Tuning the emission and morphology of cyclometalated iridium complexes
and their applications to organic light-emitting diodes
Fang-Iy Wu,
aHuei-Jen Su,
aChing-Fong Shu,*
aLiyang Luo,
aWei-Guang Diau,
aChien-Hong Cheng,
bJiun-Pey Duan
band Gene-Hsiang Lee
cReceived 11th October 2004, Accepted 25th November 2004 First published as an Advance Article on the web 10th January 2005 DOI: 10.1039/b415754k
We have synthesized two highly efficient phosphorescent iridium metal complexes,
Ir(DPQ)2(acac) and Ir(FPQ)2(acac), which are based on cyclometalated quinoline ligands, and
discuss details of their electrochemical behavior and photophysical properties (viz. absorption and photoluminescence). Single-crystal X-ray diffraction studies of Ir(DPQ)2(acac) reveal a distorted
octahedral geometry, in which the quinoline N atoms and the C atoms of the orthometalated phenyl groups are located at mutual trans and cis positions, respectively. In contrast, Ir(FPQ)2(acac) is an amorphous solid and undergoes a glass transition at 92uC, which we
attribute to the presence of the long di-n-octyl chains in the fluorenyl groups. The
phosphorescence of these Ir complexes originates from the dominant3MLCT excited state shifts to red that occur upon introducing a phenyl substituent and/or a large conjugating aromatic ring into the ligand. A polymer light-emitting diode (PLED) device that uses Ir(FPQ)2(acac) as a
phosphorescent dopant and a PVK/PBD blend as the host material produces very high efficiency (an external quantum efficiency of 8.16% at 100 mA cm22) and a pure-red emission with 1931 CIE (Commission Internationale de L’Eclairage) chromaticity coordinates of (x = 0.68, y = 0.32).
Introduction
Recently, tremendous efforts have been focused on improving the efficiency of organic light-emitting diodes (OLEDs) through either the development of better materials or improvements in device structure. During the electrical operation of OLEDs, both holes and electrons are injected from opposing electrodes and then they combine together to form singlet and triplet excitons. In a typical fluorescent OLED system, only singlet excitons provide a radiative pathway for electron–hole recombination; the radiative decay of triplet excitons is very inefficient because it is inhibited by the rule of spin conservation.1 Electrophosphorescent
mate-rials incorporating complexes of third-row transition-metal elements have attracted a great deal of attention because of their potential applications as highly efficient electrolumines-cent (EL) emitters.2–6The strong spin–orbit coupling induced by the heavy metal promotes an efficient intersystem crossing from the singlet to the triplet excited state manifold, which then facilitates strong electroluminescence by harnessing both singlet and triplet excitons after the initial charge recombina-tion. EL devices based on these phosphors allow both singlet and triplet excitons to be harvested and the internal efficiency, theoretically, can reach as high as 100%.7
For full-color display applications, red-, green-, and blue-emitting materials that have sufficient luminous efficiency and proper chromaticity must be developed. While great success has been achieved in the development of green phosphorescent materials, the design and synthesis of efficient red emitters is intrinsically more difficult because their luminescence quantum yields tend to decease as the emission wavelength increases in accordance with the energy gap law.8 The
cyclometalated iridium complexes used in EL devices are octahedral, with a 3+ oxidation state, and exhibit strong phosphorescence primarily from a triplet metal-to-ligand charge-transfer (3MLCT) or a ligand-centered 3p–p*
transi-tion.4The emission colors from these complexes, which range from blue to red, are strongly dependent on the choice of the cyclometalating ligand.4,9,10The purpose of the present study was the molecular design of highly efficient red phos-phorescent emitters based on iridium(III) phenylquinoline complexes. By extending the p-electron delocalization of the aromatic ligand chromophore, the energy gap between the ground and lowest excited states can be reduced effectively to provide emitters that have a saturated red color. In addition, these quinoline-based Ir complexes possess relatively short phosphorescence lifetimes that suppress triplet–triplet (TT) annihilation and polaron–triplet (PT) annihilation, which results in improved device quantum efficiency at a high current density.11Furthermore, the incorporation of a 9,9-di-n-octylfluorene group into the ligand gives rise to an amorphous Ir complex that improves the compatibility between the phosphorescent dopant and the polymer host and leads to highly efficient electrophosphorescent polymer light-emitting devices.12
Results and discussion
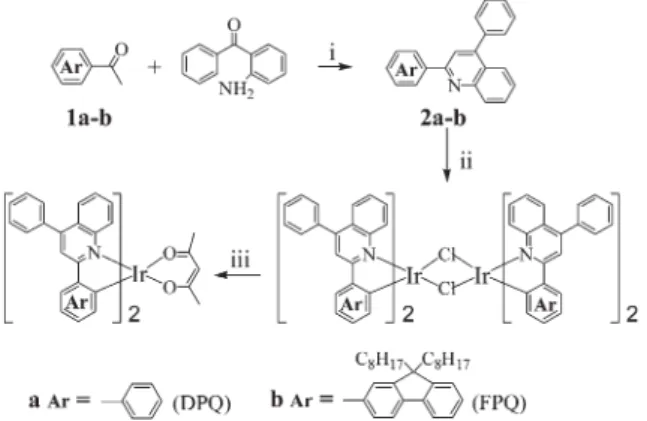
Synthesis and characterizationScheme 1 outlines the synthetic route we followed for the preparation of the iridium complexes. Two quinoline-derived ligands, 2,4-diphenylquinoline (DPQ) and 2-(9,9-di-n-octyl-fluoren-2-yl)-4-phenylquinoline (FPQ), were synthesized
conveniently from the condensation of 2-aminobenzophenone with the corresponding acetyl compounds using the acid-catalyzed Friedla¨nder reaction.13 Subsequent reactions of IrCl3?nH2O with DPQ and FPQ, respectively, in refluxing
ethoxyethanol afforded the cyclometalated chloride-bridged dimers, which were then treated with acetylacetone in the presence of Na2CO3 to give the desired Ir complexes,
Ir(DPQ)2(acac) and Ir(FPQ)2(acac). The chemical structures
of these Ir complexes were characterized by1H and13C NMR spectroscopy, mass spectrometry, and elemental analysis; the three-dimensional structure of Ir(DPQ)2(acac) was further
identified by using single-crystal X-ray crystallographic analysis.
As depicted in Fig. 1, Ir(DPQ)2(acac) possesses a distorted
octahedral geometry around the iridium center, which consists of two cyclometalated DPQ ligands and one acetylacetonate (acac) ligand. The DPQ ligands adopt a mutually eclipsed configuration with a cis-C,C trans-N,N chelate disposition, while the acac ligand is located at a unique position opposite to the carbon atoms of the DPQ ligands. This arrangement of ligands is similar to that found in other mononuclear Ir complexes that possess an ‘‘Ir(ppy)2’’ fragment.
14
Table 1 presents the selected bond lengths (A˚ ) and angles (deg) for this Ir complex. We note that the cyclometalated phenyl fragment is approximately coplanar with the quinoline skeleton—the twist angle is ca. 3.7u—whereas the substituted phenyl group at the C-4 position is strongly deformed out of the plane of the quinoline ring, as evidenced by the large dihedral angle (61.5u).
In contrast to the fact that Ir(DPQ)2(acac) is a crystalline
solid, Ir(FPQ)2(acac) is an amorphous solid, probably because
of the presence of the long and flexible n-octyl chains in the FPQ ligands.4c We investigated the thermal properties of
Ir(FPQ)2(acac) by differential scanning calorimetry (DSC).
Fig. 2 indicates that Ir(FPQ)2(acac) undergoes a glass
transition at 92uC, followed by crystallization at 154 uC and crystalline melting at 261uC. In contrast, there was no phase-transition signal observed for Ir(DPQ)2(acac) from 30 to
300uC.
Electrochemical analysis
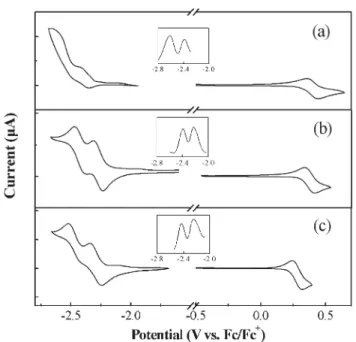
We used cyclic voltammetry, with ferrocene as the internal standard, to investigate (Fig. 3) the electrochemical behavior of these Ir metal complexes and the pristine complex iridium(III) bis(2-phenylquinolyl-N,C2) acetylacetonate [Ir(PQ)2(acac)]. During the cathodic scan in THF, we detected
two reversible reduction processes, with potentials ranging from 22.26 to 22.64 V, for all of these complexes, except for the second reduction of Ir(PQ)2(acac), which is likely to be
Scheme 1 Reagents: (i) DPP/m-cresol; (ii) IrCl3?nH2
O/2-ethoxy-ethanol; (iii) acetylacetone, Na2CO3/2-ethoxyethanol.
Fig. 1 ORTEP drawing of Ir(DPQ)2(acac) with thermal ellipsoids
representing a 50% probability level. Hydrogen atoms have been omitted for clarity.
Table 1 Selected structural parameters for Ir(DPQ)2(acac)
Bond lengths/A˚ Ir(1)–N(1) 2.072(3) Ir(1)–N(2) 2.066(3) Ir(1)–C(1) 1.963(4) Ir(1)–C(22) 1.973(4) Ir(1)–O(1) 2.176(3) Ir(1)–O(2) 2.170(3) Bond angles/deg N(1)–Ir(1)–N(2) 174.2(13) N(1)–Ir(1)–C(1) 79.7(15) N(1)–Ir(1)–O(1) 100.8(13) C(1)–Ir(1)–O(1) 91.1(15) O(1)–Ir(1)–O(2) 85.3(12) Torsion angles/deg C(5)–C(6)–C(7)–C(8) 3.1 C(26)–C(27)–C(28)–C(29) 4.2 C(8)–C(9)–C(16)–C(17) 61.4 C(29)–C(30)–C(37)–C(38) 61.5
Fig. 2 Differential scanning calorimetry (DSC) data for (a) Ir(DPQ)2(acac) and (b) Ir(FPQ)2(acac) (heating rate: 20uC min21)
under nitrogen.
overlapped with the signal for the reduction of the solvent. Additional data provided by differential pulse voltammetry (DPV) also show two distinct reduction processes. Upon the anodic sweep in CH2Cl2, each of these iridium metal
complexes exhibits a reversible oxidation with the oxidative potential falling within the range 0.28–0.40 V. As revealed previously by theoretical calculations and electrochemical studies,15,16 these reductions occur primarily on the
more-electron-accepting heterocyclic portion of the cyclometalated C‘N ligands, whereas the oxidation process is generally considered to largely involve the Ir–phenyl center. This situation is borne out by the fact that the introduction of a phenyl substituent at the C-4 position of the quinoline ring shifts the reductive potentials of Ir(PQ)2(acac) (22.41 and
22.64 V) to less-negative values (22.26 and 22.42 V) observed in Ir(DPQ)2(acac); on the other hand, the oxidative potentials
remain almost unaffected: 0.40 and 0.38 V for the former and latter, respectively. Similarly, replacement of the metalated phenyl fragment with a more-conjugated fluorenyl moiety significantly decreases the oxidative potential from the 0.38 V of Ir(DPQ)2(acac) to the 0.28 V of Ir(FPQ)2(acac), while
inducing only very minor changes in the reduction potentials. Table 2 lists the redox data. On the basis of the onset potentials of the oxidation and reduction, we can estimate the HOMO and LUMO energy levels of these Ir complexes with
regard to the energy level of ferrocene (4.8 eV below vacuum).17Because the energy level of ferrocene is determined by photoelectron spectroscopy in the solid state, this method can be considered to provide merely a rough approximation. Photophysical properties
Fig. 4 displays the absorption and emission spectra measured for Ir(DPQ)2(acac) and Ir(FPQ)2(acac) in THF solution at
298 K. We assign the strong absorption bands in the UV region to the spin-allowed1p–p* transition of the
cyclometa-lated quinoline ligands. Relative to the absorption band of Ir(DPQ)2(acac), we observe a significant bathochromic shift
for Ir(FPQ)2(acac), which results from the extended p
conjugation in the FPQ ligand. The next lowest energy absorption, with peak wavelengths in the region 440–460 nm, can be ascribed to a typical spin-allowed metal-to-ligand charge transfer (1MLCT) transition; we believe the weak bands at long wavelengths are associated with both spin–orbit coupling enhanced 3p–p* and 3MLCT transitions. It is noteworthy that the formally spin forbidden 3MLCT gains
intensity by mixing with the higher-lying 1MLCT transition through the strong spin–orbit coupling on Ir, which results in an intensity that is comparable with the allowed1MLCT.
We observed highly intense photoluminescence (PL) for Ir(DPQ)2(acac) and Ir(FPQ)2(acac) in degassed THF with
values of lmax located at 614 and 625 nm, respectively. The
broad, structureless spectral features lead us to conclude that the phosphorescence originates primarily from the 3MLCT
state.10cIt has been demonstrated that the HOMO and LUMO of cyclometalated complexes of the Ir(ppy)3 type are located
Fig. 3 Cyclic voltammograms of (a) Ir(PQ)2(acac), (b)
Ir(DPQ)2(acac), and (c) Ir(FPQ)2(acac). Inset: The corresponding
differential pulse voltammetry (DPV) in the reduction region.
Table 2 Electrochemical properties of the Ir complexes E1/2 ox /Va E1/2 red /Va Eonset ox /Va Eonset red /Va HOMO/eVb LUMO/eVc Ir(PQ)2(acac) 0.40 22.41, 22.64 0.31 22.30 25.11 22.50 Ir(DPQ)2(acac) 0.38 22.26, 22.42 0.30 22.20 25.10 22.60 Ir(FPQ)2(acac) 0.28 22.27, 22.46 0.21 22.22 25.01 22.58
aPotential values are reported versus Fc/Fc+. bDetermined from the onset oxidation potential. cDetermined from the onset reduction
potential.
Fig. 4 Absorption and PL spectra of the Ir complexes recorded in THF solutions.
mainly at the Ir–phenyl center and the electron-accepting heterocyclic portion of the ligands, respectively.15,16 With respect to Ir(PQ)2(acac), which has an emission maximum at
599 nm, the 4-phenyl substituent in the DPQ ligand leads to a bathochromic shift of ca. 15 nm in the emission peak wavelength, which can be rationalized qualitatively by considering the decrease in the LUMO energy level that results from an increase in the p-conjugation length of the quinoline moiety that is induced by the 4-phenyl group. In comparison with Ir(DPQ)2(acac), which incorporates a
cyclo-metalated phenyl group, Ir(FPQ)2(acac), which bears a
fluorenyl group, reveals an additional ca. 11 nm red shift in the emission maxima that we attribute to the extended p-conjugation raising the HOMO energy level. These observa-tions are in accordance with the results of the electrochemical analysis (vide infra). Table 3 lists the corresponding photo-physical data for the complexes we studied in the solution phase at room temperature. The emission quantum yields in degassed THF solutions excited at 360 nm are 0.14 and 0.11 for Ir(DPQ)2(acac) and Ir(FPQ)2(acac), respectively, relative
to a quinine sulfate (WFL = 0.564) standard. 18
The observed lifetimes in 2-methyltetrahydrofuran at 298 K are 1.33 ms for the former and 1.31 ms for the latter; these values are considerably shorter than that of Ir(btp)2(acac) (5.8 ms).4b
The shorter exciton lifetime, which may suppress the TT annihilation and PT annihilation, leads to an improved device quantum efficiency even at a high current density and makes these Ir complexes attractive candidates as emitting dopants for electrophosphorescent devices.11
Electrophosphorescent light-emitting devices
We have evaluated the potential of these Ir complexes as emissive materials in polymer LED applications using
devices having the configuration ITO/PEDOT/doped emitting layer/TPBI/Mg:Ag/Ag. As the host, we selected a blend of poly(vinylcarbazole) (PVK) and 30 wt% of 2-tert-butylphenyl-5-biphenyl-1,3,4-oxadiazole (PBD). PVK is a blue-emitting material that has good hole transporting properties;19 we blended it with PBD, which is a good electron transporting material, to enable the host to transport both electrons and holes.20A layer of 1,3,5-tris(N-phenylbenzimidazol-2-yl)-benzene (TPBI), an electron-injection/transport layer, was also used at the cathodic side for hole-blocking and exciton-confinement.21 As indicated in Fig. 5, the PL spectrum of
the PVK/PBD host and the absorption spectrum of Ir(FPQ)2(acac) overlap to a reasonable extent in the region
350–550 nm. This overlap should enable efficient Fo¨rster energy transfer from the singlet-excited state in the host to the MLCT band of the guest, Ir(FPQ)2(acac), followed by fast
intersystem crossing to the triplet state of Ir(FPQ)2(acac) and,
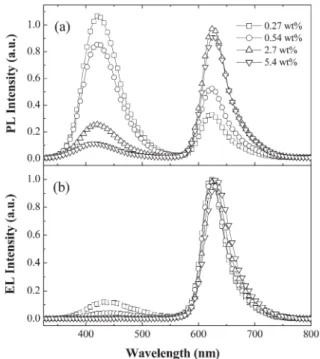
consequently, emission from its triplet state. Fig. 6a presents the extent of energy transfer between the PVK/PBD host and the Ir(FPQ)2(acac) dopant at different doping ratios. The PL
profile of the blend contains two peaks: one centered at 420 nm that originates from the exciplex emission of the PVK/PBD host,22while another peak at ca. 625 nm corresponds to the
triplet emission of Ir(FPQ)2(acac). The PVK/PBD emission at
420 nm reduced significantly upon increasing the dopant Table 3 Photophysical properties of the Ir complexes
Absorbance l/nm (log e)a l
PL,sola/nm Lifetime/msb298 K Q.Y.a,c
Ir(PQ)2(acac) 271 (4.6), 342 (4.1), 432 (3.5), 474 (3.4),553 (3.1) 599 2.0 d 0.12 Ir(DPQ)2(acac) 275 (4.7), 352 (4.3), 441 (3.9), 480 (3.7), 522 (3.6), 564 (3.4) 614 1.33 0.14 Ir(FPQ)2(acac) 313 (4.9), 382 (4.8), 456 (4.0), 500 (3.9), 547 (3.8), 600 (3.4) 625 1.31 0.11 a
Measured in THF. bMeasured in 2-methyltetrahydrofuran. cThe relative quantum yield was measured with reference to quinine sulfate (WF= 0.564 in 1 N sulfuric acid).dDatum obtained from ref. 4b.
Fig. 5 UV–Vis absorption spectrum of Ir(FPQ)2(acac) in THF
solution and PL spectrum of the PVK/PBD host.
Fig. 6 (a) PL spectra of Ir(FPQ)2(acac)-doped films. (b) EL spectra
of Ir(FPQ)2(acac)-doped devices.
concentration, but complete quenching of the emission was not achieved even at a doping concentration as high as 5.4 wt%. In contrast, the EL spectra (Fig. 6b) indicate that at a doping concentration of 2.7 wt% (corresponding to 0.5 mol% per repeating unit of PVK), the dopant emission completely dominates and results in a saturated red triplet emission from the Ir complex. The dramatic difference between the PL and EL spectra indicates that both Fo¨rster energy transfer and direct charge-trapping/recombination on the Ir(FPQ)2(acac)
guest are responsible for the observed EL.12b,23 The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) levels of Ir(FPQ)2(acac) are 25.01
and 22.58 eV, respectively, while the ionization potential and the LUMO of PVK are 25.8 and 22.2 eV,24respectively. As a
result, holes or electrons potentially can be trapped by
Ir(FPQ)2(acac) and, subsequently, they can recombine directly
with opposite charges in the dopant to form either singlet or triplet states. Through intersystem crossing, singlets in the dopants can convert efficiently into triplets and then decay radiatively by phosphorescence.
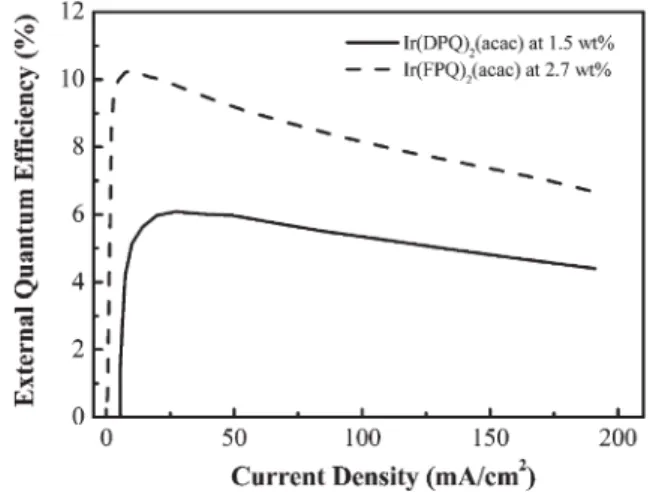
Figs. 7a and b present the current density–voltage and luminance–voltage characteristics of the Ir-doped PLEDs at various doping concentrations. The driving voltages increase as the concentration of Ir(FPQ)2(acac) increases from 0.27 to
5.4 wt%. This observation is also consistent with the charge-trapping mechanism proposed previously. Table 4 summarizes the performances of the Ir-doped devices. The device based on the 2.7 wt% blend becomes turned on at 7.0 V and reaches a maximum brightness of 13824 cd m22at 17 V. The maximum external quantum efficiency of 10.27% is achieved at a current density of 10.69 mA cm22with a brightness of 1180 cd m22.
Fig. 8 shows the external quantum efficiency of the 2.7 wt%-doped device as a function of current density. Even at a higher current density of 100 mA cm22, more than 80% of the peak
efficiency (8.16%) is sustained with a very high brightness (8746 cd m22). We attribute this high performance to the short triplet excited lifetime of Ir(FPQ)2(acac) (ca. 1.3 ms), which
may suppress the TT and PT annihilations and diminish the quenching of the triplet exciton.11A further increase in doping concentration to 5.4 wt% results in a slight decrease in the external quantum and luminance efficiencies, probably as a result of concentration quenching.2aAs indicated in Fig. 9, the EL spectrum, recorded at a bias of 9 V, of a 2.7 wt%-doped device displays a peak at 627 nm that has a full width at half maximum (FWHM) of 48 nm and Commission Internationale de L’Eclairage (CIE) color coordinates of (0.68, 0.32), which are very close to the standard red (0.67, 0.33) demanded by the National Television System Committee (NTSC). We observed no significant host emission even when the applied voltage was raised to 17 V (corresponding to the maximum brightness). Our results for the Ir(FPQ)2(acac)-doped LEDs are, to the
best of our knowledge, among the best reported to date for polymer electrophosphorescent devices that have saturated red emissions.24,25
For the sake of comparison, we also fabricated a device made from 1.5 wt% of Ir(DPQ)2(acac) (corresponding to
0.5 mol% per repeat unit of PVK). The EL spectrum at 9 V has Fig. 7 Plots of (a) current density vs. applied voltage and (b)
luminance vs. applied voltage of ITO/PEDOT/Ir-doped polymer/ TPBI/Mg:Ag devices incorporating different amounts of Ir(FPQ)2(acac).
Table 4 EL performances of devices having the structure ITO/PEDOT/EML/ TPBI/Mg:Ag Ir complexes: PVK-PBD [by weight]
Ir(FPQ)2(acac) Ir(DPQ)2(acac)
0.27 wt% 0.54 wt% 2.7 wt% 5.4 wt% 1.5 wt%
Turn-on voltage/Va 6.2 5.7 7.0 8.5 7.1
Voltage/Vb 9.1 (11.5) 10.0 (12.6) 12.3 (15.2) 15.2 (18.3) 10.5 (13.3)
Brightness/cd m22b 1216 (4087) 1739 (5993) 2153 (8746) 1183 (7186) 2282 (10176)
Luminance efficiency/cd A21b 6.12 (4.09) 8.70 (6.00) 10.77 (8.77) 8.94 (7.20) 11.41 (11.20)
External quantum efficiency (%)b 4.92 (3.29) 7.19 (4.97) 10.02 (8.16) 9.23 (7.44) 5.98 (5.35) External quantum efficiency for 200 cd m22(%) 4.51 8.68 8.66 8.44 1.81
Maximum brightness/cd m22 6725 (@ 14 V) 9739 (@ 15 V) 13824 (@ 17 V) 9025 (@ 19.5 V) 17457 (@ 16 V) Maximum luminance efficiency/cd A21 6.89 10.57 11.04 9.32 11.61
Maximum external quantum efficiency (%) 5.54 8.74 10.27 9.62 6.09
EL maximum/nmd 623 622 627 634 606
CIE coordinates, x and yd 0.60 and 0.30 0.62 and 0.31 0.68 and 0.32 0.68 and 0.32 0.61 and 0.37
aRecorded at 1 cd m22.bRecorded at 20 mA cm22.cThe data in parentheses were recorded at 100 mA cm22.dRecorded at 9 V.
a maximum peak at 606 nm with a FWHM of 59 nm and CIE color coordinates of (0.61, 0.37), which fall in the orange-red region of the CIE chromaticity diagram. The Ir(DPQ)2
(acac)-based device has a maximum brightness of 17457 cd m22; the maximum external quantum efficiency of 6.09%, however, is much lower than that (10.27%) exhibited by the Ir(FPQ)2(acac)-containing device (Fig. 8). Although the two Ir
complexes have similar PL quantum yields in solution, the device prepared from Ir(FPQ)2(acac) exhibits an external
efficiency that is nearly twice that of the Ir(DPQ)2
(acac)-based device at the same doping concentration (mol%). We ascribe this higher efficiency to the amorphous behavior of Ir(FPQ)2(acac), which leads to a more uniform distribution
of the Ir dopant in the PVK host.12 In contrast, Ir(DPQ)2(acac) is a crystalline dopant that may not disperse
well in an amorphous host polymer. Thus, the compatibility between the dopant and the polymer host is an important issue that also must be addressed when attempting to prepare high-efficiency electrophosphorescent polymer light-emitting devices.
Conclusion
We have designed cyclometalated iridium complexes, Ir(DPQ)2(acac) and Ir(FPQ)2(acac), which contain two
cyclo-metalated quinoline ligands and an ancillary acetylacetonate ligand, that are suitable for use as red-emissive materials in PLEDs. In contrast to the fact that Ir(DPQ)2(acac) is a
crystalline solid, Ir(FPQ)2(acac) is an amorphous solid and
undergoes a glass transition at 92uC, which we attribute to the presence of the long di-n-octyl chains in the fluorenyl groups. Our electrochemical studies reveal that the HOMO and LUMO of these Ir complexes are located mainly at the Ir– phenyl center and the electron-accepting heterocyclic portion of the ligands, respectively. By introducing a phenyl sub-stituent and/or a large conjugating aromatic ring into the ligand, the phosphorescence of the Ir complexes, which originates from the dominant 3MLCT excited state, shifts to red. We have demonstrated the preparation of efficient, bright-red electrophosphorescent light-emitting diodes (LEDs) employing Ir(FPQ)2(acac) doped into a blend of PVK and
30 wt% of PBD. The electroluminescence emission is characteristic of Ir(FPQ)2(acac), with a maximum at 627 nm
and CIE color coordinates of (0.68, 0.32); these coordinates are very close to those of the standard red (0.67, 0.33) demanded by the NTSC. At a current density of 10.7 mA cm22
(brightness of 1180 cd m22), the external quantum and luminous efficiencies were 10.27% and 11.0 cd A21, respec-tively. Even at a higher current density of 100 mA cm22, the device maintains a high efficiency (8.16%) and brightness (8746 cd m22).
Experimental
General directions2-Acetyl-9,9-di-n-octylfluorene,26 2,4-diphenylquinoline (DPQ),13a2-phenylquinoline (PQ),27iridium(III) bis(2-phenyl-quinolyl-N,C29) acetylacetonate [Ir(PQ)2(acac)],
4b
and the electron-transport material 1,3,5-tris(N-phenylbenzimidazol-2-yl)benzene (TPBI)28were synthesized as reported previously. Solvents were dried using standard procedures. All other reagents were used as received from commercial sources, unless otherwise stated.1H and13C NMR spectra were recorded on a
Bruker DRX 300 MHz spectrometer. Mass spectra were obtained on a JEOL JMS-SX/SX 110 mass spectrometer. Differential scanning calorimetry (DSC) was performed on a SEIKO EXSTAR 6000DSC unit at a heating rate of 20uC min21
. UV–Visible spectra were measured using an HP 8453 diode-array spectrophotometer. Photoluminescence (PL) spectra were obtained on a Hitachi F-4500 luminescence spectrometer. The emission quantum yields in THF solutions were measured by excitation at 360 nm and were compared with the solution emission of quinine sulfate/1.0 N H2SO4
(WFL = 0.564).18 Solution samples were degassed by three
freeze–pump–thaw cycles. Measurements of oxidation and reduction were undertaken, respectively, in anhydrous CH2Cl2
and anhydrous THF, containing 0.1 M TBAPF6 as the
supporting electrolyte, at scan a rate of 50 mV s21. The
potentials were measured against an Ag/Ag+(0.01 M AgNO3)
reference electrode using ferrocene as the internal standard. Fig. 8 The external quantum efficiency of Ir(FPQ)2(acac)-doped and
Ir(DPQ)2(acac)-doped devices as a function of current density. Both
devices have a doping concentration of 0.5 mol% per repeating unit of PVK.
Fig. 9 EL spectra recorded at different applied voltages of an Ir(FPQ)2(acac)-doped device having a doping concentration of
2.7 wt%.
The onset potentials were determined from the intersection of two tangents drawn at the rising current and background current of the cyclic voltammogram. Lifetime studies were performed using a Nd:YAG laser system operating at 10 Hz (NY60B-10, Continuum). The fundamental frequency at 1064 nm was tripled by a nonlinear optical crystal (THG-T15, Continuum) to generate a laser excitation pulse of 355 nm. The time-resolved emission signal at the selected frequency was collected by a monochromator (SpectraPro-300i, ARC), a photomultiplier tube (R928, Hamamatsu), and a digital oscilloscope (LT372, LeCroy).
2-(9,9-Di-n-octylfluoren-2-yl)-4-phenylquinoline (FPQ) A mixture of 2-acetyl-9,9-di-n-octylfluorene (526 mg, 1.22 mmol), 2-aminobenzophenone (264 mg, 1.34 mmol), diphenyl phosphate (DPP, 1.52 g, 6.08 mmol), and freshly distilled m-cresol (0.93 mL) was flushed with nitrogen while stirring at 25uC for ca. 20 min and then heated under nitrogen at 140uC for 3 h. After cooling, methylene chloride (15 mL) and 10% NaOH (15 mL) were added to the reaction mixture. The organic layer was separated, washed with water (3 6 20 mL), and dried (MgSO4), and then the solvent was
evaporated under vacuum. The residue was purified by column chromatography (hexane–EtOAc, 50 : 1) to afford FPQ as a yellowish liquid (608 mg, 83.9%). 1H NMR (300 MHz, CDCl3): d 0.63 (br, 4 H), 0.75 (t, J = 6.9 Hz, 6 H), 1.02–1.15 (m, 20 H), 1.95–2.07 (m, 4 H), 7.29–7.37 (m, 3 H), 7.47 (t, J = 7.5 Hz, 1 H), 7.52–7.61 (m, 6 H), 7.74–7.76 (m, 2 H), 7.83 (d, J = 7.9 Hz, 1 H), 7.87–7.90 (m, 2 H), 8.12 (s, 1 H), 8.23 (d, J = 8.0 Hz, 2 H).13C NMR (75 MHz, CDCl3): d 14.0, 14.1, 22.5, 23.8, 29.2, 30.0, 31.7, 40.4, 55.3, 119.5, 120.0, 120.1, 121.8, 122.9, 125.6, 125.7, 126.2, 126.7, 126.8, 127.4, 128.4, 128.6, 129.5, 129.6, 130.0, 138.4, 138.5, 140.6, 142.5, 148.8, 149.0, 151.3, 151.3, 157.2. HRMS [M+ + H] calcd. for C44H52N 594.4100, found 594.4099. Preparation of Ir(DPQ)2(acac)
IrCl3?3H2O (338 mg, 1.07 mmol) and H2O (4.0 mL) were
added to a solution of DPQ (750 mg, 2.66 mmol) in 2-ethoxyethanol (12 mL). The mixture was heated at 120uC under nitrogen for 24 h and then cooled to room temperature. The precipitate was washed with ethanol and purified by recrystallization from a mixture of CH2Cl2and ethanol to give
a cyclometalated chloride-bridged dimer, [Ir(DPQ)2Cl]2
(543 mg, 64.6%). A mixture of this iridium dimer (343 mg, 217 mmol), acetylacetone (48 mg, 480 mmol), Na2CO3(230 mg,
2.17 mmol), and 2-ethoxyethanol (30 mL) was heated under reflux for 12 h under a nitrogen atmosphere. After cooling to room temperature, the precipitate was filtered off and washed with water, hexane, and ether, and purified by recrystallization from a mixture of CH2Cl2and ethanol to yield Ir(DPQ)2(acac)
(338 mg, 90.9%).1H NMR (300 MHz, CDCl3): d 1.54 (s, 6 H), 4.70 (s, 1 H), 6.60 (d, J = 7.0 Hz, 2 H), 6.64 (d, J = 7.5 Hz, 2 H), 6.92 (td, J = 2.2, 8.4 Hz, 2 H), 7.38–7.45 (m, 4 H), 7.52–7.68 (m, 10 H), 7.76–7.86 (m, 4 H), 7.80 (s, 2 H), 8.57 (dd, J = 2.1, 7.8 Hz, 2 H).13C NMR (75 MHz, CDCl 3): d 28.3, 100.2, 117.1, 120.9, 122.0, 125.8, 125.8, 126.0, 126.3, 127.0, 128.7, 128.8, 129.0, 129.7, 130.2, 136.2, 137.6, 138.0, 147.2, 148.2, 149.6, 150.0, 150.8, 169.7, 185.6. HRMS [M+] calcd. for C47H36O2N2Ir 853.2406, found 853.2409. Anal. Calcd. for
C47H37O2N2Ir: C, 66.26; H, 4.14; N, 3.29. Found: C, 66.03; H,
4.13; N, 3.56%.
Preparation of Ir(FPQ)2(acac)
The cyclometalated chloride-bridged dimer, [Ir(FPQ)2Cl]2
(453 mg, 78.7%) was prepared from FPQ (462 mg, 77.7 mmol) and IrCl3?3H2O (129 mg, 408 mmol) following the procedure
described for the preparation of [Ir(DPQ)2Cl]2. A mixture of
[Ir(FPQ)2Cl]2 (453 mg, 161 mmol), acetylacetone (35.3 mg,
353 mmol), Na2CO3(170 mg, 1.60 mmol), and 2-ethoxyethanol
(25 mL) was stirred at 25uC under nitrogen for 3 min and then water (20 mL) was added. The precipitate was filtered off, washed with water, and purified by recrystallization from a mixture of CH2Cl2 and ethanol to yield Ir(FPQ)2(acac)
(281 mg, 59.4%). 1H NMR (300 MHz, CDCl 3): d 0.70–0.81 (m, 16 H), 0.96 (br, 20 H), 1.13 (br, 24 H), 1.56 (s, 6H), 1.88– 1.91 (m, 8 H), 4.75 (s, 1 H), 6.97–7.09 (m, 8 H), 1.56 (s, 6 H), 1.90 (m, 8 H), 7.20–7.24 (m, 2 H), 7.38 (br, 4 H), 7.60–7.69 (m, 6 H), 7.75–7.84 (m, 8 H), 8.08 (s, 2 H), 8.60 (m, 2 H). 13C NMR (75 MHz, CDCl3): d 14.0, 14.1,22.5, 22.6, 23.9, 24.1, 28.3, 29.0, 29.2, 29.3, 29.4, 30.0, 30.2, 31.7, 31.9, 40.2, 40.4, 54.1, 100.3, 117.3, 119.9, 120.2, 122.7, 125.4, 125.5, 125.8, 126.0, 126.5, 127.0, 127.1, 128.65, 128.70, 129.8, 130.0, 138.4, 141.0, 141.5, 143.8, 146.0, 149.1, 149.5, 149.8, 152.0, 169.9, 185.5. HRMS (m/z): [M+ + H] calcd. for C93H108O2N2Ir
1477.8040, found 1477.8036. Anal. Calcd. for C93H107O2N2Ir:
C, 75.62; H, 7.30; N, 1.90. Found: C, 75.71; H, 7.55; N, 1.72%. X-Ray structural analysis
Single-crystal X-ray diffraction data were obtained from a Bruker Smart ApexCCD diffractometer using l(Mo-Ka)
radiation (l = 0.71073 A˚ ); data collection was executed using the SMART program. Cell refinement and data reduction were undertaken using the SAINT program. The structure was determined using the SHELXTL/PC program and refined using full-matrix least-squares methods. All non-hydrogen atoms were refined anisotropically, whereas hydrogen atoms were placed at the calculated positions and included in the final stage of refinements with fixed parameters.
Selected crystal data of Ir(DPQ)2(acac): C47H35IrN2O2,
M = 851.97, triclinic, space group P1¯, a = 11.2451(5), b = 11.7178(5), c = 14.2394(6) A˚ , a = 106.876(1), b = 91.234(1), c = 100.583(1)u, V = 1759.35(13) A˚3
, Z = 2, rcalcd= 1.608 mg m23,
F(000) = 848, crystal size = 0.20 6 0.15 6 0.05 mm, l(Mo-Ka) = 0.7107 A˚ , T = 295(2) K, m = 3.839 mm21, 8059
reflections collected (Rint = 0.0505), final R1[I . 2s(I)] =
0.0347 and wR2(all data) = 0.0787.
CCDC reference number 252499. See http://www.rsc.org/ suppdata/jm/b4/b415754k/ for crystallographic data in .cif or other electronic format.
Farbricating electrophosphorescent light-emitting devices Polymer LED devices were fabricated in the configuration ITO/ poly(styrenesulfonate)-doped poly(3,4-ethylenedioxythiophene) (PEDOT) (35 nm)/doped emitting layer (50–70 nm)/TPBI
(30 nm)/Mg:Ag (100 nm)/Ag (100 nm). The PEDOT was spin-coated directly onto the ITO glass and dried at 80 uC for 12 h under vacuum to improve hole injection and sub-strate smoothness. A film of poly(N-vinylcarbazole) (PVK) and 2-(4-biphenylyl)-5-(4-tert-butylphenyl)-1,3,4-oxadiazole (PBD), having a 7 : 3 weight ratio, containing different amounts of Ir(FPQ)2(acac) (0.27, 0.54, 2.7, and 5.4 wt% in
the PVK/PBD host) was spin-coated on top of the PEDT : PSS layer using chlorobenzene as the solvent; the assembly was then dried for 3 h at 60 uC under vacuum. Prior to film casting, the polymer solution was filtered through a Teflon filter (0.45 mm). The TPBI layer was grown by thermal sublimation under vacuum (3 6 1026 Torr); it was used as an electron transporting layer that would also block holes and confine excitons. Subsequently, the cathodic Mg : Ag (10 : 1, 100 nm) alloy was deposited by coevaporation onto the TPBI layer, and then an additional Ag protection layer (100 nm) was placed onto the alloy. The current–voltage– luminance characteristics were measured under ambient conditions using a Keithley 2400 source meter and a Newport 1835C optical meter equipped with an 818ST silicon photodiode.
Acknowledgements
Financial support from the National Science Council of Taiwan is gratefully acknowledged.
Fang-Iy Wu,aHuei-Jen Su,aChing-Fong Shu,*aLiyang Luo,a Wei-Guang Diau,aChien-Hong Cheng,bJiun-Pey Duanband Gene-Hsiang Leec
aDepartment of Applied Chemistry, National Chiao Tung University,
Hsinchu, 300, Taiwan
bDepartment of Chemistry, National Tsing Hua University, Hsinchu,
300, Taiwan
c
Instrumentation Center, College of Science, National Taiwan University, Taipei, 106, Taiwan
References
1 M. Klessinger and J. Michl, Excited States and Photochemistry of Organic Molecules, VCH, New York, 1995.
2 (a) M. A. Baldo, D. F. O’Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151; (b) M. A. Baldo, M. E. Thompson and S. R. Forrest, Pure Appl. Chem., 1999, 71, 2095.
3 (a) R. C. Kwong, S. Sibley, T. Dubovoy, M. Baldo, S. R. Forrest and M. E. Thompson, Chem. Mater., 1999, 11, 3709; (b) C.-M. Che, Y.-J. Hou, M. C. W. Chan, J. Guo, Y. Liu and Y. Wang, J. Mater. Chem., 2003, 13, 1362.
4 (a) V. V. Grushin, N. Herron, D. D. LeCloux, W. J. Marshall, V. A. Petrov and Y. Wang, Chem. Commun., 2001, 1494; (b) S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H.-E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304; (c) J. C. Ostrowski, M. R. Robinson, A. J. Heeger and G. C. Bazan, Chem. Commun., 2002, 784.
5 (a) S. Bernhard, X. Gao, G. G. Malliaras and H. D. Abruna, Adv. Mater., 2002, 14, 433; (b) B. Carlson, G. D. Phelan, W. Kaminsky, L. Dalton, X. Z. Jiang, S. Liu and A. K.-Y. Jen, J. Am. Chem. Soc., 2002, 124, 14162; (c) Y.-L. Tung, P.-C. Wu, C.-S. Liu, Y. Chi, J.-K. Yu, Y.-H. Hu, P.-T. Chou, S.-M. Peng, G.-H. Lee, Y. Tao,
A. J. Carty, C.-F. Shu and F.-I. Wu, Organometallics, 2004, 23, 3745.
6 (a) S. Kan, X. Liu, F. Shen, J. Zhang, Y. Ma, G. Zhang, Y. Wang and J. Shen, Adv. Funct. Mater., 2003, 13, 603; (b) H. Xia, C. Zhang, X. Liu, S. Qiu, P. Lu, F. Shen, J. Zhang and Y. Ma, J. Phys. Chem. B, 2004, 108, 3185.
7 C. Adachi, M. A. Baldo, S. R. Forrest and M. E. Thompson, J. Appl. Phys., 2001, 90, 5048.
8 (a) J. V. Casper and T. J. Meyer, J. Phys. Chem., 1983, 87, 952; (b) S. D. Cummings and R. Eisenberg, J. Am. Chem. Soc., 1996, 118, 1949.
9 (a) C. Adachi, R. C. Kwong, P. Djurovich, V. Adamovich, M. A. Baldo, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 2001, 79, 2082; (b) B. W. D’Andrade, J. Brooks, V. Adamovich, M. E. Thompson and S. R. Forrest, Adv. Mater., 2002, 14, 1032.
10 (a) J.-P. Duan, P.-P. Sun and C.-H. Cheng, Adv. Mater., 2003, 15, 224; (b) Y.-J. Su, H.-L. Huang, C.-L. Li, C.-H. Chien, Y.-T. Tao, P.-T. Chou, S. Datta and R.-S. Liu, Adv. Mater., 2003, 15, 884; (c) A. Tsuboyama, H. Iwawaki, M. Furugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, T. Takiguchi, S. Okada, M. Hoshino and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971.
11 (a) M. A. Baldo, C. Adachi and S. R. Forrest, Phys. Rev. B, 2000, 62, 10967; (b) F.-C. Chen, Y. Yang, M. E. Thompson and J. Kido, Appl. Phys. Lett., 2002, 80, 2308.
12 (a) W. Zhu, Y. Mo, M. Yuan, W. Yang and Y. Cao, Appl. Phys. Lett., 2002, 80, 2045; (b) F.-C. Chen, S.-C. Chang, G. He, S. Pyo, Y. Yang, M. Kurotaki and J. Kido, J. Polym. Sci. Part B: Polym. Phys., 2003, 41, 2681.
13 (a) L. Lu and S. A. Jenekhe, Macromolecules, 2001, 34, 6249; (b) C.-L. Chiang and C.-F. Shu, Chem. Mater., 2002, 14, 682. 14 S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq,
R. Kwong, I. Tsyba, M. Bortz, B. Mui, R. Bau and M. E. Thompson, Inorg. Chem., 2001, 40, 1704.
15 P. J. Hay, J. Phys. Chem. A, 2002, 106, 1634.
16 (a) Y. Ohsawa, S. Sprouse, K. A. King, M. K. DeArmond, K. W. Hanck and R. J. Watts, J. Phys. Chem., 1987, 91, 1047; (b) J. Brooks, Y. Babayan, S. Lamansky, P. I. Djurovich, I. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2002, 41, 3055; (c) A. B. Tamayo, B. D. Alleyne, P. I. Djurovich, S. Lamansky, I. Tsyba, N. N. Ho, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7377.
17 J. Pommerehne, H. Vestweber, W. Guss, R. F. Mahrt, H. Ba¨ssler, M. Porsch and J. Daub, Adv. Mater., 1995, 7, 551.
18 D. F. Eaton, Pure Appl. Chem., 1988, 60, 1107. 19 W. D. Gill, J. Appl. Phys., 1972, 43, 5033.
20 (a) X. Jiang, A. K.-Y Jen, B. Carison and L. R. Dalton, Appl. Phys. Lett., 2002, 81, 3125; (b) X. Gong, J. C. Ostrowski, D. Moses, G. C. Bazan and A. J. Heeger, Adv. Funct. Mater., 2003, 13, 439; (c) X. Yang, D. Neher, D. Hertel and T. K. Da¨ubler, Adv. Mater., 2004, 16, 161.
21 S. W. Culligan, Y. Geng, S. H. Chen, K. Klubek, K. M. Vaeth and C. W. Tang, Adv. Mater., 2003, 15, 1176.
22 X. Gang, S.-H. Lim, J. C. Ostrowski, D. Moses, C. J. Bardeen and G. C. Bazan, Appl. Phys. Lett., 2004, 95, 948.
23 Y.-Y. Noh, C.-L. Lee, J.-J. Kim and K. Yase, J. Chem. Phys., 2003, 118, 2853.
24 Y. Kawamura, S. Yanagida and S. R. Forrest, J. Appl. Phys., 2002, 92, 87.
25 (a) C. Jiang, W. Yang, J. Peng, S. Xiao and Y. Cao, Adv. Mater., 2004, 16, 537; (b) I. Tanank, M. Suzuki and S. Tokito, Jpn. J. Appl. Phys., 2003, 42, 2737; (c) S. Lamansky, P. I. Djurovich, F. Abdel-Razzaq, S. Garon, D. L. Murphy and M. E. Thompson, J. Appl. Phys., 2002, 92, 1570.
26 X.-H. Zhou, J.-C. Yan and J. Pei, Org. Lett., 2003, 5, 3543. 27 H. Gilman, J. Eisch and T. Soddy, J. Am. Chem. Soc., 1957, 79,
1245.
28 J. Shi, C. W. Tang and C. H. Chen, US Patent 5,645,948, 1997.
![Table 4 EL performances of devices having the structure ITO/PEDOT/EML/ TPBI/Mg:Ag Ir complexes: PVK-PBD [by weight]](https://thumb-ap.123doks.com/thumbv2/9libinfo/7973282.158663/5.892.106.416.85.457/table-performances-devices-having-structure-pedot-complexes-weight.webp)