Metabolites with Cytotoxic Activity from the Formosan Soft Coral
Cladiella australis
Atallah F. Ahmeda,b( ), Ming-Hsuan Wua( ), Yang-Chang Wuc( ),
Chang-Feng Daid( ) and Jyh-Horng Sheua* ( ) a
Department of Marine Biotechnology and Resources, National Sun Yat-Sen University, Kaohsiung 804, Taiwan, R.O.C.
b
Department of Pharmacognosy, Faculty of Pharmacy, Mansoura University, Mansoura 35516, Egypt c
Graduate Institute of Natural Products, Kaohsiung Medical University, Kaohsiung 807, Taiwan, R.O.C.
d
Institute of Oceanography, National Taiwan University, Taipei 106, Taiwan, R.O.C.
The metabolites, (24S)-3b-acetoxyergost-5-en-21-oic acid (2), 5¢-O-acetylthymidine (3), 3¢-O-ace-tylthymidine (4), and p-vinylbenzyl alcohol (5), along with a known steroid (1) were isolated from the EtOAc extract of the Formosan soft coral Caldiella australis. The structures of new metabolites were de-termined on the basis of spectroscopic (including 1D and 2D NMR) analyses and by comparison of their NMR spectral data with those of related compounds. Except for 3, all compounds exhibited cytotoxic ac-tivity of various degrees of potency against a limited panel of human liver and breast cancer cell lines.
Keywords: Cladiella australis; Steroids; 5¢-O-acetylthymidine; 3¢-O-acetylthymidine;
p-Vinylbenzyl alcohol; Cytotoxicity; Soft coral.
INTRODUCTION
In the course of our chemical study on the octocorals, several metabolites of versatile structures possessing cyto-toxic activities have been isolated and identified.1-6 Previ-ous investigation on the EtOAc extract of Cladiella aus-tralis (Macfadyen, 1936) by our research group has re-sulted in the isolation of new eunicellin-based diterpenoids, australins A-D.7Further chromatographic purification for the rest of the fractions of the same extract has again led to the isolation of five metabolites including two steroids (1 and 2), two nucleosides (3 and 4), and
p-hydroxymethyl-styrene (5). Except for the known steroid 1, the (24S)-3b-acetoxyergost-5-en-21-oic acid (2) was obtained as a new compound, while 5¢-O-acetylthymidine (3), 3¢-O-acetyl-thymidine (4), and p-vinylbenzyl alcohol (5) were found to be new natural products. The molecular structures of these metabolites were established on the basis of spectroscopic analyses and by comparison of their NMR spectral data with those of the related compounds. Metabolites 1, 2, 4, and 5 also were found to exhibit cytotoxic activity of dif-ferent degrees of potency against the cell lines of human hepatocellular carcinoma Hep G2 and Hep 3B, human breast carcinoma MCF-7 and MDA-MB-231.
* Corresponding author. Tel: +886-7-5252000 ext. 5030; Fax: +886-7-5255020; E-mail: [email protected] O OR1 R2O N NH O O RO HOOC OH 1: R = H 2: R = Ac 3: R4: R11= H, R= Ac, R22= Ac= H 5 1 3 5 8 9 10 11 13 17 14 18 19 20 21 24 25 28 1 2 4 5 7 1' 3' 4' 5' 2' 1 2 3 4
RESULTS AND DISCUSSION
The organism was homogenized with EtOAc, fil-tered, and the organic layer was then concentrated under re-duced pressure. The residue was fractionated using silica gel column chromatography and the selected fractions were further purified by open column chromatography on silica gel and/or normal phase HPLC to afford compounds
1-5 (see Experimental). The spectral data of 1 was found to
be in full agreement with those reported previously.8 Metabolite 2 was isolated as a white solid and was found to possess a molecular formula C30H48O4, as estab-lished by the HRFABMS m/z 473.3632 (M + H)+and NMR data (Table 1). The IR absorption bands atnmax3000-3800, 1699, and 1732 cm-1revealed the presence of carboxyl and
ester carbonyl functionalities, respectively. The ion peaks appearing in the FABMS at m/z 429 and 413 suggested the elimination of carbon dioxide and acetic acid from the mol-ecule and suggested 2 as an acylated and a carboxylated compound. The13C NMR of 2 exhibited thirty carbon sig-nals which were assigned, by the aid of DEPT spectra, into six methyls, ten methylene, eight sp3methines (including one oxygenated), one sp2methine, two sp3quaternary car-bons, and three sp2quaternary carbons (including those of two carbonyls). The signals of two tertiary methyls (d 0.74 and 1.00, each 3H, s) and the three secondary methyls (d 0.85, 0.76, and 0.78, each 3H, d, J = 7.0 Hz) appeared in the 1
H NMR spectrum of 2 were assigned for H3-18, H3-19, H3-26, H3-27, and H3-28 of a 24-methylsteroid,8 respec-tively. A proton (1H,d 2.20, ddd, J = 10.0, 10.0, 3.0 Hz)
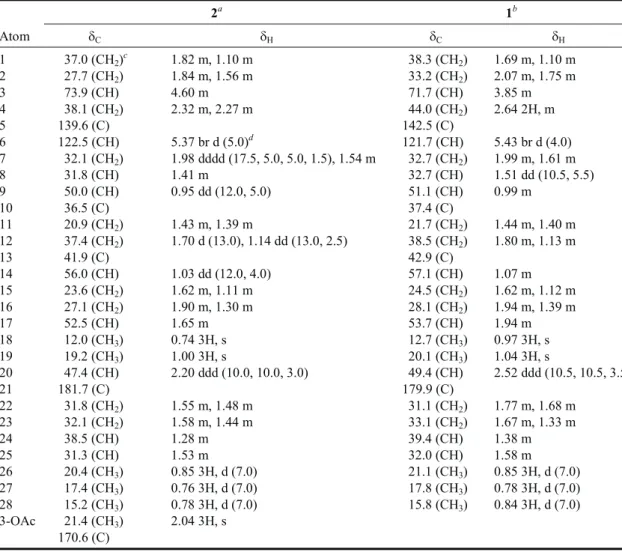
at-Table 1. 1H and13C NMR chemical shifts of 2 and 1
2a 1b Atom dC dH dC dH 1 37.0 (CH2)c 1.82 m, 1.10 m 38.3 (CH2) 1.69 m, 1.10 m 2 27.7 (CH2) 1.84 m, 1.56 m 33.2 (CH2) 2.07 m, 1.75 m 3 73.9 (CH) 4.60 m 71.7 (CH) 3.85 m 4 38.1 (CH2) 2.32 m, 2.27 m 44.0 (CH2) 2.64 2H, m 5 139.6 (C) 142.5 (C) 6 122.5 (CH) 5.37 br d (5.0)d 121.7 (CH) 5.43 br d (4.0) 7 32.1 (CH2) 1.98 dddd (17.5, 5.0, 5.0, 1.5), 1.54 m 32.7 (CH2) 1.99 m, 1.61 m 8 31.8 (CH) 1.41 m 32.7 (CH) 1.51 dd (10.5, 5.5) 9 50.0 (CH) 0.95 dd (12.0, 5.0) 51.1 (CH) 0.99 m 10 36.5 (C) 37.4 (C) 11 20.9 (CH2) 1.43 m, 1.39 m 21.7 (CH2) 1.44 m, 1.40 m 12 37.4 (CH2) 1.70 d (13.0), 1.14 dd (13.0, 2.5) 38.5 (CH2) 1.80 m, 1.13 m 13 41.9 (C) 42.9 (C) 14 56.0 (CH) 1.03 dd (12.0, 4.0) 57.1 (CH) 1.07 m 15 23.6 (CH2) 1.62 m, 1.11 m 24.5 (CH2) 1.62 m, 1.12 m 16 27.1 (CH2) 1.90 m, 1.30 m 28.1 (CH2) 1.94 m, 1.39 m 17 52.5 (CH) 1.65 m 53.7 (CH) 1.94 m 18 12.0 (CH3) 0.74 3H, s 12.7 (CH3) 0.97 3H, s 19 19.2 (CH3) 1.00 3H, s 20.1 (CH3) 1.04 3H, s 20 47.4 (CH) 2.20 ddd (10.0, 10.0, 3.0) 49.4 (CH) 2.52 ddd (10.5, 10.5, 3.5) 21 181.7 (C) 179.9 (C) 22 31.8 (CH2) 1.55 m, 1.48 m 31.1 (CH2) 1.77 m, 1.68 m 23 32.1 (CH2) 1.58 m, 1.44 m 33.1 (CH2) 1.67 m, 1.33 m 24 38.5 (CH) 1.28 m 39.4 (CH) 1.38 m 25 31.3 (CH) 1.53 m 32.0 (CH) 1.58 m 26 20.4 (CH3) 0.85 3H, d (7.0) 21.1 (CH3) 0.85 3H, d (7.0) 27 17.4 (CH3) 0.76 3H, d (7.0) 17.8 (CH3) 0.78 3H, d (7.0) 28 15.2 (CH3) 0.78 3H, d (7.0) 15.8 (CH3) 0.84 3H, d (7.0) 3-OAc 21.4 (CH3) 2.04 3H, s 170.6 (C)
Spectra recorded at 500 MHz for1H and 125 MHz for13C at 25°C,ain CDCl3andbin C5D5N,cAttached protons
tached to a methine carbon (d 47.4, CH) showed1 H-1H COSY correlation with H-17 (1H,d 1.65, m), and was as-signed as H-20. The HMBC correlations (Fig. 1) observed from H-20 to the carboxy carbon (d 181.7, C) and C-17 (d 52.5, CH) suggested the location of carboxylic acid at C-20 of the side chain. Moreover, the ion peaks of the molecular fragments at m/z 341 [M - AcOH - C5H11]+, 313 [M - AcOH - C7H15]+, and 255 [M - AcOH - C9H17O2]+also imply the presence of a carboxy group in a saturated side chain in the molecule of 2. Comparison of the NMR spectral data of 2 with those of the known metabolite 1 (Table 1), which was also isolated from the same organism by us, suggested that
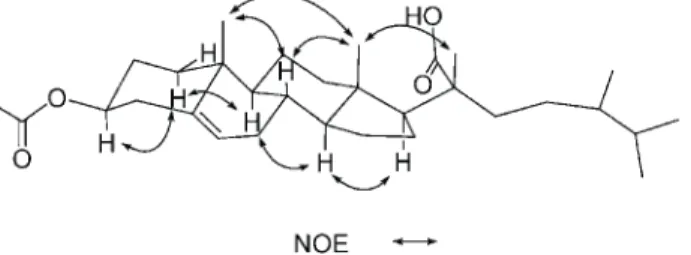
2 is the 3-O-acetyl derivative of 1. This was further sup -ported by the downfield shifts observed for H-3 (d 4.60) and C-3 (d 73.9) relative to those of 1. The planar structure of 2, including the positions of acetoxy group, carboxylate, and the olefinic double bond of this metabolite, could be further deduced from the detailed analyses of the1H-1H COSY, HMQC, and HMBC spectral correlations (Fig. 1). Finally, the relative stereochemistry of 2 was established by the analysis of the NOE correlations in NOESY spec-trum of 2, as illustrated in Fig. 2. The S-configuration at C-24 was confirmed by the close chemical shifts of the side chain carbons in comparison with those of (24S)-3b-hy-droxyergost-5-en-21-oic acid (1).8From the above findings the structure of 2 was thus determined as (24S)-3b-aceto-xyergost-5-en-21-oic acid.
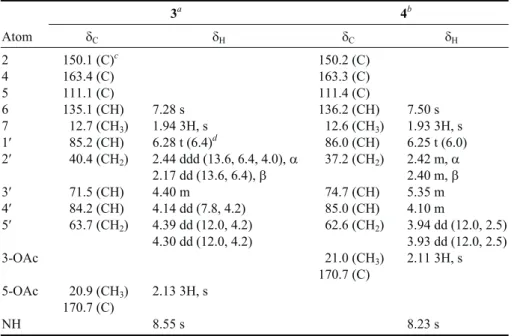
Metabolite 3 was obtained as a pale yellow gum. On the basis of its FABMS (m/z 285, [M+H]+) and NMR spec-tral data (Table 2), the molecular formula of 3 was
sug-gested as C12H16O6N2. The IR spectrum suggested the pres-ence of hydroxy (nmax3445 cm-1, broad) and carbonyl (nmax 1710 and 1690 cm-1) functionalities in 3. The ion peaks ap-pearing in the FABMS at m/z 267 ([M - H2O + H]+) and 207 ([M - H2O - AcOH + H]+) further confirmed the presence of hydroxy and acetoxy groups in 3. The1H NMR spectrum of
3 exhibited three sharp singlets atd 1.94 (3H), 7.28 (1H),
and 8.55 (1H) which were found to be the signals of a methyl substituent at C-5, H-6, and the proton of the sec-ondary amide of thymine moiety, respectively. Also, four sp2carbon signals appearing at d 163.4 (C), 150.1 (C), 135.1 (CH), 111.1 (C), together with a carbon signal atd 12.7 (CH3), were thought to be attributable to a thymine base.9,10The other ten proton signals resonating atd 2.17 and 2.44, 4.14, 4.30 and 4.39, 4.40, 6.28 (each 1H), and 2.13 (3H, s), were found by HMQC spectra to be correlated to the carbon signals atd 40.4 (CH2), 84.2 (CH), 63.7 (CH2), 71.5 (CH), 85.2 (CH), and 20.9 (CH3), respectively, of an acetylated deoxypentose. The above data, together with1H-1H COSY, HMBC and NOESY correlations (see Figs. 3 and 4) further assigned 3 as an acetyl derivative of thymidine. The downfield shifts of the 5¢-oxymethylene carbon and proton signals of 3 relative to those of normal thymidine9,10further confirmed the C-5¢ location of the acetoxy group. Therefore, the structure of compound 3 was determined as 5¢-O-acetylthymidine. It was found that al-though 5¢-O-acetylthymidine (3) has been synthesized pre-viously,11 it was isolated for the first time from natural
Fig. 1. 1H-1H COSY and HMBC correlations for 2.
Fig. 2. Key NOESY correlations of 2.
Fig. 3. 1H-1H COSY and HMBC correlations for 3-5.
sources.
Metabolite 4 was also obtained as a pale yellow gum and showed similar FABMS and IR as those of 3, suggest-ing that 4 is a geometric isomer of 3. The NMR spectral data of 4 (Table 2) also exhibited the characteristic signals of thymine and acetylated 2-deoxyribose units as those found in 3. The downfield shift observed for C-3¢ (Dd + 3.2 ppm) relative to that of 3, which was long-range correlated with H2-2¢ and H2-5¢, clearly indicated the C-3¢ location of the acetoxy group. The above observations combined with 2D-correlations revealed in the1H-1H COSY, HMBC, and NOESY spectra (Figs. 3 and 4) established the structure of
4 as 3¢-O-acetylthymidine. This is the first time that
3¢-O-acetylthymidine has been obtained from a natural source, al-though it has been obtained previously by chemical synthe-sis.12
Metabolite 5 was obtained as a pale yellow oil. This compound was found to possess the molecular formula C9H10O from the EIMS (m/z 134 [M]+) and NMR spectral data (see experimental). The IR (nmax3443 cm-1) indicated the existence of a hydroxy group. In the1H NMR spectrum, the signals appeared atd 6.83 and 7.22 (each 2H, d, J = 8.8 Hz) assigned a p-substituted phenyl moiety. The ABX sys-tem shown by the olefinic protons at d 6.49 (1H, dd, J =16.8, 9.6 Hz), 6.30 (1H, d, J =16.8 Hz), and 6.09 (1H, d, J
= 9.6 Hz) together with a 2H singlet appearing atd 4.17 in-dicated the presence of a vinyl and a hydroxymethyl, re-spectively, as the two para-substituents in the benzene ring. The above findings, together with13C NMR and 2D NMR analysis (Fig. 3), unambiguously established 5 as p-vinylbenzyl alcohol. Although 5 has been prepared pre-viously by a chemical reaction,13our present study affords
5 for the first time from a natural source.
Evaluation of in vitro cytotoxic activity of metabo-lites 1-5 was carried out, and the results are represented in Table 3. Although 3 has been shown to be not cytotoxic, the other four metabolites were found to exhibit cytotoxic ac-tivities against the four cancer cell lines with different po-tencies. The known steroidal carboxylic acid 1 exhibited significant activity against the two liver cancer cell lines Hep G2 and Hep 3B (ED50¢s 2.2 and 2.8 mg/mL, respec-Table 2. 1H and13C NMR chemical shifts of 3 and 4
3a 4b Atom dC dH dC dH 2 150.1 (C)c 150.2 (C) 4 163.4 (C) 163.3 (C) 5 111.1 (C) 111.4 (C) 6 135.1 (CH) 7.28 s 136.2 (CH) 7.50 s 7 012.7 (CH3) 1.94 3H, s 012.6 (CH3) 1.93 3H, s 1¢ 085.2 (CH) 6.28 t (6.4)d 086.0 (CH) 6.25 t (6.0) 2¢ 040.4 (CH2) 2.44 ddd (13.6, 6.4, 4.0),a 037.2 (CH2) 2.42 m,a 2.17 dd (13.6, 6.4),b 2.40 m,b 3¢ 071.5 (CH) 4.40 m 074.7 (CH) 5.35 m 4¢ 084.2 (CH) 4.14 dd (7.8, 4.2) 085.0 (CH) 4.10 m 5¢ 063.7 (CH2) 4.39 dd (12.0, 4.2) 062.6 (CH2) 3.94 dd (12.0, 2.5) 4.30 dd (12.0, 4.2) 3.93 dd (12.0, 2.5) 3-OAc 021.0 (CH3) 2.11 3H, s 170.7 (C) 5-OAc 020.9 (CH3) 2.13 3H, s 170.7 (C) NH 8.55 s 8.23 s
Spectra recordedaat 400 MHz for1H and 100 MHz for13C andbat 500 MHz for1H and 125 MHz for13C, at 25°C in CDCl3,
c
Attached protons were determined by DEPT experiments. d
The J values are in Hz in parentheses.
Table 3. Cytotoxicity (IC50mg/mL) of metabolites 1-5
Hep G2 Hep 3B MCF-7 MDA-MB-231
1 2.2 02.8 14.5 13.3 2 8.6 03.9 29.0 26.4 3 -a - - -4 7.3 27.7 - -5 9.2 16.4 29.0 15.3 a IC50> 30mg/mL.
tively), while its acetate 2 also showed significant cyto-toxicity against Hep 3B (ED503.9mg/mL) and moderate ac-tivity against Hep G2 (ED508.6mg/mL). The 3¢-O-acetyl-atedthymidine (4) exhibited moderate activity against Hep G2 (ED507.3mg/mL) but was inactive against the breast cancer cells (MCF-7 and MDA-MB-231). Metabolite 5 also showed moderate activity against Hep G2 (ED509.2 mg/mL). All of compounds 1, 2, and 5 showed only weak cytotoxicity against the breast cancer cell lines (ED50¢s 13.3-29.0mg/mL).
EXPERIMENTAL SECTION General Experimental Procedures
Melting points were determined using a Fisher-Johns melting point apparatus and were uncorrected. Optical ro-tations were measured on a Jasco DIP-1000 digital polar-imeter. Ultraviolet spectra were recorded on a Hitachi U-3210 UV spectrophotometer, and IR spectra were recorded on a Jasco FT/IR-5300 infrared spectrophotometer. EIMS was obtained with a VG Quattro GC/MS spectrometer. FABMS and HRFABMS spectra were recorded on a Jeol JMS-700 mass spectrometer. The NMR spectra were re-corded on a Bruker AMX-400 FT-NMR or on a Varian Unity INOVA 500 FT-NMR, in CDCl3using TMS as inter-nal standard, unless otherwise indicated. Si gel (Merck, 230-400 mesh) was used for column chromatography. Precoated Si gel plates (Merck, Kieselgel 60 F-254, 0.2 mm) were used for analytical TLC.
Animal Material
The soft coral C. australis was collected at a depth of 15-20 m off the coast of the southernmost tip of Taiwan in 2001, Febrauary and freezed until use. A voucher specimen was deposited at the Department of Marine Biotechnology and Resources, National Sun Yat-sen University.
Extraction and Separation
The EtOAc extract of C. australis was prepared by exhaustive homogenization of the organism (1.6 Kg) with EOAc, which was filtered and concentrated under reduced pressure. The resulting residue (23.5 g) was fractionated as previously described,7using silica gel column chromatog-raphy and EtOAc-n-hexane (stepwise, 0-100% EtOAc) fol-lowed by MeOH-EtOAc (stepwise, 0-50% MeOH) as
elu-tion systems to yield 76 fracelu-tions. Fracelu-tion 42 eluted with EtOAc-n-hexane (1:3), was purified by normal phase HPLC, using EtOAc-n-hexane (1:4) to obtain 2 (3.5 mg). Fraction 54 eluted with EtOAc-n-hexane (2:3) was further purified by column chromatography over silica gel using EtOAc-n-hexane (1:3) to afford 1 (10 mg). Fraction 57 eluted with EtOAc-n-hexane (2:3) was purified by normal phase HPLC using EtOAc-n-hexane (1:3) to give 5 (2 mg). Fraction 71 eluted with EtOAc-n-hexane (7:3) was sub-jected to normal phase HPLC using acetone-n-hexane (gra-dient, 1:4 to 1:3) to give 4 (2.5 mg). The more polar fraction 75 eluted with pure EtOAc was further separated using nor-mal phase HPLC using MeOH-EtOAc (1:99) to yield 3 (3 mg).
(24S)-3b-Hydroxyergost-5-en-21-oic acid (1)
White solid; mp 273-275 °C; [a]D25
-27° (c 1.1, pyridine) (lit.,9-30°); IR (neat) nmax 3399, 2955, 2935, 2870, 1699, 1684, 1541, 1458, 1385, 1232, 1196, 1044 cm-1;1H NMR (C5D5N, 500 MHz) and13C NMR (C5D5N, 125 MHz), see Table 1; FABMS m/z 453 (0.1, [M + Na]+), 431 (0.4, [M + H]+), 413 (0.7, [M - H2O + H]+), 366 (1.4, [M - HCOOH - H2O]+), 341 (2.6, [M - H2O - C5O11]+), 338 (2.1), 329 (0.7), 313 (1.6, [M - H2O - C7O15]+), 307 (5.7), 289 (3.4), 255 (8.5, [M - H2O - C9H17O2]+, 242 (1.6). The above data were found to be in full agreement with those re-ported previously.8
(24S)-3b-Acetoxyergost-5-en-21-oic acid (2)
White solid; mp 213-215 °C; [a]D
25 -25° (c 0.6,
pyridine); IR (neat)nmax3000-3800, 2951, 2934, 2844, 1732, 1699, 1647, 1539, 1456, 1385, 1283, 1037 cm-1;1H NMR (CDCl3, 500 MHz) and13C NMR (CDCl3, 125 MHz), see Table 1; FABMS m/z 495 (0.2, [M + Na]+), 473 (0.3, [M + H]+), 429 (0.4, [M - CO2+ H]+), 413 (9.2, [M - AcOH + H]+), 399 (8.8), 381 (1.2), 367 (1.8, [M - AcOH - HCOOH + H]+), 353 (1.7), 341 (1.8, [M - AcOH - C5O11]+), 313 (1.8, [M - AcOH - C7O15]+), 289 (2.3), 255 (8.5, [M AcOH -C9H17O2]+or/and [M - CO2- AcOH - C8H17]+), 239 (2.4); HRFABMS m/z 473.3632 (calcd for C30H49O4, 473.3633).
5¢-O-Acetylthymidine (3)
Pale yellow gum; [a]D
25-9° (c 0.6, CHCl
3); IR (neat) nmax 3445, 2951, 2930, 2870, 1710, 1690, 1645, 1474, 1375, 1275, 1086, 1049 cm-1;1H NMR (CDCl3, 400 MHz) and13C NMR (CDCl3, 100 MHz), see Table 2; FABMS m/z
285 (3.5, [M + H]+), 267 (4.3, [M - H2O + H]+), 207 (5.3, [M - AcOH - H2O + H]+), 154 (93.7), 136 (100), 107 (40.1).
3¢-O-Acetylthymidine (4)
Pale yellow gum; [a]D
25-21° (c 0.4, CHCl
3); IR (neat) nmax3443, 2951, 2930, 2870, 1710, 1690, 1647, 1474, 1375, 1244, 1096, 1049 cm-1;1H NMR (CDCl3, 500 MHz) and13C NMR (CDCl3, 125 MHz), see Table 2; FABMS m/z 307 (1.9, [M + Na]+), 285 (2.0, [M + H]+), 267 (2.6, [M -H2O + H]+), 207 (2.5, [M - AcOH - H2O + H]+), 154 (98.0), 136 (78.7), 107 (39.1).
p-Vinylbenzyl alcohol (5)
Pale yellow oil; IR (neat)nmax3443 cm-1;1H NMR (CDCl3, 400 MHz),d 7.22 (2H, d, J = 8.8 Hz, H-3,5), 6.83 (2H, d, J = 8.8 Hz, H-2,6), 6.49 (1H, dd, J = 16.8, 9.6 Hz, vi-nyl-H-2), 6.30 (1H, d, J = 16.8 Hz, vinyl-H-1a), 6.09 (1H, d, J = 9.6 Hz, vinyl-H-1b), 4.17 (2H, s) and13C NMR (CDCl3, 100 MHz),d 156.4 (C, C-4), 135.2 (CH, vinyl), 132.3 (2CH, C-2,6), 131.1 (CH2, vinyl), 119.5 (C, C-1), 115.9 (2CH, C-3,5), 60.4 (CH2, hydroxymethyl); EIMS m/z 134 (5.0, [M]+), 107 (39.3, [M - C2H3]+). Cytotoxicity Testing
Cell lines were purchased from the American Type Culture Collection (ATCC). Cytotoxicity assays of the test compounds 1-5 were performed using the MTT [3-(4,5-di-methylthiazole-2-yl)-2,5-diphenyltetrazolium bromide] colorimetric method.14,15
ACKNOWLEDGMENT
This work was supported by a grant from the National Science Council of the Republic of China (Contract No. NSC-93-2323-B-110-002) awarded to J.-H. Sheu.
Received June 10, 2005.
REFERENCES
1. Sheu, J.-H.; Chang, K.-C.; Duh, C.-Y. J. Nat. Prod. 2000, 63, 149-151.
2. Sheu, J.-H.; Hung, K.-C.; Wang, G.-H.; Duh, C.-Y. J. Nat. Prod. 2000, 63, 1603-1607.
3. Sheu, J.-H.; Wang, G.-H.; Sung, P. J.; Duh, C.-Y.; Chiang, M. Y. Tetrahedron 2001, 57, 7639-7648.
4. Wang, G.-H.; Sheu, J.-H.; Duh, C. Y.; Chiang, M. Y. J. Nat. Prod. 2002, 65, 1475-1478.
5. Ahmed, A. F.; Shiue, R.-T.; Wang, G.-H.; Dai, C.-F.; Kuo, Y.-H.; Sheu, J.-H. Tetrahedron 2003, 59, 7337-7344. 6. Ahmed, A. F.; Su, J.-H.; Shiue, R.-T.; Pan, X.-J.; Dai, C.-F.;
Kuo, Y.-H.; Sheu, J.-H. J. Nat. Prod. 2004, 67, 592-597. 7. Ahmed, A. F.; Wu, M.-H.; Wang, G.-H.; Wu, Y.-C.; Sheu,
J.-H. J. Nat. Prod. 2005, 68, 1051-1055.
8. Kobayashi, M.; Haribabu, B.; Anjaneyulu, V. Chem. Pharm. Bull. 1992, 40, 233-234, and other references cited in. 9. Pretsch, E.; Seibel, J.; Simon, W.; Clerc, T. In Spectral Data
for Structure Determination of Organic Compounds; Fresenius, W., Ed.; Springer-Verlag: New York, 1989,13C-NMR, pp C210-C212.
10. Davies, D. B.; Danyluk, S. S. Biochemistry 1974, 13, 4417-4434.
11. Nottoli, E. M.; Lambert, J. B.; Letsinger, R. L. J. Am. Chem. Soc. 1977, 99, 3486-3490.
12. Khan, A. T.; Ghosh, S.; Choudhury, L. H. Eur. J. Org. Chem. 2004, 2198-2204.
13. Abramo, J. G.; Chapin, E. C. J. Org. Chem. 1961, 26, 2671-2673.
14. Alley, M. C.; Scudiero, D. A.; Monks, A.; Hursey, M. L.; Czerwinski, M. J.; Fine, D. L.; Abbott, B. J.; Mayo, J. G.; Shoemaker, R. H.; Boyd, M. R. Cancer Res. 1988, 48, 589-601.
15. Scudiero, D. A.; Shoemaker, R. H.; Paull, K. D.; Monks, A.; Tierney, S.; Nofziger, T. H.; Currens, M. J.; Seniff, D.; Boyd, M. R. Cancer Res. 1988, 48, 4827-4833.