lecular interactions in the polymeric molecules.24 Figure 5 indicates DSC thermograms for hyper-branched poly(aryl ether oxadiazole)s P1–P5. The Tgof P1 that contains polar hydroxyl termi-nal groups is 286 °C, whereas the Tgvalues of P2 and P4 that contain less polar terminal groups, namely, ether and ester groups, are 183 and 220 °C, respectively. A further decrease in Tg to 121 and 146 °C is observed for P3 and P5, respec-tively, because of the increasing length of the alkoxyl chain of the terminal ether or ester groups.
The different chain ends also lead to differ-ences in solubility in polar and nonpolar solvents. The phenolic-terminated polymer P1 is soluble in a solution consisting of NaOH(aq)/CH3OH and in polar solvents such as DMSO and DMF. In con-trast, the ether-terminated polymers P2 and P3 are only partially soluble in DMF and insoluble in DMSO, and the ester-terminated polymers P4 and P5 are soluble in DMF and insoluble in DMSO. Conversely, polymers P2–P5 are ex-tremely soluble in relatively nonpolar solvents such as CH2Cl2and CHCl3, whereas polymer P1 is insoluble. Polymers P3 and P5, with longer alkyl chain ends, are soluble in THF, and P3 is even soluble in toluene.
SUMMARY
The synthesis of hyperbranched poly(aryl ether oxadiazole)s on the basis of
2-(4-oxyphenyl)-5-Scheme 5 Figure 4. 1H NMR spectra in CDCl
3of model com-pounds (a) 7, (b) 8, and (c) 4 as compared with (d) the hyperbranched poly(aryl ether oxadiazole) P2. * Indi-cates a signal arising from CHCl3.
(3,5-dioxyphenyl)-1,3,4-oxadiazole as the repeat-ing unit has been demonstrated. A new AB2 monomer containing two phenolic hydroxyl groups and an oxadiazole ring-activated aryl flu-oride was synthesized and used to prepare a phe-nolic-terminated hyperbranched poly(aryl ether oxadiazole). The19F NMR data and the use of the model reaction clearly demonstrated the feasibil-ity of the oxadiazole-activated aryl ether synthe-sis. Aromatic nucleophilic substitution of the aryl fluoride with the phenolates generated ether link-ages and, subsequently, the hyperbranched pol-y(aryl ether oxadiazole) P1. As determined by a combination of model compound studies and 1H NMR integration data, the DB of P1 is approxi-mately 44%. The terminal phenolic groups were readily functionalized, yielding hyperbranched polymers with a variety of functional chain ends. The nature of the chain ends was shown to have a significant effect on physical properties such as the Tg and solubility of the hyperbranched poly-(aryl ether oxadiazole)s.
The authors thank the National Science Council (ROC) (NSC 89-2113-M009-019) for their financial support.
REFERENCES AND NOTES
1. (a) Malmstro¨m, E.; Hult, A. J Macromol Sci Rev Macromol Chem Phys 1997, C37, 555; (b) Kim, Y. H. J Polym Sci Part A: Polym Chem 1998, 36, 1685; (c) Voit, B. J. J. Polym Sci Part A: Polym Chem 2000, 38, 2505, and references cited in the reviews.
2. (a) Jayakannan, M.; Ramakrishnan, S. J Polym Sci Part A: Polym Chem 2000, 38, 261; (b) Drohmann, C.; Mo¨ller, M.; Gorbatsevich, O. B.; Muzafarov, A. M. J Polym Sci Part A: Polym Chem 2000, 38, 741; (c) Gong, C.; Fre´chet, J. M. J. J Polym Sci Part A: Polym Chem 2000, 38, 2970; (d) Lin, Q.; Long, T. E. J Polym Sci Part A: Polym Chem 2000, 38, 3736; (e) Emrick, T.; Chang, H.-T.; Fre´chet, J. M. J. J Polym Sci Part A: Polym Chem 2000, 38, 4850; (f) Wu, F.-I.; Shu, C.-F. J Polym Sci Part A: Polym Chem 2001, 39, 0000; (g) Yamanaka, K.; Jikei, M.; Kakimoto, M. Macromolecules 2000, 33, 1111; (h) Paulasaari, J. K.; Weber, W. P. Macromolecules 2000, 33, 2005; (I) Magnusson, H.; Malmstro¨m, E.; Hult, A. Macromolecules 2000, 33, 3099; (j) Marko-ski, L. J.; Thompson, D. S.; Moore, J. S. Macromol-ecules 2000, 33, 5315.
3. (a) Fre´chet, J. M. J. Science 1994, 263, 1710; (b) Advances in Dendritic Molecules, Newkome, G. R., Ed.; JAI: Greenwich, CT, 1994; pp 1– 67; Vol. 1; (c) Newkome, G. R.; Moorefield, C. N.; Vo¨gtle, F. Den-dritic Molecules: Concepts, Syntheses, Perspec-tives; VCH: Weinheim, FRG, 1996; pp 1–36; (d) Zeng, F.; Zimmerman, S. C. Chem Rev 1997, 97, 1681; (e) Frey, H.; Lach, C.; Lorenz, K. Adv Mater 1998, 10, 279; (f) Crooks, R. M.; Zhao, M. Q.; Sun, L.; Chechik, V.; Yeung, L. K. Acc Chem Res 2001, 34, 181; (g) Hecht, S.; Fre´chet, J. M. J. Angew Chem Int Ed Engl 2001, 40, 74.
4. (a) Tomalia, D. A.; Baker, H.; Dewald, J.; Hall, M.; Kallos, G.; Martin, S.; Roeck, J.; Ryder, J.; Smith, P. Polym J 1985, 17, 117; (b) Newkome, G. R.; Yao, Z.-Q.; Baker, G. R.; Gupta, V. K. J Org Chem 1985, 50, 2003.
5. (a) Hawker, C. J.; Fre´chet, J. M. J. J Am Chem Soc 1990, 112, 7638; (b) Miller, T. M.; Neenan, T. X. Figure 5. DSC thermograms of hyperbranched
poly(aryl ether oxadiazole)s (a) P1, (b) P4, (c) P2, (d) P5, and (e) P3.
Table II. Thermal and Solution Properties of the Hyperbranched Poly(aryl ether oxadiazole)s
Polymer Tg(°C) Solubility Toluene CH2Cl2 CHCl3 THF DMF DMSO P1 286 ⫺ ⫺ ⫺ ⫺ ⫹ ⫹ P2 183 ⫺ ⫹ ⫹ ⫾ ⫾ ⫺ P3 121 ⫹ ⫹ ⫹ ⫹ ⫾ ⫺ P4 220 ⫺ ⫹ ⫹ ⫾ ⫹ ⫺ P5 146 ⫺ ⫹ ⫹ ⫹ ⫹ ⫺
Chem Mater 1990, 2, 346; (c) Xu, Z. F.; Moore, J. S. Angew Chem Int Ed Engl 1993, 32, 1354.
6. (a) Rose, J. B. In High Performance Polymers: Their Origin and Development; Seymour, R. B.; Kirshenbaum, G. S., Eds.; Elsevier: New York, 1986; p 197; (b) Schulz, B.; Brehmer, L. In Poly-meric Materials Encyclopedia; Salomone, J. C., Ed.; CRC: Boca Raton, FL, 1996; p 5595.
7. (a) Labadie, J. W.; Hedrick, J. L.; Ueda, M. In Step-Growth Polymers for High-Performance Ma-terials; Hedrick, J. L.; Labadie, J. W., Eds.; ACS Symposium Series 624; American Chemical Soci-ety: Washington, DC, 1996; pp 210 –225; Chapter 12; (b) Hergenrother, P. M.; Connell, J. W.; Laba-die, J. W.; Hedrick, J. L. Poly(Arylene Ether)s Con-taining Heterocyclic Units. In Advances in Polymer Science: High Performance Polymers; Springer-Verlag: Heidelberg, 1994; Vol. 117, p 67.
8. (a) Attwood, Y. E.; Dawson, P. C.; Freeman, J. L.; Hoy, L. R.; Rose, J. B.; Staniland, P. A. Polymer 1981, 22, 1096; (b) Singh, R.; Hay, A. S. Macromol-ecules 1992, 25, 1017; (c) Hay, A. S. Adv Polym Sci 1967, 4, 496.
9. (a) Hedrick, J. L.; Labadie, J. W. Macromolecules 1990, 23, 1561; (b) Hilborn, J. G.; Labadie, J. W.; Hedrick, J. L. Macromolecules 1990, 23, 2854; (c) Carter, K. R.; Miller, R. D.; Hedrick, J. L. Macro-molecules 1993, 26, 2209; (d) Strukelj, M.; Hedrick, J. C. Macromolecules 1994, 27, 7511; (e) Bottino, F. A.; Di Pasquale, G.; Leonardi, N.; Pollicino, A. J Polym Sci Part A: Polym Chem 1995, 33, 843. 10. (a) Maier, G.; Hecht, R.; Nuyken, O.; Burger, K.;
Helmreich, B. Macromolecules 1993, 26, 2583; (b) Maier, G.; Hecht, R. Macromolecules 1995, 28, 7558; (c) Maier, G.; Schneider, J. M. Macromole-cules 1998, 31, 1798.
11. (a) Hedrick, J. L.; Twieg, R. Macromolecules 1992, 25, 2021; (b) Bottino, F. A.; Di Pasquale, G.; Pol-licino, A. Macromol Rapid Commun 1999, 20, 405; (c) Bottino, F. A.; Di Pasquale, G.; Pollicino, A. Polym Bull 2000, 45, 345; (d) Bottino, F. A.; Di Pasquale, G.; Iannelli, P. Macromolecules 2001, 34,
33; (e) Hamciuc, C.; Hamciuc, E.; Bruma, M.; Klap-per, M.; Pakula, T.; Demeter, A. Polymer 2001, 42, 5955.
12. (a) Miller, T. M.; Neenan, T. X.; Kwock, E. W.; Stein, S. M. J Am Chem Soc 1993, 115, 356; (b) Chu, F.; Hawker, C. J Polym Bull 1993, 30, 265; (c) Hawker, C. J.; Chu, F. Macromolecules 1996, 29, 4370; (d) Martı´nez, C. A.; Hay, A. S. J Polym Sci Part A: Polym Chem 1997, 35, 2015; (e) Morikawa, A. Macromolecules 1998, 31, 5999.
13. (a) Srinivasan, S.; Twieg, R.; Hedrick, J. L.; Hawker, C. J. Macromolecules 1996, 29, 8543; (b) Hedrick, J. L.; Hawker, C. J.; Miller, R. D.; Twieg, R.; Srinivasan, S. A.; Trollsås, M. Macromolecules 1997, 30, 7607; (c) Gong, Z.-H.; Leu, C.-M.; Wu, F.-I.; Shu, C.-F. Macromolecules 2000, 33, 8527. 14. Klingsberg, E. J Am Chem Soc 1958, 80, 5786. 15. Huisgen, R.; Sauer, J.; Sturm, J. H.; Markgraf,
J. H. Chem Ber 1960, 93, 2106.
16. (a) Bettenhausen, J.; Strohriegl, P. Adv Mater 1996, 8, 507; (b) Greczmiel, M.; Strohriegl, P.; Meier, M.; Bru¨ tting, W. Macromolecules 1997, 30, 6042.
17. (a) Carter, K. R. Macromolecules 1995, 28, 6462; (b) Carter, K. R. In Step-Growth Polymers for High-Performance Materials; Hedrick, J. L.; Labadie, J. W., Eds.; ACS Symposium Series 624; American Chemical Society: Washington, DC, 1996; Chapter 17; pp 276 –291.
18. Hawker, C. J.; Fre´chet, J. M. J. J Chem Soc Chem Commun 1990, 1010.
19. Flory, P. J. J Am Chem Soc 1952, 74, 2718. 20. Hawker, C. J.; Lee, R.; Fre´chet, J. M. J. J Am Chem
Soc 1991, 113, 4583.
21. Ho¨lter, D.; Burgath, A.; Frey, H. Acta Polym 1997, 48, 30.
22. Mitsunobu, O. Synthesis 1981, 1.
23. Hawker, C. J.; Chu, F. Macromolecules 1996, 29, 4370.
24. Schmaljohann, D.; Ha¨uler, L.; Po¨tschke, P.; Voit, B. I.; Loontjens, T. J. A. Macromol Chem Phys 2000, 201, 49.